Selection of Membrane RNA Aptamers to Amyloid Beta Peptide: Implications for Exosome-Based Antioxidant Strategies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
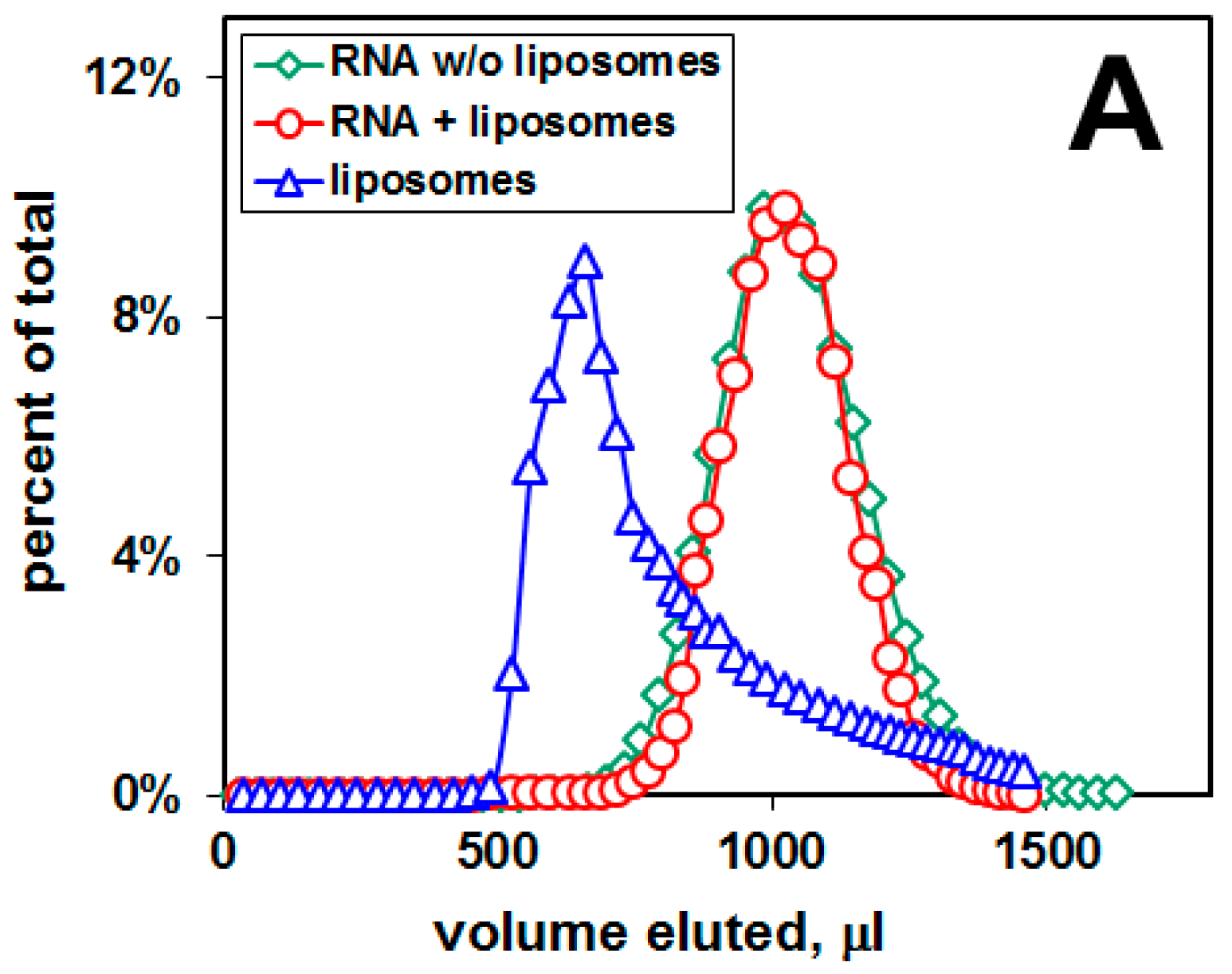
2.1. Distribution of Aβ42 and RNA between Liposomes and Buffer Solution
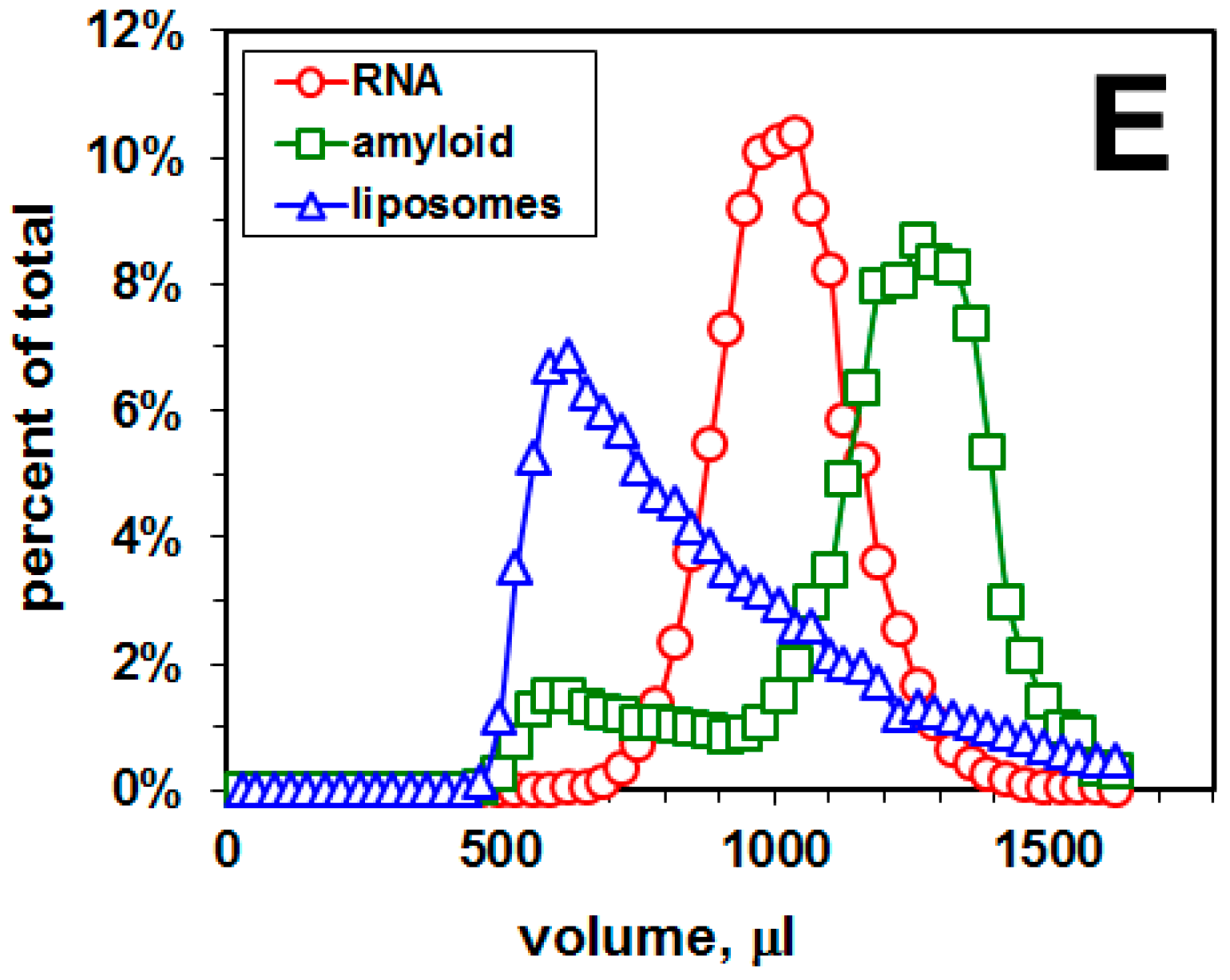
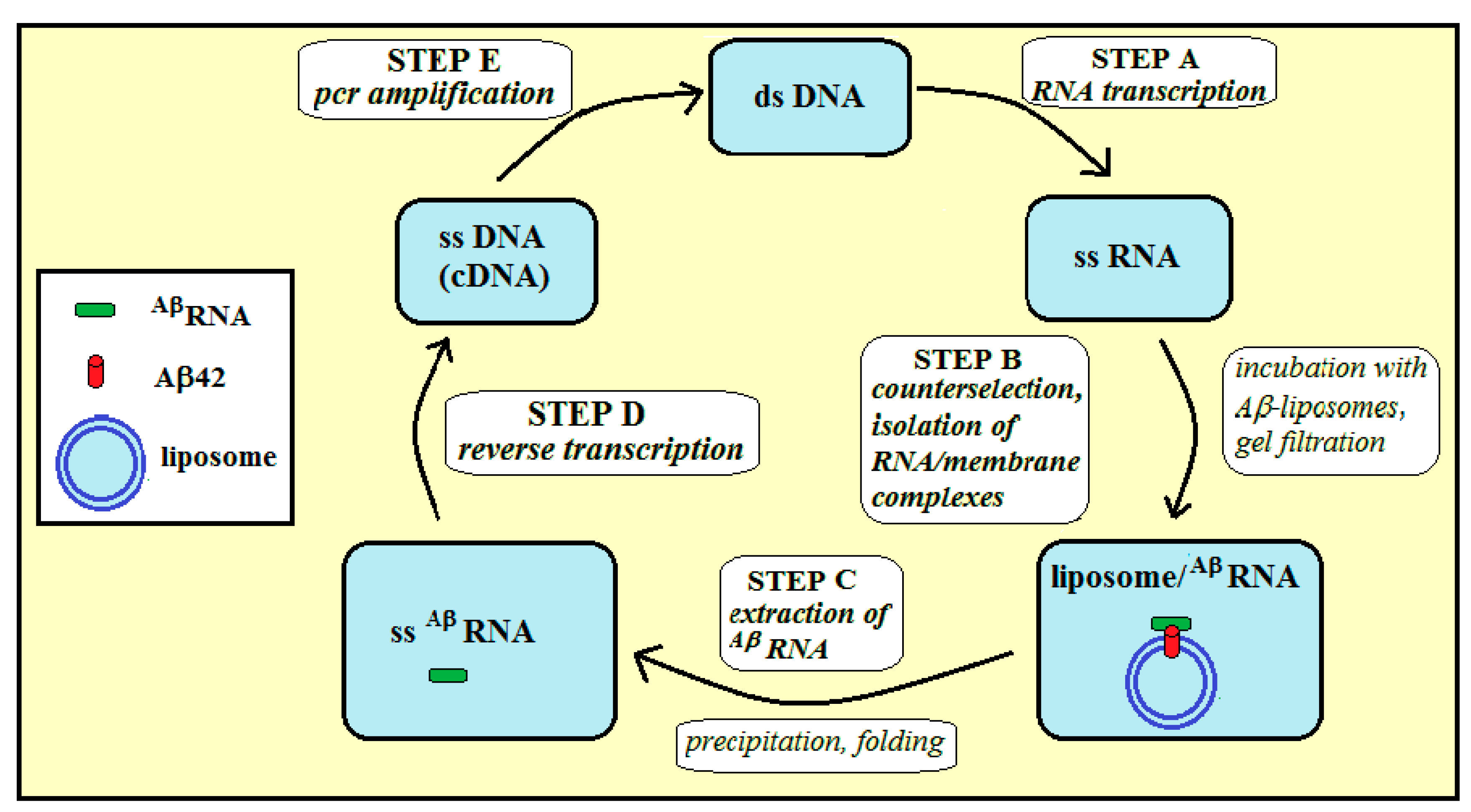
2.2. Selection of a Pool of RNA Aptamers to Model Membranes Containing Aβ42
3. Discussion
3.1. Membrane Affinity of Aβ Peptide
3.2. RNA Aptamers for Membrane Aβ42 Peptide
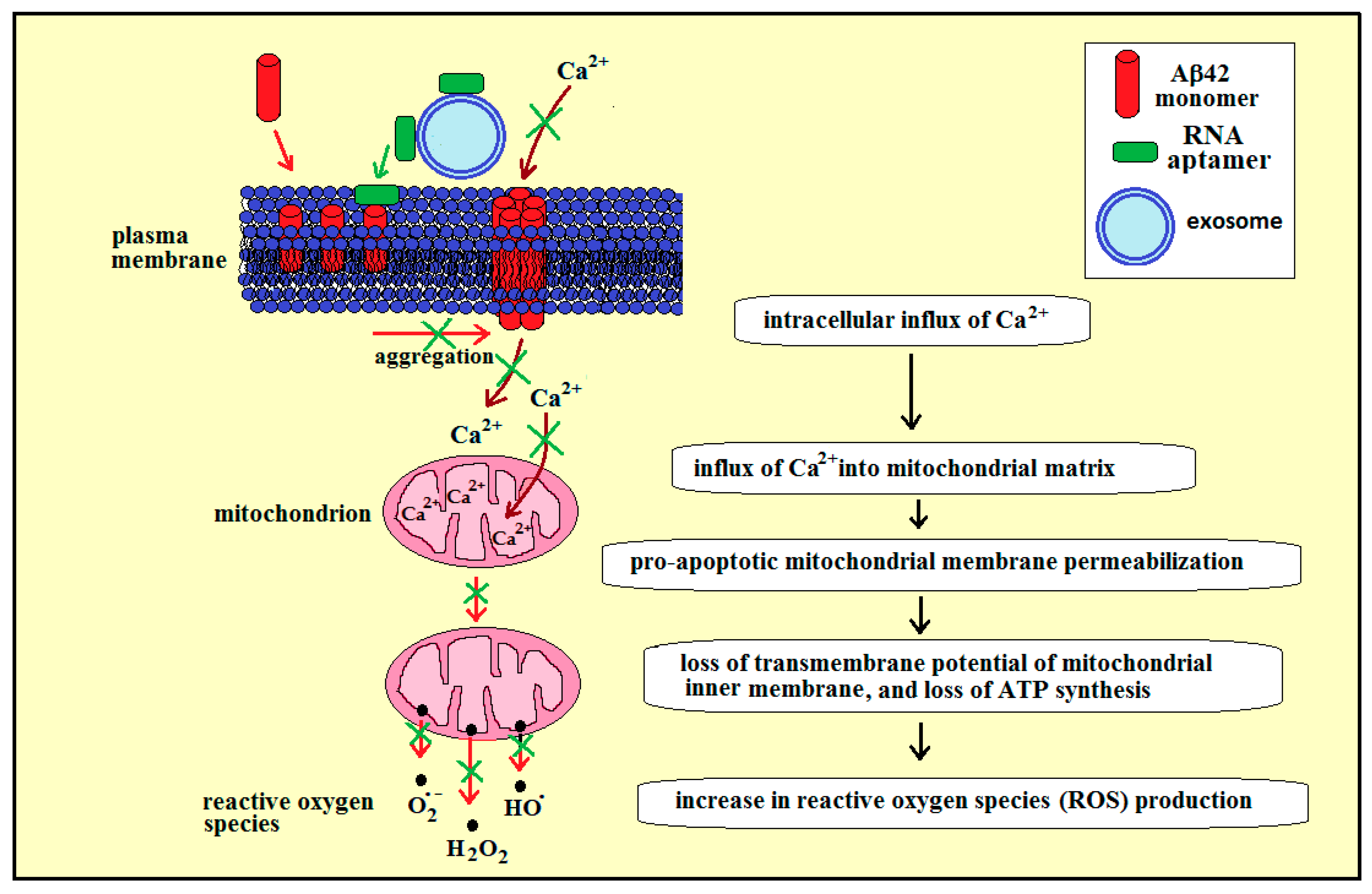
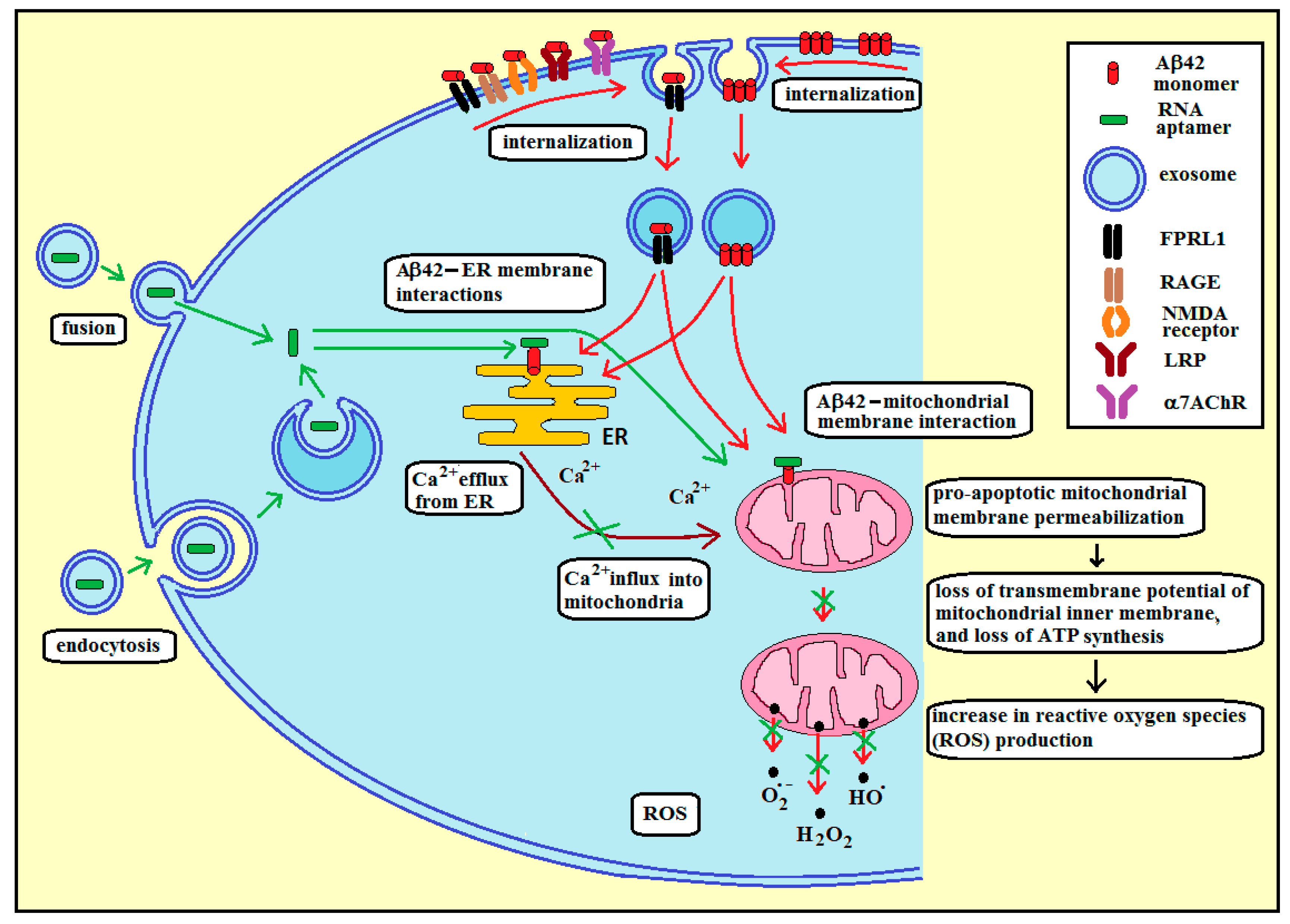
3.3. Membrane RNA—Exosome-Based Antioxidant Strategies: The Case of Membrane Amyloid Peptide Aβ42 at the Extracellular Leaflet of Plasma Membrane
3.4. Membrane RNA—Exosome-Based Antioxidant Strategies: The Case of Membrane Amyloid Peptide Aβ42 at the Cytoplasmic Leaflet of Intracellular Membranes
4. Materials and Methods
4.1. Materials
4.2. Preparation of Large Unilamellar Vesicles (LUV)
4.3. Distribution of Aβ42 between Liposomes and Buffer
4.4. Selection Procedure
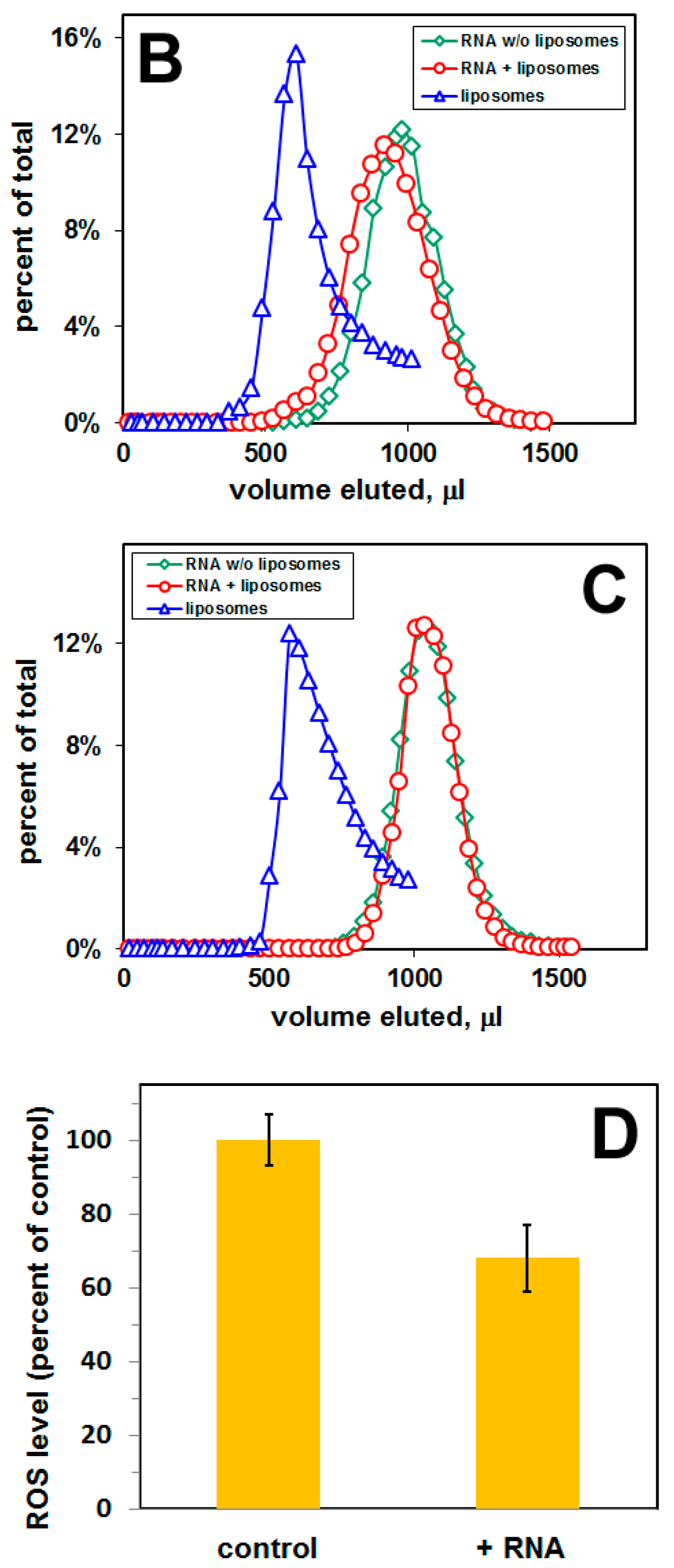
4.5. Cell Culture and Measurement of Reactive Oxygen Species (ROS)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer disease |
| Aβ | amyloid β-peptide |
| Aβ42 | 42 amino-acid-long amyloid β-peptide |
| APP | amyloid precursor protein |
| BBB | blood-brain barrier |
| CHOL | cholesterol |
| ΔΨm | transmembrane potential |
| DCF | dichlorofluorescein |
| DMSO | dimethyl sulfoxide |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| eAβ | extracellular amyloid peptide |
| ER | endoplasmic reticulum |
| iAβ | intracellular amyloid peptide |
| LUV | large unilamellar vesicles |
| MVBs | multivesicular bodies |
| PC | phosphatidylcholine |
| PS | phosphatidylserine |
| PTP | permeability transition pore |
| ROS | reactive oxygen species |
| SELEX | systematic evolution of ligands by exponential enrichment |
| siRNA | short interfering RNA |
| SM | N-stearoyl-d-erythro-sphingosylphosphorylcholine (stearoyl sphingomyelin) |
| TGN | trans-Golgi network |
References
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic protein-membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 2010, 49, 5628–5654. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, V.T. An alternative interpretation of the amyloid A hypothesis with regard to the pathogenesis of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 9093–9098. [Google Scholar] [CrossRef]
- Kotler, S.A.; Walsh, P.; Brender, J.R.; Ramamoorthy, A. Differences between amyloid- aggregation in solution and on the membrane: Insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6692–6700. [Google Scholar] [CrossRef] [PubMed]
- Klyubin, I.; Walsh, D.M.; Lemere, C.A.; Cullen, W.K.; Shankar, G.M.; Betts, V.; Spooner, E.T.; Jiang, L.; Anwyl, R.; Selkoe, D.J.; et al. Amyloid protein immunotherapy neutralizes A oligomers that disrupt synaptic plasticity in vivo. Nat. Med. 2005, 11, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Yokoseki, T.; Shibata, M.; Yamaguchi, H.; Yanagisawa, K. Suppression of A deposition in brain by peripheral administration of Fab fragments of anti-seed antibody. Biochem. Biophys. Res. Commun. 2005, 335, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Janas, T.; Yarus, M. Visualization of membrane RNAs. RNA 2003, 9, 1353–1361. [Google Scholar] [CrossRef] [Green Version]
- Janas, T.; Janas, T.; Yarus, M. A membrane transporter for tryptophan composed of RNA. RNA 2004, 10, 1541–1549. [Google Scholar] [CrossRef] [Green Version]
- Janas, T.; Janas, T.; Yarus, M. RNA, lipids and membranes. In The RNA World III.; Gesteland, R.F., Cech, T.R., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2005; pp. 207–225. ISBN 978-0879697396. [Google Scholar]
- Janas, T.; Janas, T.; Yarus, M. Specific RNA binding to ordered phospholipid bilayers. Nucleic Acids Res. 2006, 34, 2128–2136. [Google Scholar] [CrossRef] [Green Version]
- Janas, T.; Janas, T. The selection of aptamers specific for membrane molecular targets. Cell. Mol. Biol. Lett. 2011, 16, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Janas, T.; Janas, T.; Yarus, M. Human tRNASec associates with Hela membranes, cell lipid liposomes and synthetic lipid bilayers. RNA 2012, 18, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Araki, W.; Tamaoka, A. Amyloid beta-protein and lipid rafts: Focused on biogenesis and catabolism. Front. Biosci. 2015, 20, 314–324. [Google Scholar] [CrossRef]
- Simakova, O.; Arispe, N.J. The cell-selective neurotoxicity of the Alzheimer’s A peptide is determined by surface phosphatidylserine and cytosolic ATP levels. Membrane binding is required for A toxicity. J. Neurosci. 2007, 27, 13719–13729. [Google Scholar] [CrossRef] [PubMed]
- Bokvist, M.; Lindstrom, F.; Watts, A.; Grobner, G. Two types of Alzheimer’s -Amyloid (1–40) peptide membrane interactions: Aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 2004, 335, 1039–1049. [Google Scholar] [CrossRef]
- Nasica-Labouze, J.; Nguyen, P.H.; Sterpone, F.; Berthoumieu, O.; Buchete, N.V.; Coté, S.; Simone, A.D.; Doig, A.J.; Faller, P.; Garcia, A. Amyloid β protein and Alzheimer’s disease: When computer simulations complement experimental studies. Chem. Rev. 2015, 115, 3518–3563. [Google Scholar] [CrossRef]
- Ylera, F.; Lurz, R.; Erdmann, V.A.; Furste, J.P. Selection of RNA aptamers to the Alzheimer’s disease amyloid peptide. Biochem. Biophys. Res. Commun. 2002, 290, 1583–1588. [Google Scholar] [CrossRef]
- Rahimi, F.; Murakami, K.; Summers, J.L.; Chen, C.H.B.; Bitan, G. RNA aptamers generated against oligomeric A 40 recognize common amyloid aptatopes with low specificity but high sensitivity. PLoS ONE 2009, 4, e7694. [Google Scholar] [CrossRef]
- Takahashi, T.; Tada, K.; Mihara, H. RNA aptamers selected against amyloid-peptide (A) inhibit the aggregation of A. Mol. Biosyst. 2009, 5, 986–991. [Google Scholar] [CrossRef]
- Rahimi, F. Aptamers selected for recognizing amyloid-Protein—A Case for Cautious Optimism. Int. J. Mol. Sci. 2018, 19, 668. [Google Scholar] [CrossRef]
- Janas, T.; Janas, M.M.; Sapoń, K.; Janas, T. Mechanisms of RNA loading into exosomes. FEBS Lett. 2015, 589, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Janas, A.M.; Sapoń, K.; Janas, T.; Stowell, M.H.; Janas, T. Exosomes and other extracellular vesicles in neural cells and neurodegenerative diseases. Biochim. Biophys. Acta 2016, 1858, 1139–1151. [Google Scholar] [CrossRef]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Sarko, D.K.; McKinnney, C.E. Exosomes: Origins and therapeutic potential for neurodegenerative disease. Front. Neurosci. 2017, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Romagnoli, A.; Paolo Pinton, P.; Rizzuto, R. Ca2+ signaling, mitochondria and cell death. Curr. Mol. Med. 2008, 8, 119–130. [Google Scholar] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Magi, S.; Castaldo, P.; Macri, M.L.; Maiolino, M.; Matteucci, A.; Bastioli, G.; Gratteri, S.; Amoroso, S.; Lariccia, V. Intracellular calcium dysregulation: Implications for Alzheimer’s disease. Biomed. Res. Int. 2016, 2016, 6701324. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef]
- Llorente, A.; Skotland, T.; Sylvanne, T.; Kauhanen, D.; Rog, T.; Orlowski, A.; Vattulainen, I.; Ekroos, K.; Sandvig, K. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim. Biophys. Acta 2014, 1831, 1302–1309. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.F.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Kojima, R.; Bojar, D.; Rizzi, G.; Charpin-El Hamri, G.; El-Baba, M.D.; Saxena, P.; Ausländer, S.; Tan, K.R.; Fussenegger, M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Milner, T.A.; Li, F.; Nam, E.E.; Edgar, M.A.; Yamaguchi, H.; Beal, M.F.; Xu, H.; Greengard, P.; Gouras, G.K. Intraneuronal Alzheimer A 42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 2002, 161, 1869–1879. [Google Scholar] [CrossRef]
- Umeda, T.; Tomiyama, T.; Sakama, N.; Tanaka, S.; Lambert, M.P.; Klin, W.L.; Mori, H. Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 2011, 89, 1031–1042. [Google Scholar] [CrossRef]
- Nair, S.; Trainib, M.; Dawes, I.W.; Perrone, G.G. Genome-wide analysis of Saccharomyces cerevisiae identifies cellular processes affecting intracellular aggregation of Alzheimer’s amyloid-β42: Importance of lipid homeostasis. Mol. Biol. Cell 2014, 25, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Cruz, E.; Kumar, S.; Yuan, L.; Arikkath, J.; Batra, S.K. Intracellular amyloid beta expression leads to dysregulation of the mitogen-activated protein kinase and bone morphogenetic protein-2 signaling axis. PLoS ONE 2018, 13, e0191696. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Zhao, X.; Lu, W.; Zhang, Q.; Hua, Z. Intracellular Aβ and its pathological role in Alzheimer’s disease: Lessons from the cellular to animal model. Curr. Alzheimer Res. 2016, 13, 621–630. [Google Scholar] [CrossRef]
- Jin, S.; Kedia, N.; Illes-Toth, E.; Haralampiev, I.; Prisner, S.; Herrmann, A.; Wanker, E.E.; Bieschke, J. Amyloid-(1–42) aggregation initiates its cellular uptake and cytotoxicity. J. Biol. Chem. 2016, 291, 19590–19606. [Google Scholar] [CrossRef]
- Ma, K.G.; Lv, J.; Yang, W.N.; Chang, K.W.; Hu, X.D.; Shi, L.L.; Zhai, W.Y.; Zong, H.F.; Qian, Y.H. The p38 mitogen activated protein kinase regulates β-amyloid protein internalization through the α7 nicotinic acetylcholine receptor in mouse brain. Brain Res. Bull. 2018, 137, 41–52. [Google Scholar] [CrossRef]
- Gibson, G.E.; Thakkar, A. Interactions of mitochondria/metabolism and calcium regulation in Alzheimer’s disease: A calcinist point of view. Neurochem. Res. 2017, 42, 1636–1648. [Google Scholar] [CrossRef]
- Muller, M.; Ahumada-Castro1, U.; Sanhueza, M.; Gonzalez-Billault, C.; Felipe, A.; Court, F.A.; Cardenas, C. Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Y.G.; Tikhonova, L.A.; Kosenko, E.A. Critical analysis of Alzheimer’s amyloid-beta toxicity to mitochondria. Front. Biosci. 2015, 20, 173–197. [Google Scholar] [CrossRef]
- Head, E.; Lott, I.T. Down Syndrome and beta-amyloid deposition. Curr. Opin. Neurol. 2004, 17, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Schupf, N. Genetic and host factors for dementia in Down’s syndrome. Brit. J. Psychiatry 2002, 180, 405–410. [Google Scholar] [CrossRef]
- Zana, M.; Janka, Z.; Kalman, J. Oxidative stress: A bridge between Down’s syndrome and Alzheimer’s disease. Neurobiol. Aging 2007, 28, 648–676. [Google Scholar] [CrossRef] [PubMed]
- Gehman, J.D.; O’Brien, C.C.; Shabanpoor, F.; Wade, J.D.; Separovic, F. Metal effects on the membrane interactions of amyloid-b peptides. Eur. Biophys. J. 2008, 37, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Vlassov, A.; Khvorova, A.; Yarus, M. Binding and disruption of phospholipid bilayers by supramolecular RNA complexes. Proc. Natl Acad. Sci. USA 2001, 98, 7706–7711. [Google Scholar] [CrossRef] [Green Version]
- Uhlmann, E.; Peyman, A.; Ryte, A.; Schmidt, A.; Budecke, E. Use of minimally modified antisense oligonucleotides for specific inhibition of gene expression. Methods Enzymol. 2000, 313, 268–284. [Google Scholar]
- Sapoń, K.; Janas, T.; Sikorski, A.F.; Janas, T. Polysialic acid chains exhibit enhanced affinity for ordered regions of membranes. Biochim. Biophys. Acta Biomembr. 2019, 1861, 245–255. [Google Scholar] [CrossRef]
- Lazniewska, J.; Milowska, K.; Zablocka, M.; Mignani, S.; Caminade, A.M.; Majoral, J.P.; Bryszewska, M.; Gabryelak, T. Mechanism of cationic phosphorus dendrimer toxicity against murine neural cell lines. Mol. Pharm. 2013, 10, 3484–3496. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janas, T.; Sapoń, K.; Stowell, M.H.B.; Janas, T. Selection of Membrane RNA Aptamers to Amyloid Beta Peptide: Implications for Exosome-Based Antioxidant Strategies. Int. J. Mol. Sci. 2019, 20, 299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020299
Janas T, Sapoń K, Stowell MHB, Janas T. Selection of Membrane RNA Aptamers to Amyloid Beta Peptide: Implications for Exosome-Based Antioxidant Strategies. International Journal of Molecular Sciences. 2019; 20(2):299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020299
Chicago/Turabian StyleJanas, Teresa, Karolina Sapoń, Michael H. B. Stowell, and Tadeusz Janas. 2019. "Selection of Membrane RNA Aptamers to Amyloid Beta Peptide: Implications for Exosome-Based Antioxidant Strategies" International Journal of Molecular Sciences 20, no. 2: 299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020299