Analysis of Procollagen C-Proteinase Enhancer-1/Glycosaminoglycan Binding Sites and of the Potential Role of Calcium Ions in the Interaction

 , ,
, ,  and
and
Abstract
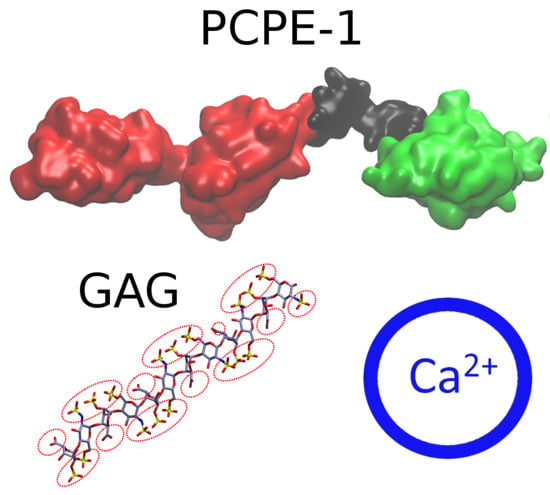
:
1. Introduction
2. Results and Discussion
2.1. Experimental Results
2.2. Modeling the Full Structure of PCPE-1
2.3. PCPE-1 Interactions with Glycosaminoglycans
2.3.1. The NTR-Domain
2.3.2. CUB1-CUB2 Domains
2.3.3. Full PCPE-1
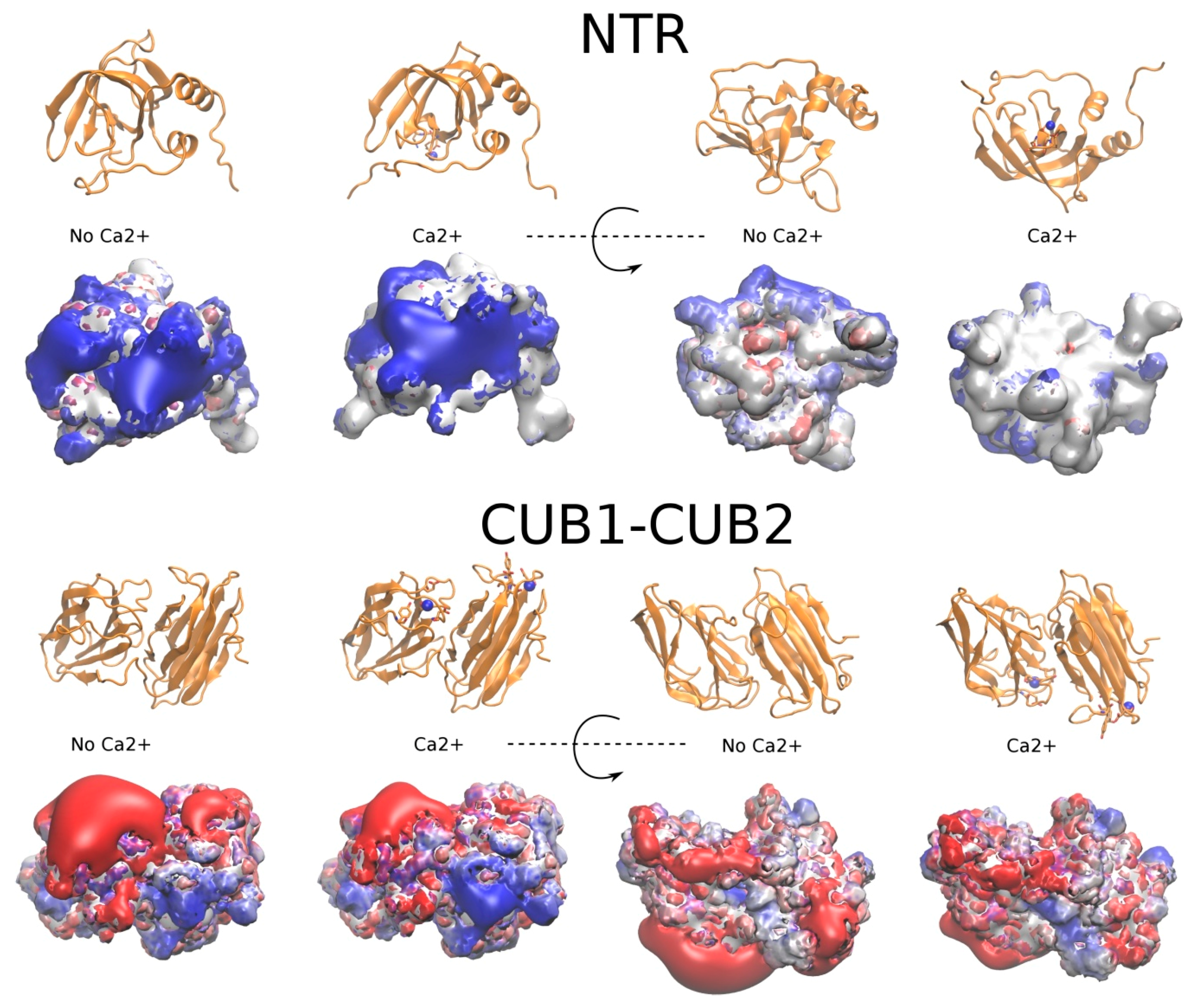
2.4. The Potential Role of Ca2+ in PCPE-1 Interactions with Glycosaminoglycans
2.4.1. Prediction of Ca2+ Binding Sites
2.4.2. PCPE-1 Interactions with Glycosaminoglycans in the Presence of Ca2+ Ions
2.4.3. Predicting Longer GAG Binding Poses Using the Fragment-Based Approach
3. Materials and Methods
3.1. Surface Plasmon Resonance (SPR) Binding Assays
3.2. Structures
3.2.1. Protein Structures
3.2.2. Glycosaminoglycan Structures
3.3. Electrostatic Potential Calculations
3.4. Coarse-Grained MD Simulations
3.5. Molecular Docking
3.5.1. Autodock 3
3.5.2. Fragment-Based Approach
3.6. All-Atom MD Simulations and MM-GBSA Free Energy Calculations
3.7. Ca2+ Ion Position Prediction
3.7.1. Molecular Dynamics Approach
3.7.2. FoldX and IonCom
3.8. Visualization and Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD3 | Autodock 3 |
| BMP-1 | Bone morphogenetic protein-1 |
| CG | Coarse-grained |
| CS | Chondroitin sulfate |
| CUB | Complement, sea urchin protein Uegf, BMP-1 |
| DS | Dermatan sulfate |
| GAG | Glycosaminoglycan |
| HP | Heparin |
| HS | Heparan sulfate |
| MD | Molecular dynamics |
| MM-GBSA | Molecular mechanics-generalized born surface area |
| mTLD | Mammalian tolloid |
| NTR | Netrin-like domain |
| PBSA | Poisson‒Boltzmann surface area |
| PCPE-1 | Procollagen C-proteinase enhancer-1 |
| SAXS | Small angle X-ray scattering |
| UNRES | United residue force field |
References
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; pp. 1–784. [Google Scholar]
- Pomin, V.H.; Mulloy, B. Glycosaminoglycans and Proteoglycans. Pharmaceuticals 2018, 11, 27. [Google Scholar] [CrossRef]
- Proudfoot, A.E. Chemokines and Glycosaminoglycans. Front. Immunol. 2015, 6, 246. [Google Scholar] [CrossRef]
- Shute, J. Glycosaminoglycan and chemokine/growth factor interactions. Handb. Exp. Pharmacol. 2012, 207, 307–324. [Google Scholar]
- Iozzo, R.V.; Zoellerm, J.J.; Nyströmm, A. Basement membrane proteoglycans: Modulators Par Excellence of cancer growth and angiogenesis. Mol. Cells 2009, 27, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Almond, A. Multiscale modeling of glycosaminoglycan structure and dynamics: Current methods and challenges. Curr. Opin. Struct. Biol. 2018, 50, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Pichert, A.; Samsonov, S.A.; Theisgen, S.; Thomas, L.; Baumann, L.; Schiller, J.; Beck-Sickinger, A.G.; Huster, D.; Pisabarro, M.T. Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology 2012, 22, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Penk, A.; Baumann, L.; Huster, D.; Samsonov, S.A. NMR and Molecular Modeling Reveal Specificity of the Interactions between CXCL14 and Glycosaminoglycans. Glycobiology 2019. [Google Scholar] [CrossRef] [PubMed]
- Bojarski, K.K.; Sieradzan, A.K.; Samsonov, S.A. Molecular Dynamics Insights into Protein-Glycosaminoglycan Systems from Microsecond-Scale Simulations. Biopolymers 2019. [Google Scholar] [CrossRef]
- Uciechowska-Kaczmarzyk, U.; Chauvot de Beauchene, I.; Samsonov, S.A. Docking software performance in protein-glycosaminoglycan systems. J. Mol. Graph. Mod. 2019, 90, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Rother, S.; Samsonov, S.A.; Hofmann, T.; Blaszkiewicz, J.; Köhling, S.; Schnabelrauch, M.; Möller, S.; Rademann, J.; Kalkhof, S.; von Bergen, M.; et al. Structural and functional insights into the interaction of sulfated glycosaminoglycans with tissue inhibitor of metalloproteinase-3—A possible regulatory role on extracellular matrix homeostasis. Acta Biomater. 2016, 45, 143–154. [Google Scholar] [CrossRef]
- Vallet, S.D.; Miele, A.E.; Uciechowska-Kaczmarzyk, U.; Liwo, A.; Duclos, D.; Samsonov, S.A.; Ricard-Blum, S. Insights into the structure and dynamics of lysyl oxidase propeptide, a flexible protein with numerous partners. Sci. Rep. 2018, 8, 11768. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S.; Ruggiero, F.; van der Rest, M. The collagen superfamily. Top. Curr. Chem. 2005, 247, 35–84. [Google Scholar]
- Vadon-Le Goff, S.; Hulmes, D.J.S.; Moali, C. BMP-1/tolloid-like proteinases synchronize matrix assembly with growth factor activation to promote morphogenesis and tissue remodeling. Matrix Biol. 2015, 44–46, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Takahara, K.; Lyons, G.E.; Greenspan, D.S. Bone morphogenetic protein-1 and a mammalian tolloid homologue (mTld) are encoded by alternatively spliced transcripts which are differentially expressed in some tissues. J. Biol. Chem. 1994, 269, 32572–32578. [Google Scholar]
- Kessler, E.; Takahara, K.; Biniaminov, L.; Brusel, M.; Greenspan, D.S. Bone Morphogenetic Protein-1: The Type I Procollagen C-Proteinase. Science 1996, 271, 360–362. [Google Scholar] [CrossRef]
- Li, S.W.; Sieron, A.L.; Fertala, A.; Hojima, Y.; Arnold, W.V.; Prockop, D.J. The C-proteinase that processes procollagens to fibrillar collagens is identical to the protein previously identified as bone morphogenic protein-1. Proc. Natl. Acad. Sci. USA 1996, 93, 5127–5130. [Google Scholar] [CrossRef]
- Kessler, E.; Hassoun, E. Procollagen C-Proteinase Enhancer 1 (PCPE-1) in Liver Fibrosis. Methods Mol. Biol. 2019, 1944, 189–201. [Google Scholar]
- Hassoun, E.; Safrin, M.; Ziv, H.; Pri-Chen, S.; Kessler, E. Procollagen C-proteinase enhancer 1 (PCPE-1) as a plasma marker of muscle and liverf in mice. PLoS ONE 2016, 11, e0159606. [Google Scholar]
- Takahara, K.; Kessler, E.; Biniaminov, L.; Brusel, M.; Eddy, R.L.; Jani-Saitfl, S.; Shows, T.B.; Greenspan, D.S. Type I procollagen COOH-terminal proteinase enhancer protein: Identification, primary structure, and chromosomal localization of the cognate human gene (PCOLCE). J. Biol. Chem. 1994, 269, 26280–26285. [Google Scholar]
- Bányai, L.; Patthy, L. The NTR module: Domains of netrins, secreted frizzled related proteins, and type I procollagen C-proteinase enhancer protein are homologous with tissue inhibitors of metalloproteases. Protein Sci. 1999, 8, 1636–1642. [Google Scholar] [CrossRef] [Green Version]
- Liepinsh, E.; Banyai, L.; Pintacuda, G.; Trexler, M.; Patthy, L.; Otting, G. NMR structure of the netrin-like domain (NTR) of human type I procollagen C-proteinase enhancer defines structural consensus of NTR domains and assesses potential proteinase inhibitory activity and ligand binding. J. Biol. Chem. 2003, 278, 25982–25989. [Google Scholar] [CrossRef] [PubMed]
- Pulido, D.; Sharma, U.; Vadon-Le Goff, S.; Hussain, S.A.; Cordes, S.; Mariano, N.; Bettler, E.; Moali, C.; Aghajari, N.; Hohenester, E.; et al. Structural Basis for the Acceleration of Procollagen Processing by Procollagen C-Proteinase Enhancer-1. Structure 2018, 26, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Gaboriaud, C.; Gregory-Pauron, L.; Teillet, F.; Thielens, N.M.; Bally, I.; Arlaud, G.J. Structure and properties of the Ca2+-binding CUB domain, a widespread ligand-recognition unit involved in major biological functions. Biochem. J. 2011, 439, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Font, B.; Eichenberger, D.; Moreau, C.; Ricard-Blum, S.; Hulmes, D.J.S.; Moali, C. Insights into how CUB domains can exert specific functions while sharing a common fold. Conserved and specific features of the CUB1 domain contribute to the molecular basis of procollagen C-proteinase enhancer-1 activity. J. Biol. Chem. 2007, 282, 16924–16933. [Google Scholar] [CrossRef]
- Bernocco, S.; Steiglitz, B.M.; Svergun, D.I.; Petoukhov, M.V.; Ruggiero, F.; Ricard-Blum, S.; Ebel, C.; Geourjon, C.; Deleage, G.; Font, B.; et al. Low resolution structure determination shows procollagen C-proteinase enhancer to be an elongated multidomain glycoprotein. J. Biol. Chem. 2003, 278, 7199–7205. [Google Scholar] [CrossRef]
- Moschcovich, L.; Bernocco, S.; Font, B.; Rivkin, H.; Eichenberger, D.; Chejanovsky, N.; Hulmes, D.J.; Kessler, E. Folding and activity of recombinant human procollagen C-proteinase enhancer. Eur. J. Biochem. 2001, 268, 2991–2996. [Google Scholar] [CrossRef]
- Weiss, T.; Ricard-Blum, S.; Moschcovich, L.; Wineman, E.; Mesilaty, S.; Kessler, E. Binding of procollagen C-proteinase enhancer-1 (PCPE-1) to heparin/heparan sulfate: Properties and role in PCPE-1 interaction with cells. J. Biol. Chem. 2010, 285, 33867–33874. [Google Scholar] [CrossRef]
- Bekhouche, M.; Kronenberg, D.; Vadon-Le Goff, S.; Bijakowski, C.; Lim, N.H.; Font, B.; Kessler, E.; Colige, A.; Nagase, H.; Murphy, G.; et al. Role of the netrin-like domain of procollagen C-proteinase enhancer-1 in the control of metalloproteinase activity. J. Biol. Chem. 2010, 285, 15950–15959. [Google Scholar] [CrossRef]
- Sankaranarayanan, N.V.; Nagarajan, B.; Desai, U.R. So you think computational approaches to understanding glycosaminoglycan-protein interactions are too dry and too rigid? Think again! Curr. Opin. Struct. Biol. 2018, 50, 91–100. [Google Scholar] [CrossRef]
- Samsonov, S.A.; Pisabarro, M.T. Computational analysis of interactions in structurally available protein-glycosaminoglycan complexes. Glycobiology 2016, 26, 850–861. [Google Scholar] [CrossRef]
- Möbius, K.; Nordsieck, K.; Pichert, A.; Samsonov, S.A.; Thomas, L.; Schiller, J.; Kalkhof, S.; Pisabarro, M.T.; Beck-Sickinger, A.G.; Huster, D. Investigation of lysine side chain interactions of Interleukin-8 with Heparin and other glycosaminoglycans studied by a methylation-NMR approach. Glycobiology 2013, 23, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Ester, M.; Kriegel, H.; Sander, J.; Xu, X. A density-based algorithm for discovering clusters in large spatial databases with noise. In Proceedings of the International Conference on Knowledge Discovery and Data Mining, Portland, OR, USA, 2–4 August 1996; pp. 226–231. [Google Scholar]
- Imberty, A.; Lortat-Jacob, H.; Pérez, S. Structural view of glycosaminoglycan-protein interactions. Carbohydr. Res. 2007, 342, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Capila, I.; Hernáiz, M.J.; Mo, Y.D.; Mealy, T.R.; Campos, B.; Dedman, J.R.; Linhardt, R.J.; Seaton, B.A. Annexin V—Heparin oligosaccharide complex suggests heparan sulfate—Mediated assembly on cell surfaces. Structure 2001, 9, 57–64. [Google Scholar] [CrossRef]
- Woodhead, N.E.; Long, W.F.; Willamson, F.B. Binding of zinc ions to heparin. Analysis by equilibrium dialysis suggests the occurrence of two, entropy-driven, processes. Biochem J. 1986, 237, 281–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevic, I.; Parmar, N.; Paredes, N.; Berry, L.R.; Chan, A.K.C. Binding of heparin to metals. Cell Biochem. Biophys. 2011, 59, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Multhaup, G.; Bush, A.I.; Pollwein, P.; Masters, C.L. Binding of heparin to metals. FEBS Lett. 1994, 335, 151–154. [Google Scholar] [CrossRef]
- Eckert, R.; Ragg, H. Zinc ions promote the interaction between heparin and heparin cofactor II. FEBS Lett. 2003, 541, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Ricard-Blum, S.; Féraud, O.; Lortat-Jacob, H.; Rencurosi, A.; Fukai, N.; Dkhissi, F.; Vittet, D.; Imberty, A.; Olsen, B.R.; van der Rest, M. Characterization of endostatin binding to heparin and heparan sulfate by surface plasmon resonance and molecular modeling: Role of divalent cations. J. Biol. Chem. 2004, 279, 2927–2936. [Google Scholar] [CrossRef]
- Han, Q.; Fu, Y.; Zhou, H.; He, Y.; Luo, Y. Contributions of Zn(II)-binding to the structural stability of endostatin. FEBS Lett. 2007, 581, 3027–3032. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Liang, X.; Beaudet, J.M.; Lee, Y.; Linhardt, R.J. The Effects of Metal Ions on Heparin/Heparin Sulfate-Protein Interactions. J. Biomed. Technol. Res. 2014, 1, 10. [Google Scholar] [CrossRef]
- Seo, Y.; Schenauer, M.R.; Leary, J.A. Biologically Relevant Metal-Cation Binding Induces Conformational Changes in Heparin Oligosaccharides as Measured by Ion Mobility Mass Spectrometry. Int. J. Mass Spectrom. 2011, 303, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Lerner, L.; Torchia, D.A. A multinuclear NMR study of the interactions of cations with proteoglycans, heparin, and Ficoll. J. Biol. Chem. 1986, 261, 12706–12714. [Google Scholar] [PubMed]
- Parrish, R.F.; Fair, W.R. Selective binding of zinc ions to heparin rather than to other glycosaminoglycans. Biochem. J. 1981, 193, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grushka, E.; Cohen, A.S. The Binding of Cu(II) and Zn(II) Ions by Heparin. Anal. Lett. 1982, 15, 1277–1288. [Google Scholar] [CrossRef]
- Grant, D.; Long, W.F.; Williamson, F.B. Infrared spectroscopy of heparin-cation complexes. Biochem. J. 1987, 244, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Sattelle, B.; Shakeri, J.; Almond, A. Does microsecond sugar ring flexing encode 3D-shape and bioactivity in the heparanome? Biomacromolecules 2013, 14, 1149–1159. [Google Scholar] [CrossRef]
- Samsonov, S.; Bichmann, L.; Pisabarro, M.T. Coarse-grained model of glycosaminglycans. J. Chem. Inf. Mod. 2015, 55, 114–124. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER16; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Rhee, Y.M.; Pande, V.S. Multiplexed-replica exchange molecular dynamics method for protein folding simulation. Biophys. J. 2003, 84, 775–786. [Google Scholar] [CrossRef]
- Hansmann, U.H.E.; Okamoto, Y. Comparative study of multicanonical and simulated annealing algorithms in the protein folding problem. Physica A 1994, 212, 415–437. [Google Scholar] [CrossRef] [Green Version]
- Liwo, A.; Baranowski, M.; Czaplewski, C.; Gołaś, E.; He, Y.; Jagieła, D.; Krupa, P.; Maciejczyk, M.; Makowski, M.; Mozolewska, M.A.; et al. A unified coarse-grained model of biological macromolecules based on mean-field multipole-multipole interactions. J. Mol. Model. 2014, 20, 2306. [Google Scholar] [CrossRef] [PubMed]
- Liwo, A.; Czaplewski, C.; Ołdziej, S.; Scheraga, H.A. Computational techniques for efficient conformational sampling of proteins. Curr. Opin. Struct. Biol. 2008, 18, 134–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczyńska, A.S.; Mozolewska, M.A.; Krupa, P.; Giełdoń, A.; Liwo, A.; Czaplewski, C. Prediction of protein structure with the coarse-grained UNRES force field assisted by small X-ray scattering data and knowledge-based information. Proteins 2018, 86, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Murtagh, F.; Heck, A. Multivariate Data Analysis. In Astrophysics and Space Science Library; Kluwer Academic Publishers: Strasbourg, France, 1987; pp. 55–109. [Google Scholar]
- Rotkiewicz, P.; Skolnick, J. Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 2008, 29, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarcklan algorithm an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Samsonov, S.; Gehrcke, J.-P.; Pisabarro, M.T. Flexibility and explicit solvent in molecular dynamics-based docking of protein-glycosaminoglycan systems. J. Chem. Inf. Mod. 2014, 54, 582–592. [Google Scholar] [CrossRef]
- Joseph, R.R.; Mosier, P.D.; Desai, U.R.; Rajarathnam, K. Solution NMR characterization of chemokine CXCL8/IL-8 monomer and dimer binding to glycosaminoglycans: Structural plasticity mediates differential binding interactions. Biochem. J. 2015, 472, 121–133. [Google Scholar] [CrossRef]
- Nivedha, A.K.; Makeneni, S.; Foley, B.L.; Tessier, M.B.; Woods, R.J. Importance of ligand conformational energies in carbohydrate docking: Sorting the wheat from the chaff. J. Comput. Chem. 2014, 35, 526–539. [Google Scholar] [CrossRef]
- Samsonov, S.A.; Zacharias, M.; Chauvot de Beauchene, I. Modeling large protein-glycosaminoglycan complexes using a fragment-based approach. J. Comput. Chem. 2019, 40, 1429–1439. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 4, 622–655. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Case, D.A.; Bashford, D. Effective Born radii in the generalized Born approximation: The importance of being perfect. J. Comput. Chem. 2002, 23, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inf. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. Free energy calculations of glycosaminoglycan-protein interactions. Glycobiology 2009, 19, 1103–1115. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef]
- Schymkowitz, J.W.; Rousseau, F.; Martins, I.C.; Ferkinghoff-Borg, J.; Stricher, F.; Serrano, L. Prediction of water and metal binding sites and their affinities by using the Fold-X force field. Proc. Natl. Acad. Sci. USA 2005, 102, 10147–10152. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Dong, Q.; Yang, J.; Zhang, Y. Recognizing metal and acid radical ion binding sites by integrating ab initio modeling with template-based transferals. Bioinformatics 2016, 32, 3260–3269. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.2r3pre; Schrödinger, LLC: New York, NY, USA, 2002.
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; ISBN 3-900051-07-0. Available online: http://www.R-project.org/ (accessed on 1 May 2017).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Restraints | Probability | Radius of Gyration (Å) |
|---|---|---|---|
| 1 | CUB1-CUB2, NTR domains | 34 | 22.2 |
| 2 | 32 | 24.8 | |
| 3 | 18 | 22.6 | |
| 4 | 8 | 22.8 | |
| 5 | 8 | 22.6 | |
| 1 | CUB1-CUB2, NTR domains + SAXS-based | 39 | 43.5 |
| 2 | 21 | 44.5 | |
| 3 | 17 | 43.5 | |
| 4 | 14 | 43.3 | |
| 5 | 9 | 43.9 |
| GAG | 1 m, ε | 2 # | 3 Size | 4 ΔG (kcal/mol) | 5 Polarity |
|---|---|---|---|---|---|
| CS6, dp4 | 3, 2 | 1 | 19 | −42.0 ± 6.6; −48.3 ± 7.7; −41.3 ± 6.6 | 17/2 |
| 2 | 6 | −30.1 ± 16.0; −63.3 ± 7.1 | 6/0 | ||
| 3 | 4 | −34.4 ± 9.6; −38.4 ± 8.6 | 2/2 | ||
| 4 | 3 | −46.7 ± 10.5 | 3/0 | ||
| CS6, dp6 | 3, 2 | 1 | 3 | −56.6 ± 9.0 | 3/0 |
| 2 | 3 | −33.9 ± 9.2 | 3/0 | ||
| 3 | 3 | −36.8 ± 7.1; −64.2 ± 11.8 | 3/0 | ||
| DS, dp6 | 3, 2 | 1 | 6 | −35.5 ± 6.3; −41.5 ± 6.8 | 5/1 |
| 2 | 4 | −36.7 ± 6.6 | 4/0 | ||
| 3 | 3 | −63.7 ± 8.3 | 3/0 | ||
| 4 | 3 | −37.8 ± 8.2 | 2/1 | ||
| HP, dp2 | 3, 2 | 1 | 25 | −44.9 ± 9.3; −41.1 ± 7.3; −23.1 ± 7.6 | 25/0 |
| 2 | 12 | −27.9 ± 9.2 | 12/0 | ||
| 3 | 9 | −42.0 ± 9.0 | 9/0 | ||
| 4 | 3 | −27.7 ± 8.9; −28.7 ± 5.6 | 3/0 | ||
| HP, dp4 | 3, 2 | 1 | 32 | −39.0 ± 7.2; −29.4 ± 10.4 | 21/11 |
| 2 | 3 | −53.9 ± 7.2 | 3/0 | ||
| 3 | 3 | −50.6 ± 11.5; −57.4 ± 8.6 | 2/1 | ||
| HP, dp6 | 3, 2 | 1 | 15 | −69.5 ± 7.8; −56.7 ± 7.4; −43.5 ± 9.7; −54.0 ± 14.8; −80.5 ± 10.7 | 9/6 |
| 2 | 7 | −68.3 ± 11.0; −44.7 ± 7.5; −57.1 ± 10.6; −55.4 ± 9.1 | 4/3 | ||
| 3 | 6 | −50.1 ± 10.0; −44.6 ± 9.5; −65.7 ± 11.5; −61.8 ± 13.6 | 4/2 |
| GAG | CS6, dp4 | CS6, dp6 | DS, dp6 | HP, dp2 | HP, dp4 | HP, dp6 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CS6, dp4 | 10 | 7 | 7 | 6 | 6 | 4 | 7 | 5 | 6 | 6 | 4 | 7 | 5 | 5 | 5 | 6 | 6 | 6 | 7 | 7 | 7 |
| 7 | 10 | 6 | 9 | 3 | 2 | 6 | 6 | 6 | 7 | 5 | 6 | 7 | 6 | 6 | 5 | 8 | 7 | 8 | 7 | 9 | |
| 7 | 6 | 10 | 5 | 4 | 3 | 5 | 6 | 5 | 7 | 4 | 7 | 4 | 6 | 5 | 4 | 5 | 7 | 7 | 7 | 6 | |
| 6 | 9 | 5 | 10 | 3 | 2 | 5 | 6 | 6 | 6 | 4 | 5 | 7 | 5 | 5 | 6 | 9 | 6 | 7 | 6 | 8 | |
| CS6, dp6 | 6 | 3 | 4 | 3 | 10 | 7 | 5 | 2 | 5 | 3 | 1 | 4 | 3 | 1 | 1 | 5 | 3 | 2 | 5 | 5 | 4 |
| 4 | 2 | 3 | 2 | 7 | 10 | 5 | 2 | 4 | 2 | 1 | 5 | 2 | 1 | 1 | 4 | 2 | 1 | 4 | 3 | 3 | |
| 7 | 6 | 5 | 5 | 5 | 5 | 10 | 5 | 5 | 4 | 5 | 6 | 5 | 4 | 4 | 7 | 5 | 4 | 7 | 6 | 7 | |
| DS, dp6 | 5 | 6 | 6 | 6 | 2 | 2 | 5 | 10 | 5 | 6 | 6 | 6 | 6 | 7 | 6 | 5 | 6 | 8 | 7 | 7 | 7 |
| 6 | 6 | 5 | 6 | 5 | 4 | 5 | 5 | 10 | 5 | 5 | 4 | 6 | 4 | 4 | 6 | 7 | 5 | 7 | 6 | 6 | |
| 6 | 7 | 7 | 6 | 3 | 2 | 4 | 6 | 5 | 10 | 6 | 6 | 6 | 6 | 6 | 3 | 5 | 7 | 7 | 8 | 6 | |
| 4 | 5 | 4 | 4 | 1 | 1 | 5 | 6 | 5 | 6 | 10 | 4 | 4 | 5 | 5 | 3 | 4 | 6 | 5 | 5 | 5 | |
| HP, dp2 | 7 | 6 | 7 | 5 | 4 | 5 | 6 | 6 | 4 | 6 | 4 | 10 | 5 | 5 | 5 | 5 | 5 | 6 | 7 | 7 | 7 |
| 5 | 7 | 4 | 7 | 3 | 2 | 5 | 6 | 6 | 6 | 4 | 5 | 10 | 5 | 5 | 7 | 7 | 5 | 7 | 7 | 7 | |
| 5 | 6 | 6 | 5 | 1 | 1 | 4 | 7 | 4 | 6 | 5 | 5 | 5 | 10 | 6 | 3 | 5 | 7 | 6 | 6 | 6 | |
| 5 | 6 | 5 | 5 | 1 | 1 | 4 | 6 | 4 | 6 | 5 | 5 | 5 | 6 | 10 | 3 | 5 | 6 | 6 | 6 | 6 | |
| HP, dp4 | 6 | 5 | 4 | 6 | 5 | 4 | 7 | 5 | 6 | 3 | 3 | 5 | 7 | 3 | 3 | 10 | 7 | 3 | 6 | 5 | 6 |
| 6 | 8 | 5 | 9 | 3 | 2 | 5 | 6 | 7 | 5 | 4 | 5 | 7 | 5 | 5 | 7 | 10 | 6 | 7 | 6 | 8 | |
| 6 | 7 | 7 | 6 | 2 | 1 | 4 | 8 | 5 | 7 | 6 | 6 | 5 | 7 | 6 | 3 | 6 | 10 | 6 | 7 | 7 | |
| HP, dp6 | 7 | 8 | 7 | 7 | 5 | 4 | 7 | 7 | 7 | 7 | 5 | 7 | 7 | 6 | 6 | 6 | 7 | 6 | 10 | 9 | 9 |
| 7 | 7 | 7 | 6 | 5 | 3 | 6 | 7 | 6 | 8 | 5 | 7 | 7 | 6 | 6 | 5 | 6 | 7 | 9 | 10 | 8 | |
| 7 | 9 | 6 | 8 | 4 | 3 | 7 | 7 | 6 | 6 | 5 | 7 | 7 | 6 | 6 | 6 | 8 | 7 | 9 | 8 | 10 | |
| GAG | CS6, dp4 | CS6, dp6 | DS, dp6 | HP, dp2 | HP, dp4 | HP, dp6 |
|---|---|---|---|---|---|---|
| CS6, dp4 | 10 | 5 | 7 | 6 | 9 | 8 |
| CS6, dp6 | 5 | 10 | 4 | 4 | 6 | 6 |
| DS, dp6 | 7 | 4 | 10 | 6 | 7 | 7 |
| HP, dp2 | 6 | 4 | 6 | 10 | 7 | 7 |
| HP, dp4 | 9 | 6 | 7 | 7 | 10 | 9 |
| HP, dp6 | 8 | 6 | 7 | 7 | 9 | 10 |
| 1 m, ε | 2 # | 3 Size | 4 ΔG, kcal/mol | 5 TopMM-GBSA 10 Residues for GAG Binding | 6 Polarity |
|---|---|---|---|---|---|
| 2, 2.64 | 1 | 4 | −62.4.8 ± 19.0; −54.9 ± 9.1 −49.6 ± 18.6 | R435, K436, R275, R288, K279, K299, K365, K434, N331, K295 | 4/0 |
| 2 | 3 | −50.1 ± 9.7; −79.0 ± 17.0; −38.1 ± 9.4 | K436, R435, K365, K299, K434, K271, K295, R288, K165, K279 | 3/0 | |
| 3 | 3 | −30.8 ± 10.7; −36.0 ± 7.8; −42.3 ± 10.6 | K299, K436, K279, K365, K271, K434, K295, K165, Q282, R435 | 2/1 |
| Protein | PDB ID | Experimental Structure | Method | ||
|---|---|---|---|---|---|
| FoldX | IonCom | 1 MD | |||
| Annexin V | 1G5N | 9 | 6 | 8 | 6 |
| NTR | 1UAP | 0 | 0 | 0 | 2 |
| 3 | |||||
| 1 | |||||
| 1 | |||||
| 1 | |||||
| CUB1-CUB2 | 6FZV | 2 | 2 | 2 | 2 |
| 2 | |||||
| 1 | |||||
| 2 | |||||
| 1 | |||||
| Ca2+ Number (X-Ray) | 1 ΔG, kcal/mol | 2 ΔGele, kcal/mol | 3 FoldX | 3 IonCom | 3 MD |
|---|---|---|---|---|---|
| 319 | 57.2 ± 4.7 | −310.4 ± 10.3 | + | + | + |
| 320 | 47.5 ± 4.8 | −264.8 ± 15.7 | + | + | – |
| 321 | 36.5 ± 3.5 | −296.0 ± 10.9 | – | + | + |
| 322 | 59.7 ± 4.9 | −380.5 ± 9.5 | + | + | – |
| 323 | 36.4 ± 3.5 | −332.4 ± 7.9 | – | – | + |
| 324 | 62.4 ± 4.4 | −376.6 ± 8.1 | + | + | + |
| 325 | 47.5 ± 6.1 | −413.2 ± 13.0 | + | + | + |
| 326 | 39.3 ± 3.7 | −312.2 ± 9.2 | – | + | + |
| 327 | 59.2 ± 4.7 | −302.3 ± 8.6 | + | + | – |
| PCPE-1 Domain | Ca2+ Site | 2 ΔGele, kcal/mol | Site Occupancy, ns |
|---|---|---|---|
| NTR, MD1 | E405, E406, N407 G367, D370 | −116.5 ± 20.4 −58.3 ± 15.7 | 65 40 |
| NTR, MD2 | E405, E406, N407 D314/N-terminus of NTR G367, D370 | −125.8 ± 14.7 −38.6 ± 19.0 −49.6 ± 14.2 | 85 35 90 |
| NTR, MD3 | E405, E406, N407 | −120.3 ± 15.9 | 75 |
| NTR, MD4 | E405, E406, N407 | −123.4 ± 12.2 | 45 |
| NTR, MD5 | E405, E406, N407 1 E405, E406, N407/G367, D370 | −51.7 ± 13.7; −156.2 ± 33.6 | 65 25 |
| CUB1-CUB2 (X-ray, PDB ID: 6FZV) | E85, Y92, D93, D134 Y180, E208, D216, D258 | −363.2 ± 10.8 −466.5 ± 12.5 | 100 100 |
| CUB1-CUB2, MD1 | E85, Y92, D93, D134 Y180, E208, D216, D258 | −389.9 ± 19.3 −302.9 ± 24.3 | 25 90 |
| CUB1-CUB2, MD2 | E85, Y92, D93, D134 Y180, E208, D216, D258 | −368.3 ± 18.7 −371.9 ± 11.8 | 85 90 |
| CUB1-CUB2, MD3 | Y180, E208, D216, D258 | −389.2 ± 18.6 | 85 |
| CUB1-CUB2, MD4 | E85, Y92, D93, D134 Y180, E208, D216, D258 | −293.7 ± 9.0 −374.0 ± 16.1 | 75 95 |
| CUB1-CUB2, MD5 | Y180, E208, D216, D258 | −521.4 ± 10.8 | 95 |
| 1 m, ε | 2 # | 3 Size | 4 ΔG, kcal/mol | 5 TopMM-GBSA 10 Residues for GAG Binding | 6 Polarity |
|---|---|---|---|---|---|
| 2, 2.8 | 1 | 6 | −58.8 ± 12.2; −56.0 ± 19.7; −58.6 ± 13.7 | R435, K436, K434, K365, K299, K279, K295, R288, P438, K271 | 5/1 |
| 2 | 4 | −79.5 ± 15.6; −32.6 ± 11.0; −42.8 ± 10.0 | K436, R435, K279, R288, K365, K299, K434, Q282, G281, K287 | 3/1 | |
| 3 | 3 | −30.8 ± 10.7; −70.7 ± 13.2; −56.6 ± 18.4 | R435, K299, K436, K365, K434, R275, K279, K295, K271, K305 | 3/0 |
| 1 # | 2 Ca2+ | 3 ΔG, kcal/mol | 4 TopMM-GBSA 10 Residues for GAG Binding |
|---|---|---|---|
| 1 | – | −65.6 ± 12.3 | K299, R288, R435, K436, K295, K293, K305, K287, K365, P298 |
| 2 | – | −64.2 ± 11.2 | K436, K434, R275, K279, K295, R435, K365, R288, K299, K287 |
| 3 | – | −94.7 ± 12.4 | K295, K436, R435, K365, K434, K293, K299, R288, K305, V294 |
| 1 | + | −93.3 ± 12.3 | R435, K279, K295, K436, K305, K299, K434, N331, K271, R324 |
| 2 | + | −73.8 ± 11.9 | R435, K434, K436, K299, K295, K279, P298, K293, K287, K305 |
| 3 | + | −102.5 ± 14.7 | K436, K299, K295, R435, R275, K293, K279, K434, K305, P441 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potthoff, J.; Bojarski, K.K.; Kohut, G.; Lipska, A.G.; Liwo, A.; Kessler, E.; Ricard-Blum, S.; Samsonov, S.A. Analysis of Procollagen C-Proteinase Enhancer-1/Glycosaminoglycan Binding Sites and of the Potential Role of Calcium Ions in the Interaction. Int. J. Mol. Sci. 2019, 20, 5021. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205021
Potthoff J, Bojarski KK, Kohut G, Lipska AG, Liwo A, Kessler E, Ricard-Blum S, Samsonov SA. Analysis of Procollagen C-Proteinase Enhancer-1/Glycosaminoglycan Binding Sites and of the Potential Role of Calcium Ions in the Interaction. International Journal of Molecular Sciences. 2019; 20(20):5021. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205021
Chicago/Turabian StylePotthoff, Jan, Krzysztof K. Bojarski, Gergely Kohut, Agnieszka G. Lipska, Adam Liwo, Efrat Kessler, Sylvie Ricard-Blum, and Sergey A. Samsonov. 2019. "Analysis of Procollagen C-Proteinase Enhancer-1/Glycosaminoglycan Binding Sites and of the Potential Role of Calcium Ions in the Interaction" International Journal of Molecular Sciences 20, no. 20: 5021. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205021