Human Germ Cell Tumors are Developmental Cancers: Impact of Epigenetics on Pathobiology and Clinic

 ,
, 
 ,
,  and
and
Abstract
:1. Introduction: Germ Cell Tumors in General
1.1. Epidemiology
1.2. The ‘Genvironmental’ Model
1.2.1. Genetic Risk Factors
1.2.2. Environmental Risk Factors
Internal Risk Factors
External Risk Factors
1.2.3. Interplay between Environmental and Genetic Risk Factors: The ‘Genvironment’
1.3. Classification
2. Pathobiology of Germ Cell Tumors and their Developmental Potential
2.1. Normal Physiology of Embryonic and Germ Cell Development
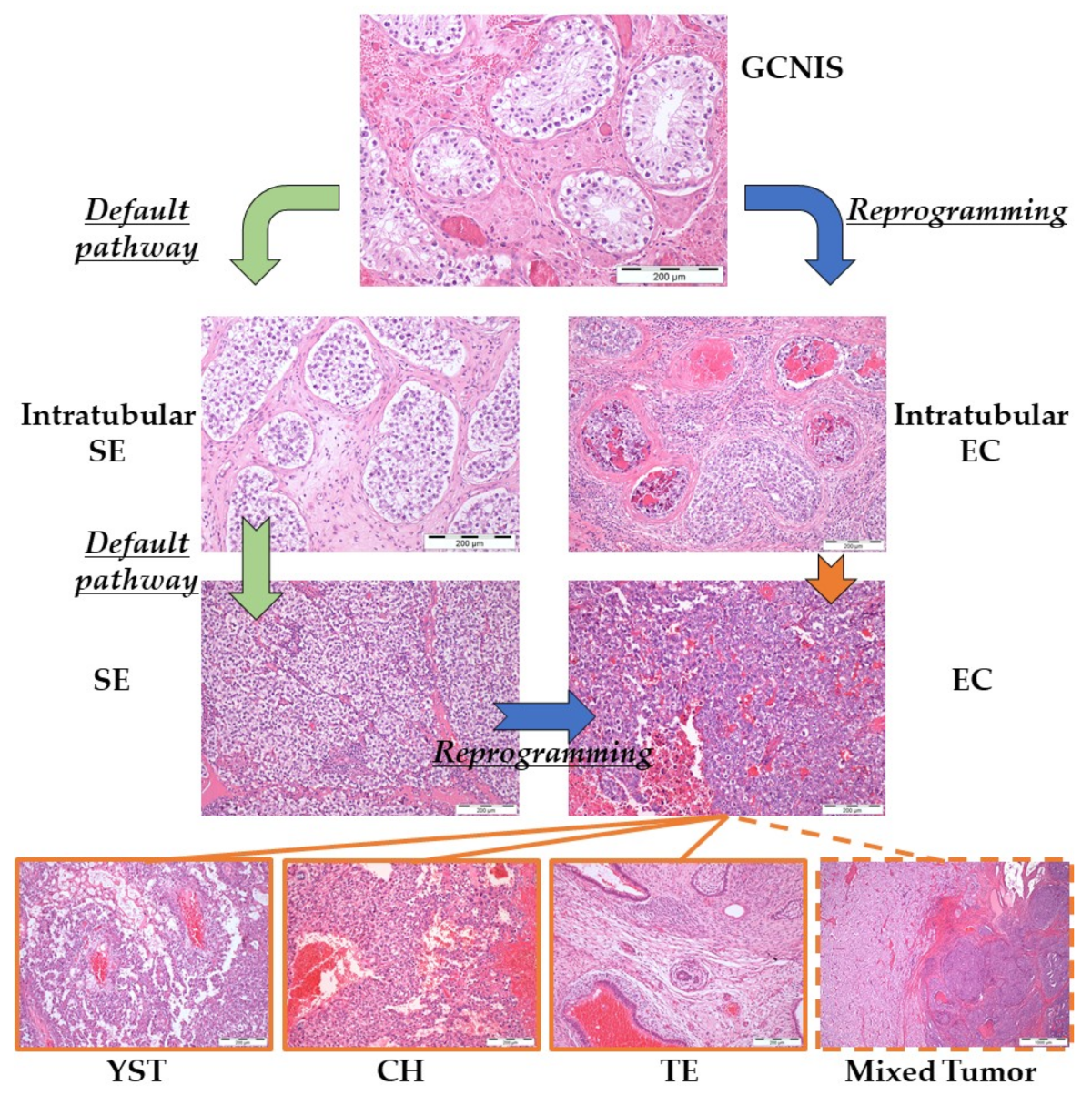
2.2. Type II Germ Cell Tumors of the Testis
2.2.1. Developmental Potential
2.2.2. Brief Pathogenetic Overview
3. Taking Advantage of the Developmental Model: Biomarkers for Clinical Implementation
3.1. Use of High-Throughput Methodologies
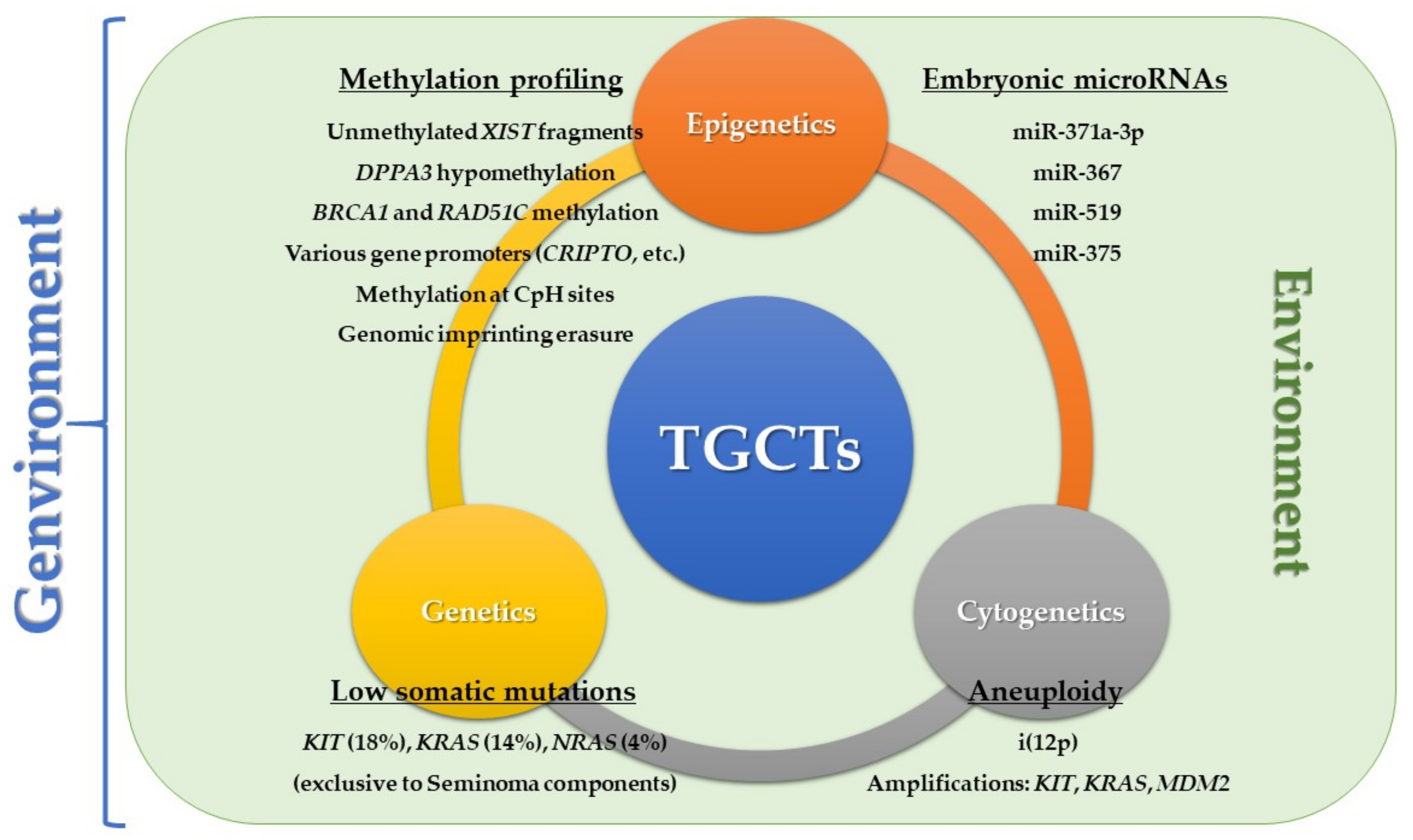
3.2. The Role of Epigenetics
3.2.1. Methylation-Based Biomarkers Relating to the Developmental Model
3.2.2. MicroRNAs Relating to the Developmental Model
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AFP | alpha-fetoprotein |
| AR | androgen receptor |
| β-HCG | human chorionic gonadotropin, subunit β |
| CH | choriocarcinoma |
| CNV | copy number variation |
| COBRA | combined bisulfite restriction analysis |
| DMR | differentially methylated regions |
| DSD | disorders of sex development |
| EC | embryonal carcinoma |
| FTGCT | familial TGCT |
| GCNIS | germ cell neoplasia in situ |
| GCT | germ cell tumor |
| GI | genomic imprinting |
| GWAS | genome wide association |
| hPSC | human pluripotent stem cell |
| HR | homologous recombination |
| LC | lymphoid compensation |
| LDH | lactate dehydrogenase |
| lncRNA | long non-coding RNA |
| MeDIP | methylated DNA immunoprecipitation |
| miR | microRNA |
| ncRNA | non-coding RNA |
| NST | non-seminomatous tumor |
| PCR | polymerase chain reaction |
| PGC | primordial germ cells |
| RNA-seq | RNA sequencing |
| SCNA | somatic copy number aberrations |
| SE | seminoma |
| sncRNA | small non-coding RNA |
| SNP | single nucleotide polymorphisms |
| snRNA | small nuclear RNA |
| ST | spermatocytic tumor |
| TDS | testicular dysgenesis syndrome |
| TE | teratoma |
| TGCT | testicular germ cell tumor |
| TSmiR | targeted serum miR |
| WES | whole exome sequencing |
| WGS | whole genome sequencing |
| XIC | X-chromosome inactivation center |
| XIST | X-inactive specific transcript |
| YST | yolk sac tumor |
References
- Moch, H.; Humphrey, P.; Ulbright, T.; Reuter, V. WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th ed.; IARC: Lyon, France, 2016. [Google Scholar]
- De Angelis, R.; Sant, M.; Coleman, M.P.; Francisci, S.; Baili, P.; Pierannunzio, D.; Trama, A.; Visser, O.; Brenner, H.; Ardanaz, E.; et al. Cancer survival in Europe 1999-2007 by country and age: Results of EUROCARE--5-a population-based study. Lancet Oncol. 2014, 15, 23–34. [Google Scholar] [CrossRef]
- Stang, A.; Trabert, B.; Wentzensen, N.; Cook, M.B.; Rusner, C.; Oosterhuis, J.W.; McGlynn, K.A. Gonadal and extragonadal germ cell tumours in the United States, 1973–2007. Int. J. Androl. 2012, 35, 616–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabert, B.; Chen, J.; Devesa, S.S.; Bray, F.; McGlynn, K.A. International patterns and trends in testicular cancer incidence, overall and by histologic subtype, 1973–2007. Andrology 2015, 3, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [PubMed]
- Surveillance, Epidemiology, and End Results (SEER) Program. Research Data (1973–2015); National Cancer Institute, DCCPS, Surveillance Research Program: Rockville, MD, USA, 2018. [Google Scholar]
- Shah, M.N.; Devesa, S.S.; Zhu, K.; McGlynn, K.A. Trends in testicular germ cell tumours by ethnic group in the United States. Int. J. Androl. 2007, 30, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, S.M.; Lowrance, W.T. Epidemiology and Diagnosis of Testis Cancer. Urol. Clin. N. Am. 2015, 42, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Greiman, A.K.; Rosoff, J.S.; Prasad, S.M. Association of Human Development Index with global bladder, kidney, prostate and testis cancer incidence and mortality. BJU Int. 2017, 120, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Beyer, J.; Albers, P.; Altena, R.; Aparicio, J.; Bokemeyer, C.; Busch, J.; Cathomas, R.; Cavallin-Stahl, E.; Clarke, N.W.; Classen, J.; et al. Maintaining success, reducing treatment burden, focusing on survivorship: Highlights from the third European consensus conference on diagnosis and treatment of germ-cell cancer. Ann. Oncol. 2013, 24, 878–888. [Google Scholar] [CrossRef]
- Curreri, S.A.; Fung, C.; Beard, C.J. Secondary malignant neoplasms in testicular cancer survivors. Urol. Oncol. 2015, 33, 392–398. [Google Scholar] [CrossRef]
- Ostrowski, K.A.; Walsh, T.J. Infertility with Testicular Cancer. Urol. Clin. N. Am. 2015, 42, 409–420. [Google Scholar] [CrossRef]
- Honecker, F.; Aparicio, J.; Berney, D.; Beyer, J.; Bokemeyer, C.; Cathomas, R.; Clarke, N.; Cohn-Cedermark, G.; Daugaard, G.; Dieckmann, K.P.; et al. ESMO Consensus Conference on testicular germ cell cancer: Diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, 1658–1686. [Google Scholar] [CrossRef]
- O’Shaughnessy, M.J.; Feldman, D.R.; Carver, B.S.; Sheinfeld, J. Late Relapse of Testicular Germ Cell Tumors. Urol. Clin. N. Am. 2015, 42, 359–368. [Google Scholar] [CrossRef]
- Hanna, N.H.; Einhorn, L.H. Testicular cancer--discoveries and updates. N. Engl. J. Med. 2014, 371, 2005–2016. [Google Scholar] [CrossRef]
- Rijlaarsdam, M.A.; Looijenga, L.H. An oncofetal and developmental perspective on testicular germ cell cancer. Semin. Cancer Biol. 2014, 29, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Levy, M.; Orlando, G.; Loveday, C.; Law, P.J.; Migliorini, G.; Holroyd, A.; Broderick, P.; Karlsson, R.; Haugen, T.B.; et al. Identification of 19 new risk loci and potential regulatory mechanisms influencing susceptibility to testicular germ cell tumor. Nat. Genet. 2017, 49, 1133–1140. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Albers, P.; Berney, D.M.; Feldman, D.R.; Daugaard, G.; Gilligan, T.; Looijenga, L.H.J. Testicular cancer. Nat. Rev. Dis. Primers 2018, 4, 29. [Google Scholar] [CrossRef]
- Wang, Z.; McGlynn, K.A.; Rajpert-De Meyts, E.; Bishop, D.T.; Chung, C.C.; Dalgaard, M.D.; Greene, M.H.; Gupta, R.; Grotmol, T.; Haugen, T.B.; et al. Meta-analysis of five genome-wide association studies identifies multiple new loci associated with testicular germ cell tumor. Nat. Genet. 2017, 49, 1141–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharazmi, E.; Hemminki, K.; Pukkala, E.; Sundquist, K.; Tryggvadottir, L.; Tretli, S.; Olsen, J.H.; Fallah, M. Cancer Risk in Relatives of Testicular Cancer Patients by Histology Type and Age at Diagnosis: A Joint Study from Five Nordic Countries. Eur. Urol. 2015, 68, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Gundy, S.; Babosa, M.; Baki, M.; Bodrogi, I. Increased predisposition to cancer in brothers and offspring of testicular tumor patients. Pathol. Oncol. Res. 2004, 10, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Kratz, C.P.; Mai, P.L.; Greene, M.H. Familial testicular germ cell tumours. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Rorth, M.; Grigor, K.M.; Jorgensen, N.; Skakkebaek, N.E.; Rajpert-De Meyts, E. Contralateral biopsy in the management of testicular cancer: What we have learned and what we need to improve. Andrology 2015, 3, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, J.W.; Stoop, H.; Dohle, G.; Boellaard, W.; van Casteren, N.; Wolffenbuttel, K.; Looijenga, L.H. A pathologist’s view on the testis biopsy. Int. J. Androl. 2011, 34, e14–e19. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.H.; Mai, P.L.; Loud, J.T.; Pathak, A.; Peters, J.A.; Mirabello, L.; McMaster, M.L.; Rosenberg, P.; Stewart, D.R. Familial testicular germ cell tumors (FTGCT)—Overview of a multidisciplinary etiologic study. Andrology 2015, 3, 47–58. [Google Scholar] [CrossRef]
- Cools, M.; Looijenga, L.H. Tumor risk and clinical follow-up in patients with disorders of sex development. Pediatr. Endocrinol. Rev. 2011, 9, 519–524. [Google Scholar]
- Looijenga, L.H.; Hersmus, R.; Oosterhuis, J.W.; Cools, M.; Drop, S.L.; Wolffenbuttel, K.P. Tumor risk in disorders of sex development (DSD). Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 480–495. [Google Scholar] [CrossRef] [Green Version]
- Pleskacova, J.; Hersmus, R.; Oosterhuis, J.W.; Setyawati, B.A.; Faradz, S.M.; Cools, M.; Wolffenbuttel, K.P.; Lebl, J.; Drop, S.L.; Looijenga, L.H. Tumor risk in disorders of sex development. Sex. Dev. 2010, 4, 259–269. [Google Scholar] [CrossRef]
- Hersmus, R.; van Bever, Y.; Wolffenbuttel, K.P.; Biermann, K.; Cools, M.; Looijenga, L.H. The biology of germ cell tumors in disorders of sex development. Clin. Genet. 2017, 91, 292–301. [Google Scholar] [CrossRef]
- Van der Zwan, Y.G.; Biermann, K.; Wolffenbuttel, K.P.; Cools, M.; Looijenga, L.H. Gonadal maldevelopment as risk factor for germ cell cancer: Towards a clinical decision model. Eur. Urol. 2015, 67, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Looijenga, L.H.; Stoop, H.; Biermann, K. Testicular cancer: Biology and biomarkers. Virchows Arch. 2014, 464, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zwan, Y.G.; Stoop, H.; Rossello, F.; White, S.J.; Looijenga, L.H. Role of epigenetics in the etiology of germ cell cancer. Int. J. Dev. Biol. 2013, 57, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.C.; Kanetsky, P.A.; Wang, Z.; Hildebrandt, M.A.; Koster, R.; Skotheim, R.I.; Kratz, C.P.; Turnbull, C.; Cortessis, V.K.; Bakken, A.C.; et al. Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat. Genet. 2013, 45, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Ruark, E.; Seal, S.; McDonald, H.; Zhang, F.; Elliot, A.; Lau, K.; Perdeaux, E.; Rapley, E.; Eeles, R.; Peto, J.; et al. Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat. Genet. 2013, 45, 686–689. [Google Scholar] [CrossRef] [Green Version]
- Kratz, C.P.; Han, S.S.; Rosenberg, P.S.; Berndt, S.I.; Burdett, L.; Yeager, M.; Korde, L.A.; Mai, P.L.; Pfeiffer, R.; Greene, M.H. Variants in or near KITLG, BAK1, DMRT1, and TERT-CLPTM1L predispose to familial testicular germ cell tumour. J. Med. Genet. 2011, 48, 473–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbull, C.; Rapley, E.A.; Seal, S.; Pernet, D.; Renwick, A.; Hughes, D.; Ricketts, M.; Linger, R.; Nsengimana, J.; Deloukas, P.; et al. Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. Nat. Genet. 2010, 42, 604–607. [Google Scholar] [CrossRef] [Green Version]
- Mayer, F.; Stoop, H.; Sen, S.; Bokemeyer, C.; Oosterhuis, J.W.; Looijenga, L.H. Aneuploidy of human testicular germ cell tumors is associated with amplification of centrosomes. Oncogene 2003, 22, 3859–3866. [Google Scholar] [CrossRef] [PubMed]
- Basten, S.G.; Davis, E.E.; Gillis, A.J.; van Rooijen, E.; Stoop, H.; Babala, N.; Logister, I.; Heath, Z.G.; Jonges, T.N.; Katsanis, N.; et al. Mutations in LRRC50 predispose zebrafish and humans to seminomas. PLoS Genet. 2013, 9, e1003384. [Google Scholar] [CrossRef]
- Litchfield, K.; Levy, M.; Dudakia, D.; Proszek, P.; Shipley, C.; Basten, S.; Rapley, E.; Bishop, D.T.; Reid, A.; Huddart, R.; et al. Rare disruptive mutations in ciliary function genes contribute to testicular cancer susceptibility. Nat. Commun. 2016, 7, 13840. [Google Scholar] [CrossRef] [Green Version]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Main, K.M. Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects. Hum. Reprod. 2001, 16, 972–978. [Google Scholar] [CrossRef]
- Cook, M.B.; Akre, O.; Forman, D.; Madigan, M.P.; Richiardi, L.; McGlynn, K.A. A systematic review and meta-analysis of perinatal variables in relation to the risk of testicular cancer—Experiences of the mother. Int. J. Epidemiol. 2009, 38, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Rajpert-De Meyts, E.; McGlynn, K.A.; Okamoto, K.; Jewett, M.A.; Bokemeyer, C. Testicular germ cell tumours. Lancet 2016, 387, 1762–1774. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Cook, M.B. Etiologic factors in testicular germ-cell tumors. Future Oncol. 2009, 5, 1389–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannandrea, F.; Fargnoli, S. Environmental Factors Affecting Growth and Occurrence of Testicular Cancer in Childhood: An Overview of the Current Epidemiological Evidence. Children 2017, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Trabert, B.; Sigurdson, A.J.; Sweeney, A.M.; Strom, S.S.; McGlynn, K.A. Marijuana use and testicular germ cell tumors. Cancer 2011, 117, 848–853. [Google Scholar] [CrossRef]
- Daling, J.R.; Doody, D.R.; Sun, X.; Trabert, B.L.; Weiss, N.S.; Chen, C.; Biggs, M.L.; Starr, J.R.; Dey, S.K.; Schwartz, S.M. Association of marijuana use and the incidence of testicular germ cell tumors. Cancer 2009, 115, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Friedman, N.B.; Moore, R.A. Tumors of the testis; a report on 922 cases. Mil. Surg. 1946, 99, 573–593. [Google Scholar] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Costa, A.L.; Vilela-Salgueiro, B.; Rodrigues, A.; Guimaraes, R.; Cantante, M.; Lopes, P.; Antunes, L.; Jeronimo, C.; Henrique, R. Testicular germ cell tumors: Revisiting a series in light of the new WHO classification and AJCC staging systems, focusing on challenges for pathologists. Hum. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nogales, F.F.; Jimenez, R.E. Pathology and Biology of Human Germ Cell Tumors; Springer-Verlag GmbH: Berlin, Germany, 2017. [Google Scholar]
- Oosterhuis, J.W.; Looijenga, L.H. Testicular germ-cell tumours in a broader perspective. Nat. Rev. Cancer 2005, 5, 210–222. [Google Scholar] [CrossRef]
- Slack, J.M. Origin of stem cells in organogenesis. Science 2008, 322, 1498–1501. [Google Scholar] [CrossRef]
- Dong, W.L.; Tan, F.Q.; Yang, W.X. Wnt signaling in testis development: Unnecessary or essential? Gene 2015, 565, 155–165. [Google Scholar] [CrossRef]
- Windley, S.P.; Wilhelm, D. Signaling Pathways Involved in Mammalian Sex Determination and Gonad Development. Sex. Dev. 2015, 9, 297–315. [Google Scholar] [CrossRef] [PubMed]
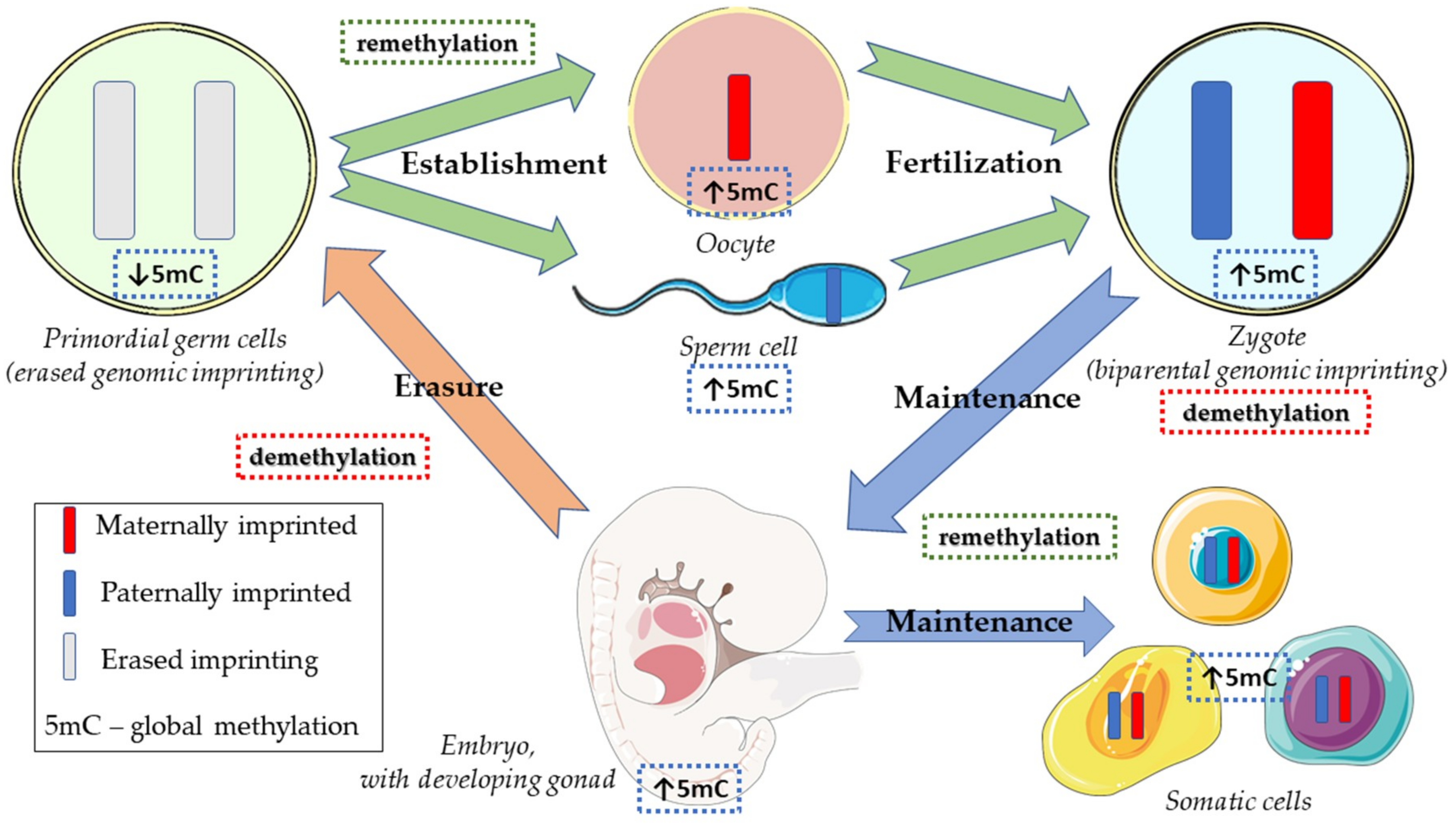
- Ferguson-Smith, A.C.; Bourc’his, D. The discovery and importance of genomic imprinting. eLife 2018, 7, e42368. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [CrossRef]
- Surani, M.A.; Barton, S.C.; Norris, M.L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984, 308, 548–550. [Google Scholar] [CrossRef]
- Barton, S.C.; Surani, M.A.; Norris, M.L. Role of paternal and maternal genomes in mouse development. Nature 1984, 311, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, R.; Feil, R. Genomic imprinting and human disease. Essays Biochem. 2010, 48, 187–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messerschmidt, D.M. Should I stay or should I go: Protection and maintenance of DNA methylation at imprinted genes. Epigenetics 2012, 7, 969–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popp, C.; Dean, W.; Feng, S.; Cokus, S.J.; Andrews, S.; Pellegrini, M.; Jacobsen, S.E.; Reik, W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 2010, 463, 1101–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Duan, J.; Gao, X.; Zhu, W.; Lu, X.; Yang, L.; Zhang, J.; Li, G.; Ci, W.; et al. Programming and inheritance of parental DNA methylomes in mammals. Cell 2014, 157, 979–991. [Google Scholar] [CrossRef]
- Hackett, J.A.; Sengupta, R.; Zylicz, J.J.; Murakami, K.; Lee, C.; Down, T.A.; Surani, M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 2013, 339, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Payer, B.; Lee, J.T.; Namekawa, S.H. X-inactivation and X-reactivation: Epigenetic hallmarks of mammalian reproduction and pluripotent stem cells. Hum. Genet. 2011, 130, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Ohhata, T.; Wutz, A. Reactivation of the inactive X chromosome in development and reprogramming. Cell. Mol. Life Sci. 2013, 70, 2443–2461. [Google Scholar] [CrossRef]
- Seisenberger, S.; Peat, J.R.; Hore, T.A.; Santos, F.; Dean, W.; Reik, W. Reprogramming DNA methylation in the mammalian life cycle: Building and breaking epigenetic barriers. Phil. Trans. R. Soc. B Biol. Sci. 2013, 368, 20110330. [Google Scholar] [CrossRef] [PubMed]
- Buljubasic, R.; Buljubasic, M.; Bojanac, A.K.; Ulamec, M.; Vlahovic, M.; Jezek, D.; Bulic-Jakus, F.; Sincic, N. Epigenetics and testicular germ cell tumors. Gene 2018, 661, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lyu, B.; Roth, L.M. Perspectives on testicular germ cell neoplasms. Hum. Pathol. 2017, 59, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Delahunt, B.; Magi-Galluzzi, C.; Algaba, F.; Egevad, L.; Ulbright, T.M.; Tickoo, S.K.; Srigley, J.R.; Epstein, J.I.; Berney, D.M.; et al. The World Health Organization 2016 classification of testicular germ cell tumours: A review and update from the International Society of Urological Pathology Testis Consultation Panel. Histopathology 2017, 70, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Berney, D.M. Tumors of the Testis: Morphologic Features and Molecular Alterations. Surg. Pathol. Clin. 2015, 8, 687–716. [Google Scholar] [CrossRef] [PubMed]
- Honecker, F.; Stoop, H.; de Krijger, R.R.; Chris Lau, Y.F.; Bokemeyer, C.; Looijenga, L.H. Pathobiological implications of the expression of markers of testicular carcinoma in situ by fetal germ cells. J. Pathol. 2004, 203, 849–857. [Google Scholar] [CrossRef]
- Spoor, J.A.; Oosterhuis, J.W.; Hersmus, R.; Biermann, K.; Wolffenbuttel, K.P.; Cools, M.; Kazmi, Z.; Ahmed, S.F.; Looijenga, L.H.J. Histological Assessment of Gonads in DSD: Relevance for Clinical Management. Sex. Dev. 2018, 12, 106–122. [Google Scholar] [CrossRef]
- Skakkebaek, N.E. Possible carcinoma-in-situ of the testis. Lancet 1972, 2, 516–517. [Google Scholar] [CrossRef]
- Kier, M.G.; Lauritsen, J.; Almstrup, K.; Mortensen, M.S.; Toft, B.G.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Rorth, M.; von der Maase, H.; Agerbaek, M.; et al. Screening for carcinoma in situ in the contralateral testicle in patients with testicular cancer: A population-based study. Ann. Oncol. 2015, 26, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, J.W.; Looijenga, L.H. The biology of human germ cell tumours: Retrospective speculations and new prospectives. Eur. Urol. 1993, 23, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Li, X. Familial risk in testicular cancer as a clue to a heritable and environmental aetiology. Br. J. Cancer 2004, 90, 1765–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oosterhuis, J.W.; Castedo, S.M.; de Jong, B.; Cornelisse, C.J.; Dam, A.; Sleijfer, D.T.; Schraffordt Koops, H. Ploidy of primary germ cell tumors of the testis. Pathogenetic and clinical relevance. Lab. Investig. 1989, 60, 14–21. [Google Scholar] [PubMed]
- Atkin, N.B.; Baker, M.C. Specific chromosome change, i(12p), in testicular tumours? Lancet 1982, 2, 1349. [Google Scholar] [CrossRef]
- Looijenga, L.H.; Zafarana, G.; Grygalewicz, B.; Summersgill, B.; Debiec-Rychter, M.; Veltman, J.; Schoenmakers, E.F.; Rodriguez, S.; Jafer, O.; Clark, J.; et al. Role of gain of 12p in germ cell tumour development. APMIS 2003, 111, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Summersgill, B.; Yost, S.; Sultana, R.; Labreche, K.; Dudakia, D.; Renwick, A.; Seal, S.; Al-Saadi, R.; Broderick, P.; et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat. Commun. 2015, 6, 5973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemmer, K.; Corless, C.L.; Fletcher, J.A.; McGreevey, L.; Haley, A.; Griffith, D.; Cummings, O.W.; Wait, C.; Town, A.; Heinrich, M.C. KIT mutations are common in testicular seminomas. Am. J. Pathol. 2004, 164, 305–313. [Google Scholar] [CrossRef]
- Looijenga, L.H.; de Leeuw, H.; van Oorschot, M.; van Gurp, R.J.; Stoop, H.; Gillis, A.J.; de Gouveia Brazao, C.A.; Weber, R.F.; Kirkels, W.J.; van Dijk, T.; et al. Stem cell factor receptor (c-KIT) codon 816 mutations predict development of bilateral testicular germ-cell tumors. Cancer Res. 2003, 63, 7674–7678. [Google Scholar]
- Dorssers, L.C.; Gillis, A.J.; Stoop, H.; van Marion, R.; Nieboer, M.M.; van Riet, J.; van de Werken, H.J.; Oosterhuis, J.W.; de Ridder, J.; Looijenga, L.H. Molecular Heterogeneity and Early Metastatic Clone Selection in Testicular Germ Cell Cancer Development. bioRxiv 2018. [Google Scholar] [CrossRef]
- Levine, H.; Afek, A.; Shamiss, A.; Derazne, E.; Tzur, D.; Zavdy, O.; Barchana, M.; Kark, J.D. Risk of germ cell testicular cancer according to origin: A migrant cohort study in 1,100,000 Israeli men. Int. J. Cancer 2013, 132, 1878–1885. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic mechanisms in mammals. Cell. Mol. Life Sci. 2009, 66, 596–612. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Peschansky, V.J.; Wahlestedt, C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics 2014, 9, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M. The gift of observation: An interview with Mary Lyon. Interview by Jane Gitschier. PLoS Genet. 2010, 6, e1000813. [Google Scholar]
- Chaligne, R.; Heard, E. X-chromosome inactivation in development and cancer. FEBS Lett. 2014, 588, 2514–2522. [Google Scholar] [CrossRef] [Green Version]
- Gendrel, A.V.; Heard, E. Noncoding RNAs and epigenetic mechanisms during X-chromosome inactivation. Annu. Rev. Cell Dev. Biol. 2014, 30, 561–580. [Google Scholar] [CrossRef]
- Looijenga, L.H.; Gillis, A.J.; van Gurp, R.J.; Verkerk, A.J.; Oosterhuis, J.W. X inactivation in human testicular tumors. XIST expression and androgen receptor methylation status. Am. J. Pathol. 1997, 151, 581–590. [Google Scholar]
- Kawakami, T.; Okamoto, K.; Sugihara, H.; Hattori, T.; Reeve, A.E.; Ogawa, O.; Okada, Y. The roles of supernumerical X chromosomes and XIST expression in testicular germ cell tumors. J. Urol. 2003, 169, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Smiraglia, D.J.; Szymanska, J.; Kraggerud, S.M.; Lothe, R.A.; Peltomaki, P.; Plass, C. Distinct epigenetic phenotypes in seminomatous and nonseminomatous testicular germ cell tumors. Oncogene 2002, 21, 3909–3916. [Google Scholar] [CrossRef]
- Wermann, H.; Stoop, H.; Gillis, A.J.; Honecker, F.; van Gurp, R.J.; Ammerpohl, O.; Richter, J.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Global DNA methylation in fetal human germ cells and germ cell tumours: Association with differentiation and cisplatin resistance. J. Pathol. 2010, 221, 433–442. [Google Scholar] [CrossRef]
- Netto, G.J.; Nakai, Y.; Nakayama, M.; Jadallah, S.; Toubaji, A.; Nonomura, N.; Albadine, R.; Hicks, J.L.; Epstein, J.I.; Yegnasubramanian, S.; et al. Global DNA hypomethylation in intratubular germ cell neoplasia and seminoma, but not in nonseminomatous male germ cell tumors. Mod. Pathol. 2008, 21, 1337–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, T.; Okamoto, K.; Ogawa, O.; Okada, Y. XIST unmethylated DNA fragments in male-derived plasma as a tumour marker for testicular cancer. Lancet 2004, 363, 40–42. [Google Scholar] [CrossRef]
- Looijenga, L.H.; Oosterhuis, J.W. Clinical value of the X chromosome in testicular germ-cell tumours. Lancet 2004, 363, 6–8. [Google Scholar] [CrossRef]
- Ushida, H.; Kawakami, T.; Minami, K.; Chano, T.; Okabe, H.; Okada, Y.; Okamoto, K. Methylation profile of DNA repetitive elements in human testicular germ cell tumor. Mol. Carcinog. 2012, 51, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Beland, F.A. DNA hypomethylation in the origin and pathogenesis of human diseases. Cell. Mol. Life Sci. 2009, 66, 2249–2261. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Vives, L.; Jorda, M.; Morales, C.; Munoz, M.; Vendrell, E.; Peinado, M.A. Genome-wide tracking of unmethylated DNA Alu repeats in normal and cancer cells. Nucleic Acids Res. 2008, 36, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Gonda, T.A.; Gamble, M.V.; Salas, M.; Seshan, V.; Tu, S.; Twaddell, W.S.; Hegyi, P.; Lazar, G.; Steele, I.; et al. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 2008, 68, 9900–9908. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.L.; Lobo, J.; Jeronimo, C.; Henrique, R. The epigenetics of testicular germ cell tumors: Looking for novel disease biomarkers. Epigenomics 2017, 9, 155–169. [Google Scholar] [CrossRef]
- Costa, A.L.; Moreira-Barbosa, C.; Lobo, J.; Vilela-Salgueiro, B.; Cantante, M.; Guimaraes, R.; Lopes, P.; Braga, I.; Oliveira, J.; Antunes, L.; et al. DNA methylation profiling as a tool for testicular germ cell tumors subtyping. Epigenomics 2018. [Google Scholar] [CrossRef]
- Spiller, C.M.; Gillis, A.J.; Burnet, G.; Stoop, H.; Koopman, P.; Bowles, J.; Looijenga, L.H. Cripto: Expression, epigenetic regulation and potential diagnostic use in testicular germ cell tumors. Mol. Oncol. 2016, 10, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Smith-Sorensen, B.; Lind, G.E.; Skotheim, R.I.; Fossa, S.D.; Fodstad, O.; Stenwig, A.E.; Jakobsen, K.S.; Lothe, R.A. Frequent promoter hypermethylation of the O6-Methylguanine-DNA Methyltransferase (MGMT) gene in testicular cancer. Oncogene 2002, 21, 8878–8884. [Google Scholar] [CrossRef] [PubMed]
- Koul, S.; Houldsworth, J.; Mansukhani, M.M.; Donadio, A.; McKiernan, J.M.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.; Murty, V.V. Characteristic promoter hypermethylation signatures in male germ cell tumors. Mol. Cancer 2002, 1, 8. [Google Scholar] [CrossRef]
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol. Cancer 2004, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, C.; Lengert, A.V.H.; Carcano, F.M.; Silva, E.C.A.; Brait, M.; Lopes, L.F.; Vidal, D.O. MGMT and CALCA promoter methylation are associated with poor prognosis in testicular germ cell tumor patients. Oncotarget 2017, 8, 50608–50617. [Google Scholar] [CrossRef] [PubMed]
- Lind, G.E.; Skotheim, R.I.; Fraga, M.F.; Abeler, V.M.; Esteller, M.; Lothe, R.A. Novel epigenetically deregulated genes in testicular cancer include homeobox genes and SCGB3A1 (HIN-1). J. Pathol. 2006, 210, 441–449. [Google Scholar] [CrossRef]
- Cheung, H.H.; Yang, Y.; Lee, T.L.; Rennert, O.; Chan, W.Y. Hypermethylation of genes in testicular embryonal carcinomas. Br. J. Cancer 2016, 114, 230–236. [Google Scholar] [CrossRef]
- Amatruda, J.F.; Ross, J.A.; Christensen, B.; Fustino, N.J.; Chen, K.S.; Hooten, A.J.; Nelson, H.; Kuriger, J.K.; Rakheja, D.; Frazier, A.L.; et al. DNA methylation analysis reveals distinct methylation signatures in pediatric germ cell tumors. BMC Cancer 2013, 13, 313. [Google Scholar] [CrossRef]
- Kato, N.; Tamura, G.; Fukase, M.; Shibuya, H.; Motoyama, T. Hypermethylation of the RUNX3 gene promoter in testicular yolk sac tumor of infants. Am. J. Pathol. 2003, 163, 387–391. [Google Scholar] [CrossRef]
- Kato, N.; Shibuya, H.; Fukase, M.; Tamura, G.; Motoyama, T. Involvement of adenomatous polyposis coli (APC) gene in testicular yolk sac tumor of infants. Hum. Pathol. 2006, 37, 48–53. [Google Scholar] [CrossRef]
- Van der Zwan, Y.G.; Rijlaarsdam, M.A.; Rossello, F.J.; Notini, A.J.; de Boer, S.; Watkins, D.N.; Gillis, A.J.; Dorssers, L.C.; White, S.J.; Looijenga, L.H. Seminoma and embryonal carcinoma footprints identified by analysis of integrated genome-wide epigenetic and expression profiles of germ cell cancer cell lines. PLoS ONE 2014, 9, e98330. [Google Scholar] [CrossRef]
- De Jong, J.; Weeda, S.; Gillis, A.J.; Oosterhuis, J.W.; Looijenga, L.H. Differential methylation of the OCT3/4 upstream region in primary human testicular germ cell tumors. Oncol. Rep. 2007, 18, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Biermann, K.; Gillis, A.J.; Steger, K.; Looijenga, L.H.; Schorle, H. NANOG promoter methylation and expression correlation during normal and malignant human germ cell development. Epigenetics 2011, 6, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brait, M.; Maldonado, L.; Begum, S.; Loyo, M.; Wehle, D.; Tavora, F.F.; Looijenga, L.H.; Kowalski, J.; Zhang, Z.; Rosenbaum, E.; et al. DNA methylation profiles delineate epigenetic heterogeneity in seminoma and non-seminoma. Br. J. Cancer 2012, 106, 414–423. [Google Scholar] [CrossRef]
- Nettersheim, D.; Arndt, I.; Sharma, R.; Riesenberg, S.; Jostes, S.; Schneider, S.; Holzel, M.; Kristiansen, G.; Schorle, H. The cancer/testis-antigen PRAME supports the pluripotency network and represses somatic and germ cell differentiation programs in seminomas. Br. J. Cancer 2016, 115, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Markulin, D.; Vojta, A.; Samarzija, I.; Gamulin, M.; Beceheli, I.; Jukic, I.; Maglov, C.; Zoldos, V.; Fucic, A. Association Between RASSF1A Promoter Methylation and Testicular Germ Cell Tumor: A Meta-analysis and a Cohort Study. Cancer Genom. Proteom. 2017, 14, 363–372. [Google Scholar]
- Rijlaarsdam, M.A.; Tax, D.M.; Gillis, A.J.; Dorssers, L.C.; Koestler, D.C.; de Ridder, J.; Looijenga, L.H. Genome wide DNA methylation profiles provide clues to the origin and pathogenesis of germ cell tumors. PLoS ONE 2015, 10, e0122146. [Google Scholar] [CrossRef] [PubMed]
- Killian, J.K.; Dorssers, L.C.; Trabert, B.; Gillis, A.J.; Cook, M.B.; Wang, Y.; Waterfall, J.J.; Stevenson, H.; Smith, W.I., Jr.; Noyes, N.; et al. Imprints and DPPA3 are bypassed during pluripotency- and differentiation-coupled methylation reprogramming in testicular germ cell tumors. Genome Res. 2016, 26, 1490–1504. [Google Scholar] [CrossRef] [Green Version]
- Von Meyenn, F.; Reik, W. Forget the Parents: Epigenetic Reprogramming in Human Germ Cells. Cell 2015, 161, 1248–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveros-Etter, M.; Li, Z.; Nee, K.; Hosohama, L.; Hargan-Calvopina, J.; Lee, S.A.; Joti, P.; Yu, J.; Clark, A.T. PGC Reversion to Pluripotency Involves Erasure of DNA Methylation from Imprinting Control Centers followed by Locus-Specific Re-methylation. Stem Cell Rep. 2015, 5, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leao, R.; Nayan, M.; Punjani, N.; Jewett, M.A.S.; Fadaak, K.; Garisto, J.; Lewin, J.; Atenafu, E.G.; Sweet, J.; Anson-Cartwright, L.; et al. A New Model to Predict Benign Histology in Residual Retroperitoneal Masses After Chemotherapy in Nonseminoma. Eur. Urol. Focus. 2018. [Google Scholar] [CrossRef] [PubMed]
- Noor, D.A.M.; Jeyapalan, J.N.; Alhazmi, S.; Carr, M.; Squibb, B.; Wallace, C.; Tan, C.; Cusack, M.; Hughes, J.; Reader, T.; et al. Genome-wide methylation analysis identifies genes silenced in non-seminoma cell lines. NPJ Genom. Med. 2016, 1, 15009. [Google Scholar] [CrossRef]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef] [PubMed]
- Eini, R.; Dorssers, L.C.; Looijenga, L.H. Role of stem cell proteins and microRNAs in embryogenesis and germ cell cancer. Int. J. Dev. Biol. 2013, 57, 319–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfossi, S.; Babayan, A.; Pantel, K.; Calin, G.A. Clinical utility of circulating non-coding RNAs—An update. Nat. Rev. Clin. Oncol. 2018, 15, 541–563. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.; Li, T.; Huang, Y.; Sun, J.; Zhu, Y.; Yang, Y.; Lu, Z.J. RNA Biomarkers: Frontier of Precision Medicine for Cancer. Noncoding RNA 2017, 3, e9. [Google Scholar] [CrossRef]
- Dominguez-Vigil, I.G.; Moreno-Martinez, A.K.; Wang, J.Y.; Roehrl, M.H.A.; Barrera-Saldana, H.A. The dawn of the liquid biopsy in the fight against cancer. Oncotarget 2018, 9, 2912–2922. [Google Scholar] [CrossRef]
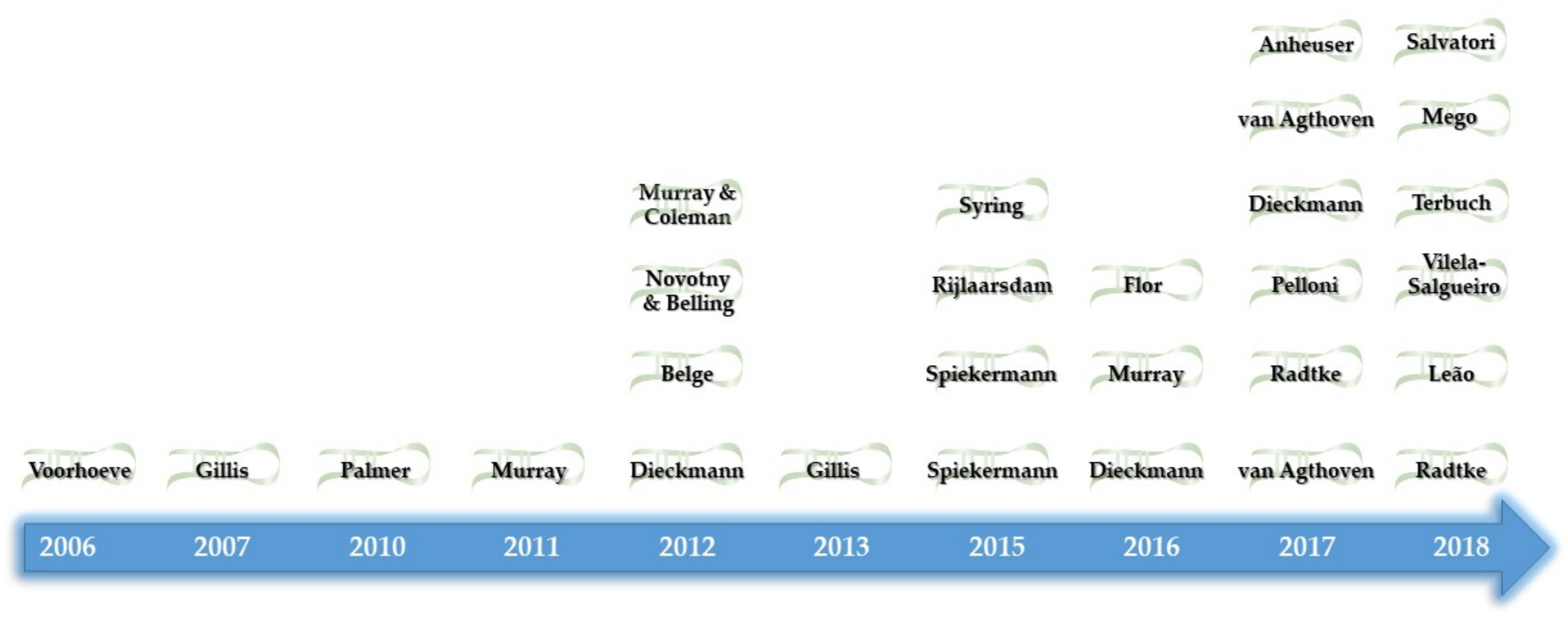
- Henrique, R.; Jeronimo, C. Testicular Germ Cell Tumors Go Epigenetics: Will miR-371a-3p Replace Classical Serum Biomarkers? Eur. Urol. 2017, 71, 221–222. [Google Scholar] [CrossRef]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.J.; Stoop, H.J.; Hersmus, R.; Oosterhuis, J.W.; Sun, Y.; Chen, C.; Guenther, S.; Sherlock, J.; Veltman, I.; Baeten, J.; et al. High-throughput microRNAome analysis in human germ cell tumours. J. Pathol. 2007, 213, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.D.; Murray, M.J.; Saini, H.K.; van Dongen, S.; Abreu-Goodger, C.; Muralidhar, B.; Pett, M.R.; Thornton, C.M.; Nicholson, J.C.; Enright, A.J.; et al. Malignant germ cell tumors display common microRNA profiles resulting in global changes in expression of messenger RNA targets. Cancer Res. 2010, 70, 2911–2923. [Google Scholar] [CrossRef]
- Novotny, G.W.; Belling, K.C.; Bramsen, J.B.; Nielsen, J.E.; Bork-Jensen, J.; Almstrup, K.; Sonne, S.B.; Kjems, J.; Rajpert-De Meyts, E.; Leffers, H. MicroRNA expression profiling of carcinoma in situ cells of the testis. Endocr. Relat. Cancer 2012, 19, 365–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilela-Salgueiro, B.; Barros-Silva, D.; Lobo, J.; Costa, A.L.; Guimaraes, R.; Cantante, M.; Lopes, P.; Braga, I.; Oliveira, J.; Henrique, R.; et al. Germ cell tumour subtypes display differential expression of microRNA371a-3p. Phil. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170338. [Google Scholar] [CrossRef]
- Van Agthoven, T.; Looijenga, L.H.J. Accurate primary germ cell cancer diagnosis using serum based microRNA detection (ampTSmiR test). Oncotarget 2017, 8, 58037–58049. [Google Scholar] [CrossRef] [PubMed]
- Van Agthoven, T.; Eijkenboom, W.M.H.; Looijenga, L.H.J. microRNA-371a-3p as informative biomarker for the follow-up of testicular germ cell cancer patients. Cell. Oncol. 2017, 40, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, M.J.; Halsall, D.J.; Hook, C.E.; Williams, D.M.; Nicholson, J.C.; Coleman, N. Identification of microRNAs From the miR-371–373 and miR-302 clusters as potential serum biomarkers of malignant germ cell tumors. Am. J. Clin. Pathol. 2011, 135, 119–125. [Google Scholar] [CrossRef]
- Belge, G.; Dieckmann, K.P.; Spiekermann, M.; Balks, T.; Bullerdiek, J. Serum levels of microRNAs miR-371-3: A novel class of serum biomarkers for testicular germ cell tumors? Eur. Urol. 2012, 61, 1068–1069. [Google Scholar] [CrossRef]
- Murray, M.J.; Coleman, N. Testicular cancer: A new generation of biomarkers for malignant germ cell tumours. Nat. Rev. Urol. 2012, 9, 298–300. [Google Scholar] [CrossRef]
- Syring, I.; Bartels, J.; Holdenrieder, S.; Kristiansen, G.; Muller, S.C.; Ellinger, J. Circulating serum miRNA (miR-367-3p, miR-371a-3p, miR-372-3p and miR-373-3p) as biomarkers in patients with testicular germ cell cancer. J. Urol. 2015, 193, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Flor, I.; Spiekermann, M.; Loning, T.; Dieckmann, K.P.; Belge, G.; Bullerdiek, J. Expression of microRNAs of C19MC in Different Histological Types of Testicular Germ Cell Tumour. Cancer Genom. Proteom. 2016, 13, 281–289. [Google Scholar]
- Dieckmann, K.P.; Spiekermann, M.; Balks, T.; Flor, I.; Loning, T.; Bullerdiek, J.; Belge, G. MicroRNAs miR-371-3 in serum as diagnostic tools in the management of testicular germ cell tumours. Br. J. Cancer 2012, 107, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, A.J.; Rijlaarsdam, M.A.; Eini, R.; Dorssers, L.C.; Biermann, K.; Murray, M.J.; Nicholson, J.C.; Coleman, N.; Dieckmann, K.P.; Belge, G.; et al. Targeted serum miRNA (TSmiR) test for diagnosis and follow-up of (testicular) germ cell cancer patients: A proof of principle. Mol. Oncol. 2013, 7, 1083–1092. [Google Scholar] [CrossRef]
- Anheuser, P.; Radtke, A.; Wulfing, C.; Kranz, J.; Belge, G.; Dieckmann, K.P. Serum Levels of MicroRNA371a-3p: A Highly Sensitive Tool for Diagnosing and Staging Testicular Germ Cell Tumours: A Clinical Case Series. Urol. Int. 2017, 99, 98–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rijlaarsdam, M.A.; Van Agthoven, T.; Gillis, A.J.; Patel, S.; Hayashibara, K.; Lee, K.Y.; Looijenga, L.H. Identification of known and novel germ cell cancer-specific (embryonic) miRs in serum by high-throughput profiling. Andrology 2015, 3, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Spiekermann, M.; Dieckmann, K.P.; Balks, T.; Bullerdiek, J.; Belge, G. Is relative quantification dispensable for the measurement of microRNAs as serum biomarkers in germ cell tumors? Anticancer Res. 2015, 35, 117–121. [Google Scholar] [PubMed]
- Spiekermann, M.; Belge, G.; Winter, N.; Ikogho, R.; Balks, T.; Bullerdiek, J.; Dieckmann, K.P. MicroRNA miR-371a-3p in serum of patients with germ cell tumours: Evaluations for establishing a serum biomarker. Andrology 2015, 3, 78–84. [Google Scholar] [CrossRef]
- Pelloni, M.; Coltrinari, G.; Paoli, D.; Pallotti, F.; Lombardo, F.; Lenzi, A.; Gandini, L. Differential expression of miRNAs in the seminal plasma and serum of testicular cancer patients. Endocrine 2017, 57, 518–527. [Google Scholar] [CrossRef]
- Dieckmann, K.P.; Spiekermann, M.; Balks, T.; Ikogho, R.; Anheuser, P.; Wosniok, W.; Loening, T.; Bullerdiek, J.; Belge, G. MicroRNA miR-371a-3p—A Novel Serum Biomarker of Testicular Germ Cell Tumors: Evidence for Specificity from Measurements in Testicular Vein Blood and in Neoplastic Hydrocele Fluid. Urol. Int. 2016, 97, 76–83. [Google Scholar] [CrossRef]
- Murray, M.J.; Bell, E.; Raby, K.L.; Rijlaarsdam, M.A.; Gillis, A.J.; Looijenga, L.H.; Brown, H.; Destenaves, B.; Nicholson, J.C.; Coleman, N. A pipeline to quantify serum and cerebrospinal fluid microRNAs for diagnosis and detection of relapse in paediatric malignant germ-cell tumours. Br. J. Cancer 2016, 114, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Dieckmann, K.P.; Radtke, A.; Spiekermann, M.; Balks, T.; Matthies, C.; Becker, P.; Ruf, C.; Oing, C.; Oechsle, K.; Bokemeyer, C.; et al. Serum Levels of MicroRNA miR-371a-3p: A Sensitive and Specific New Biomarker for Germ Cell Tumours. Eur. Urol. 2017, 71, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radtke, A.; Cremers, J.F.; Kliesch, S.; Riek, S.; Junker, K.; Mohamed, S.A.; Anheuser, P.; Belge, G.; Dieckmann, K.P. Can germ cell neoplasia in situ be diagnosed by measuring serum levels of microRNA371a-3p? J. Cancer Res. Clin. Oncol. 2017, 143, 2383–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terbuch, A.; Adiprasito, J.B.; Stiegelbauer, V.; Seles, M.; Klec, C.; Pichler, G.P.; Resel, M.; Posch, F.; Lembeck, A.L.; Stoger, H.; et al. MiR-371a-3p Serum Levels Are Increased in Recurrence of Testicular Germ Cell Tumor Patients. Int. J. Mol. Sci. 2018, 19, 3130. [Google Scholar] [CrossRef] [PubMed]
- Leao, R.; van Agthoven, T.; Figueiredo, A.; Jewett, M.A.S.; Fadaak, K.; Sweet, J.; Ahmad, A.E.; Anson-Cartwright, L.; Chung, P.; Hansen, A.; et al. Serum miRNA Predicts Viable Disease after Chemotherapy in Patients with Testicular Nonseminoma Germ Cell Tumor. J. Urol. 2018, 200, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Radtke, A.; Hennig, F.; Ikogho, R.; Hammel, J.; Anheuser, P.; Wulfing, C.; Belge, G.; Dieckmann, K.P. The Novel Biomarker of Germ Cell Tumours, Micro-RNA-371a-3p, Has a Very Rapid Decay in Patients with Clinical Stage 1. Urol. Int. 2018, 100, 470–475. [Google Scholar] [CrossRef]
- Mego, M.; Agthoven, T.V.; Gronesova, P.; Chovanec, M.; Miskovska, V.; Mardiak, J.; Looijenga, L. Clinical utility of plasma miR-371a-3p in testicular germ cell tumors. J. Clin. Oncol. 2018, 36, e16540. [Google Scholar] [CrossRef]
- Mego, M.; Agthoven, T.V.; Gronesova, P.; Chovanec, M.; Miskovska, V.; Mardiak, J.; Looijenga, L. Clinical utility of plasma miR-371a-3p in germ cell tumors. J. Cell. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Salvatori, D.C.F.; Dorssers, L.C.J.; Gillis, A.J.M.; Perretta, G.; van Agthoven, T.; Gomes Fernandes, M.; Stoop, H.; Prins, J.B.; Oosterhuis, J.W.; Mummery, C.; et al. The MicroRNA-371 Family as Plasma Biomarkers for Monitoring Undifferentiated and Potentially Malignant Human Pluripotent Stem Cells in Teratoma Assays. Stem Cell Rep. 2018, 11, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.J.; Goldmann, J.; Loser, P.; Loring, J.F. A call to standardize teratoma assays used to define human pluripotent cell lines. Cell Stem Cell. 2010, 6, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Damjanov, I.; Andrews, P.W. Teratomas produced from human pluripotent stem cells xenografted into immunodeficient mice—A histopathology atlas. Int. J. Dev. Biol. 2016, 60, 337–419. [Google Scholar] [CrossRef] [PubMed]
- International Stem Cell Initiative. Assessment of established techniques to determine developmental and malignant potential of human pluripotent stem cells. Nat. Commun. 2018, 9, 1925. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics | Context | Source |
|---|---|---|
| Age adjusted incidence rates: 64/1,000,000 (males) versus 4/1,000,000 (females) | Germ cell tumors | Europe (EUROCARE) |
| Incidence rates: 0.8% rise/year Estimated new cases: 5.7/100,000/year (all males, 2011–2015) | Testicular cancer | United States of America (SEER) |
| Age-standardized incidence rate: 1.7/100,000 (all males) versus 2.7/100,000 (males aged 15–39 years) 5-year prevalence: 150,377 cases (males aged 15–39 years) Estimated new cases (85,635) and deaths (13,288) in 2040 (all males) | Testicular cancer | World (Globocan) |
| Factor | Relative Risk OR |
|---|---|
| Genetic | |
| Familial risk Brother with TGCT Father with TGCT | 8–10 xs 4–6 xs |
| Studies in twins Monozygotic twins Dizygotic twins | 76 xs 35 xs |
| Contralateral tumor | 24.8–27.6 |
| Various SNPs KITLG-related | OR >2 or <0.5 |
| Environmental | |
| Internal | |
| Cryptorchidism | 3.5–17.1 |
| Infertility | 1.16–6.72 |
| Hypospadias | 1.26–3.61 |
| Atrophy | 20.5 |
| Previous inguinal hernia | 1.63 |
| Microlithiasis | 3.42–13.2 |
| Disturbed hormonal conditions in utero (maternal bleeding, first born child, low and high birthweight, short gestational age) | ~1.3 |
| Low birthweight | OR 1.28 |
| Number of siblings ≥5 | OR 0.71 |
| External | |
| High body mass index | ↑/↓/- |
| High stature | ↑/- |
| Late onset of puberty | ↓ |
| Diet high in fat and dairy products | ↑ |
| Low physical exercise | ↑/↓/- |
| Firefighters, metal/leather/agricultural workers | ↑ |
| Testicular trauma | ↑ |
| Marijuana smoking | OR 1.7 |
| GCT Type | Age Group | Sex | Site | Phenotype | Developmental State | Precursor Cell | GI |
|---|---|---|---|---|---|---|---|
| 0 | Neonates | F/M | Midline | Included and parasitic twins | Omnipotent (2C state) | Blastomere | BiP |
| I | <6 years | F/M | Gonads, midline | TE, YST | Pluripotent (primed state) | Methylated PGC/gonocyte | BiP to partially erased |
| II | Postpubertal | >>M | Gonads, midline | SE/Dysg, NST | Totipotent (naïve state) | Hypomethylated PGC/gonocyte | Erased |
| III | >55 years | M | Testis | ST | Spermatogonium to premeiotic spermatocyte | Spermatogonium/spermatocyte | Partially to complete paternal |
| IV | Postpubertal | F | Ovary | Dermoid cyst | Maternally imprinted 2C state | Oogonia / oocyte | Partially to complete Maternal |
| V | Postpubertal | F | Placenta, uterus | Hydatidiform mole | Paternally imprinted 2C state | Empty ovum / spermatozoa | Completely paternal |
| VI | >60 years | F/M | Ovary, atypical sites | Resembling type I or NST components of type II | Primed state or NST lineages of naïve state | Somatic cell induced to pluripotency | Pattern of originating cell |
| Methodology | Sample Type | Major Findings | Year | Author |
|---|---|---|---|---|
| Methylation | ||||
| Bisulfite sequencing; PCR | Tissues (n = 31 TGCTs) and plasma (n = 25 TGCT samples, n = 24 non-TGCT samples) | XIST region IV frequently unmethylated in TGCTs, especially in SEs | 2004 | Kawakami et al. |
| Bisulfite sequencing; COBRA | Tissues (n = 14 TGCTs, n = 10 adjacent testicular parenchyma, n = 3 non-TGCTs) and TGCT cell lines | LINE1 hypomethylated in both SEs and NSTs; XIST and CDH1 mainly hypomethylated in SEs and methylated in NSTs | 2011 | Ushida et al. |
| qMS-PCR | Tissues (n = 161 TGCTs, n = 16 controls) | Differential methylation of CRIPTO, HOXA9, MGMT, RASSF1A and SCGB3A1 gene promoters among TGCT subtypes | 2018 | Costa et al. |
| Genome-wide DNA methylation analysis | Tissues (n = 130 TGCTs, n = 128 benign neighboring testes) | DPPA3 is hypomethylated in both SEs and NSTs; hypermethylation of HM13 in NSTs and subtype-specific hypermethylation of H19 in TEs | 2016 | Killian et al. |
| Genome-wide DNA methylation analysis | Tissues (n = 91 GCTs) and GCT cell lines | SEs, dysgerminomas and STs are globally hypomethylated, while ECs, NSTs and type I TEs are hypermethylated | 2015 | Rijlaarsdam et al. |
| Genome-wide DNA methylation analysis; RT-qPCR | GCT cell lines | Localized hypermethylation status in YSTs vs. disperse hypermethylation status in ECs and TEs | 2015 | Noor et al. |
| MeDIP; DNA-tiling hybridization; RT-qPCR; IHC | Tissues (n = 6 ECs) | Hypermethylated DMRs in ECs (X- and Y-linked genes, genes related to metabolism) | 2016 | Cheung et al. |
| Genome-wide DNA methylation analysis | Tissues (n = 137 TGCTs) | ECs display methylation at CpH sites; methylation of BRCA1 and RAD51C silencing in NSTs | 2018 | Shen et al. |
| MicroRNAs | ||||
| miR library | NA | miR-372 and miR-373 netralize p53 (oncomiRs) | 2006 | Voorhoeve et al. |
| High-throughput screening of 156 miRs; qPCR | GCT tissues (n = 69) and cell lines | Relevance of miR-371–373 cluster; association with differentiation | 2007 | Gillis et al. |
| High-throughput screening of 615 miRs; RT-qPCR | Pediatric malignant GCTs, controls and GCT cell lines (n = 48) | Overexpression of miR-371~373 and miR-372 clusters in all tumor subtypes | 2010 | Palmer et al. |
| Multiplex PCR | Serum (n = 1) of a four-year-old boy | First report of utility of serum miRs in GCTs (miR-371–373 and miR-302 clusters); decrease after treatment | 2011 | Murray et al. |
| RT-qPCR | Serum (n = 12 patients, n = 11 controls) | Overexpression of miR-371-3 in patients and decrease after treatment | 2012 | Belge et al. |
| RT-qPCR | Serum (n = 8 malignant GCTs) | Additional specificity of using miR-367-3p | 2012 | Murray and Coleman |
| RT-qPCR | Serum (n = 24 GCTs, n = 17 controls) and GCT tissues (n = 15) | miR-371~373 measured in TVB in 6 patients (higher levels); no correlation with levels in tissues | 2012 | Dieckmann et al. |
| miR array; RT-qPCR | GCNIS tissue samples (n = 12) | Identification of miRs unique to GCNIS cells | 2012 | Novotny and Belling et al. |
| TSmiR | Serum (n = 80 GCTs, n = 47 controls, n = 12 non-GCT masses) | miR-371/372/373/367 panel with 98% sensitivity in diagnosis; higher expression levels in metastatic patients | 2013 | Gillis et al. |
| RT-qPCR | Serum (testing cohort: n = 30 patients and n = 18 controls; validation cohort: n = 76 patients, n = 84 controls) | miR-367-3p, miR-371a-3p, miR372-3p and miR-373-3p overexpressed in patients; miR-371-a-3p showing 84.7% sensitivity and 99% specificity in diagnosis | 2015 | Syring et al. |
| RT-qPCR | Serum (n = 25 GCTs, 6 GCNIS, n = 24 non-testicular malignancies, n = 20 controls), seminal plasma (n = 5), urine (n = 3) and pleural effusions (n = 1) | miR-371a-3p detected in seminal plasma and pleural effusions, but not in urine; confirmation of its value in serum | 2015 | Spiekermann et al. |
| High-throughput screening of 750 miRs; RT-qPCR | Serum (n = 14 GCTs, n = 11 controls) | Confirmation of the relevance of miR-371–373 cluster; novel relevant miRs identified | 2015 | Rijlaarsdam et al. |
| RT-qPCR | Serum (n=25 TGCTs, n=4 non-TGCTs, n = 17 controls) | Suggestion that normalization (relative quantification) is not required when quantifying miR-371-3 | 2015 | Spiekermann |
| RT-qPCR | Serum and cerebral spinal fluid (n = 45 each) of 25 pediatric patients | Four serum microRNA panel (miR-371a-3p, miR-372-3p, miR-373-30 and miR-367-3p) with high sensitivity and specificity in discriminating intracranial GCT vs. non-GCT malignancies; first demonstration of relapse detection | 2016 | Murray et al. |
| RT-qPCR | GCT tissues and serum (n = 25 patients) | C19MC cluster overexpressed in aggressive subtypes | 2016 | Flor et al. |
| RT-qPCR | Tumor surrounding hydroceles (n = 9) and serum (n = 64 GCTs) | Hydroceles showing high levels of miR-371a-3p; association with tumor size; confirmed the value of miR-371a-3p in follow-up (relapse detection) | 2016 | Dieckmann et al. |
| ampTSmiR | Serum (n = 250 TGCTs, n = 60 non-TGCTs, n = 104 controls) | Largest series tested; panel composed of miR-371a-3p, miR-373-3p and miR-367-3p with 90% sensitivity and 91% specificity | 2017 | van Agthoven et al. |
| RT-qPCR | Serum (n = 312 consecutive patients with testicular disease) | Elevated levels aided in detection of clinically silent GCTs and metastases | 2017 | Anheuser et al. |
| RT-qPCR | Serum and seminal plasma (n = 48 patients, n = 28 controls) | miR-371a-3p suggested as a poor biomarker in seminal plasma, contrarily to miR-142 | 2017 | Peloni et al. |
| RT-qPCR | Serum (n = 166 GCTs, n = 106 controls) | miR-371a-3p shows the best performance in TGCT detection (88.7% sensitivity, 93.4% specificity) | 2017 | Dieckmann et al. |
| RT-qPCR | Serum (n = 27 GCNIS) | miR-371a-3p overexpressed in GCNIS patients | 2017 | Radtke et al. |
| ampTSmiR | Serum (n = 1 SE, n = 5 NST) of patients with relapse/residual disease | miR-371a-3p outperformed classical protein markers in detection of disease relapse, except for mature TE | 2017 | van Agthoven et al. |
| RT-qPCR | Tissues (n = 119 TGCTs, n = 15 controls) | miR-371a-3p discriminated TGCTs from controls with 92% sensitivity and 93% specificity; decreasing expression with tumor differentiation; TEs discriminated from controls | 2018 | Vilela-Salgueiro et al. |
| ampTSmiR | Serum (n = 82 TGCTs) | miR-371a-3p discriminates viable disease post-chemotherapy (AUC = 0.87) | 2018 | Leão et al. |
| RT-qPCR | Serum (24 TGCTs, clinical stage I) | miR-371a-3p has a very short half-life (<12 h) | 2018 | Radtke et al. |
| RT-qPCR | Serum (n = 10 TGCT patients with relapse) | Confirmed miR-371a-3p value in detecting relapses | 2018 | Terbuch et al. |
| ampTSmiR | Plasma (n = 199 TGCTs, before chemotherapy) | miR-371a-3p predicts prognosis in chemotherapy naïve patients | 2018 | Mego et al. |
| Teratoma assay (mouse model) | Plasma of mice | Value of miR-371 family members in detecting undifferentiated and potentially malignant elements present in xenografts | 2018 | Salvatori et al. |
| miR-sequencing data | Tissues (n = 137 TGCTs) | miR-519 cluster overexpressed in ECs; miR-375 overexpressed in TEs and YSTs | 2018 | Shen et al. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobo, J.; Gillis, A.J.M.; Jerónimo, C.; Henrique, R.; Looijenga, L.H.J. Human Germ Cell Tumors are Developmental Cancers: Impact of Epigenetics on Pathobiology and Clinic. Int. J. Mol. Sci. 2019, 20, 258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020258
Lobo J, Gillis AJM, Jerónimo C, Henrique R, Looijenga LHJ. Human Germ Cell Tumors are Developmental Cancers: Impact of Epigenetics on Pathobiology and Clinic. International Journal of Molecular Sciences. 2019; 20(2):258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020258
Chicago/Turabian StyleLobo, João, Ad J. M. Gillis, Carmen Jerónimo, Rui Henrique, and Leendert H. J. Looijenga. 2019. "Human Germ Cell Tumors are Developmental Cancers: Impact of Epigenetics on Pathobiology and Clinic" International Journal of Molecular Sciences 20, no. 2: 258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020258