Prion Protein in Glioblastoma Multiforme

 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
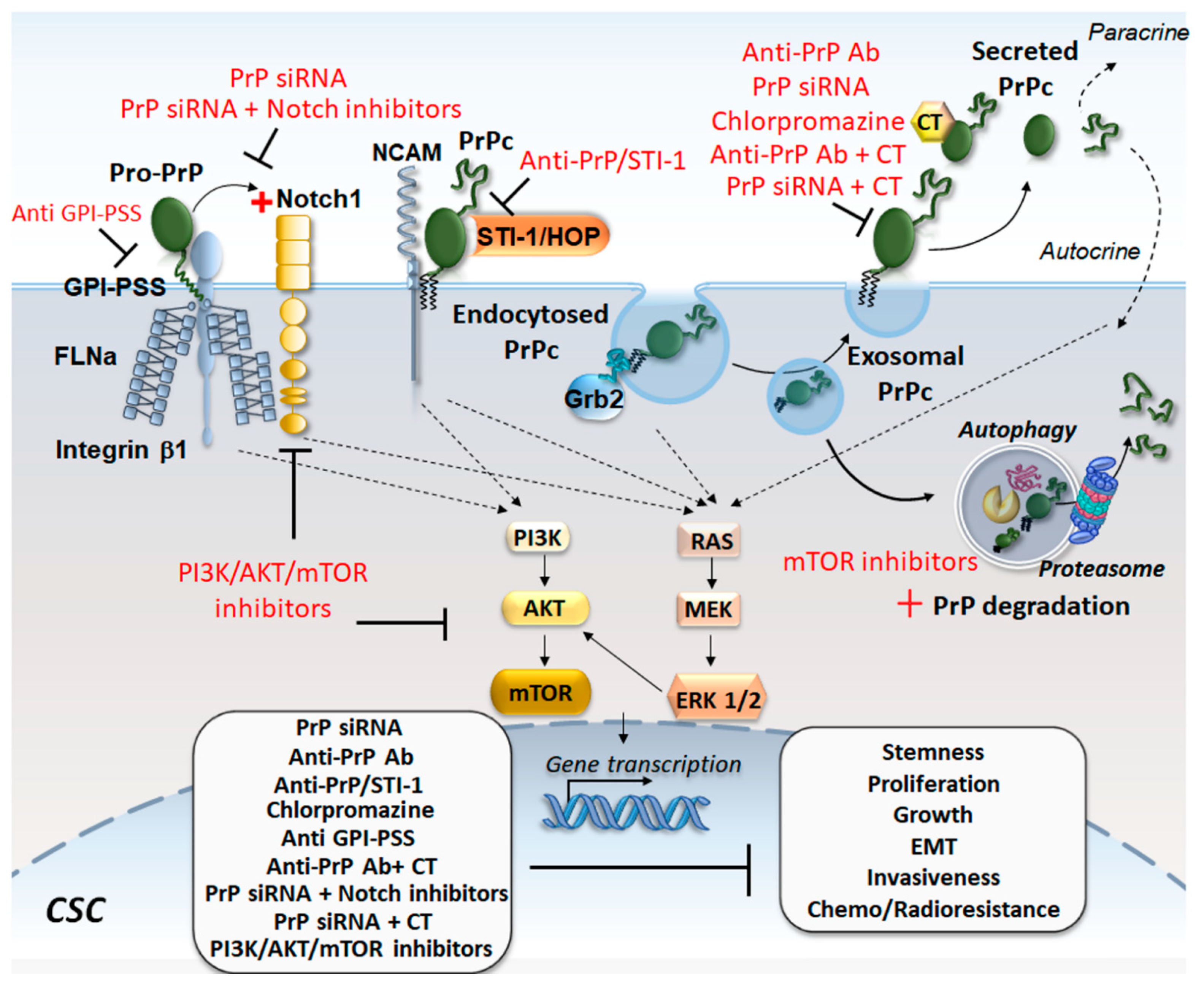{kind=link}
1. Introduction
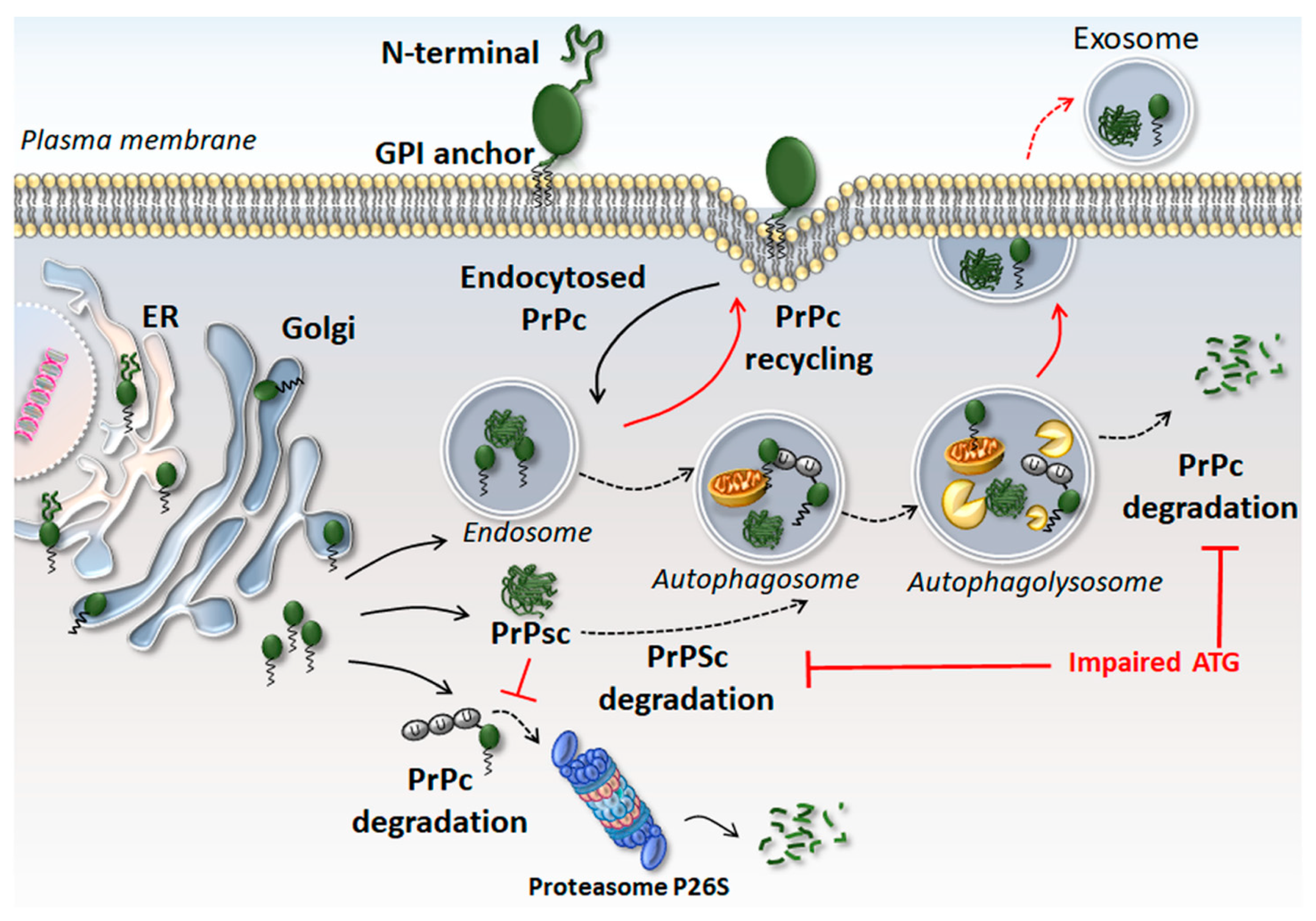
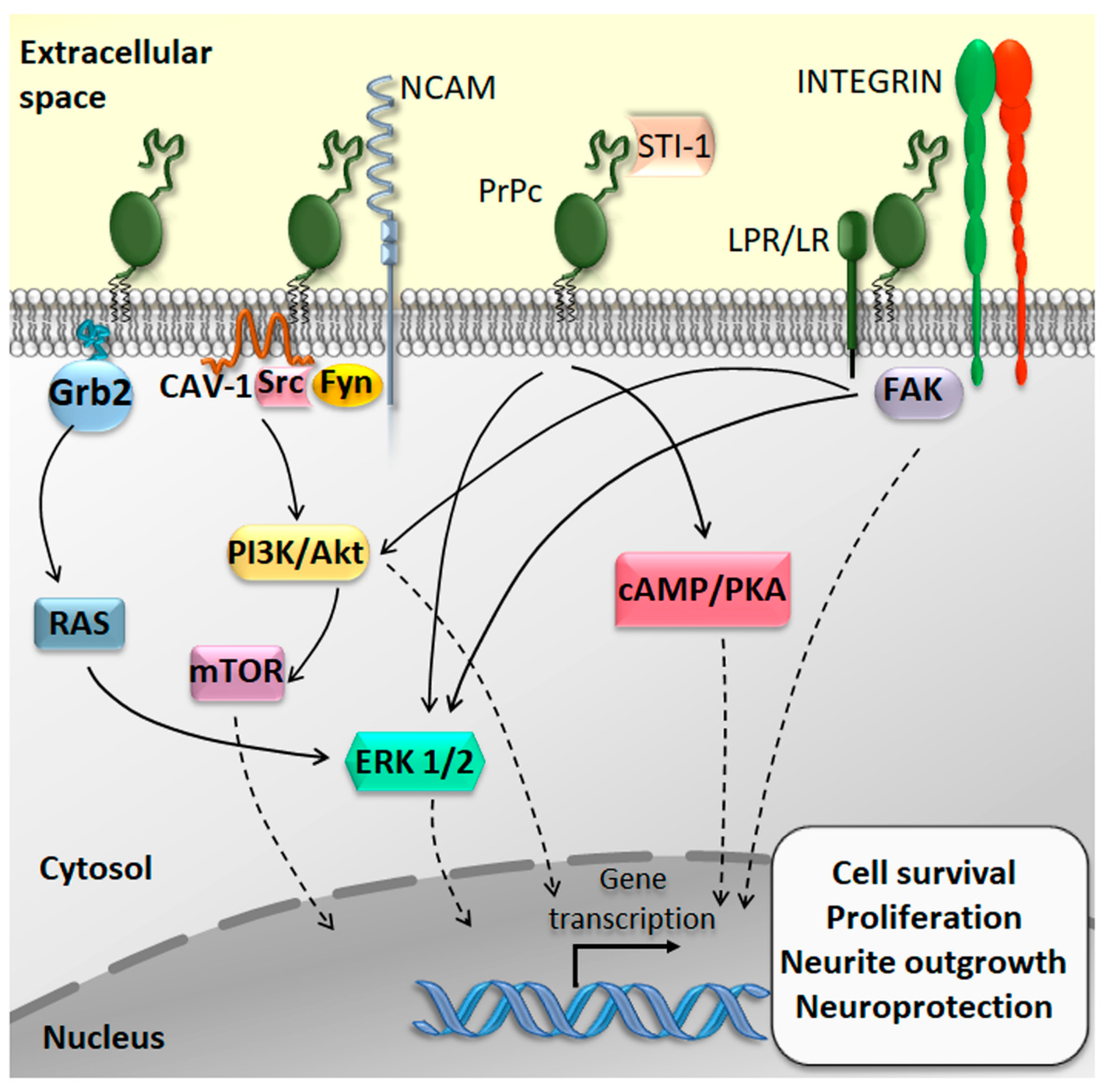
2. The Physiological Function of the Cellular Prion Protein (PrPc)
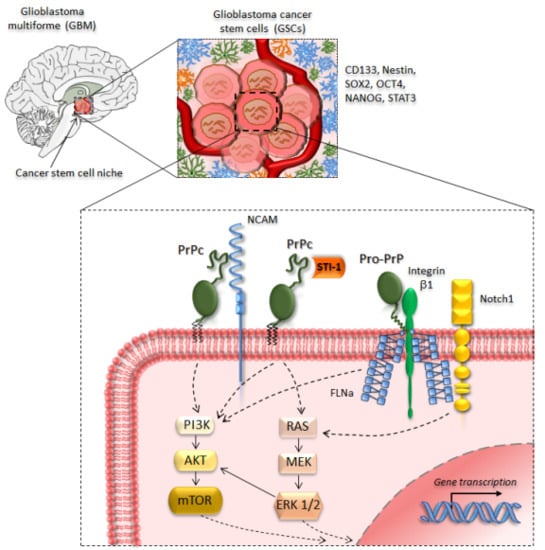
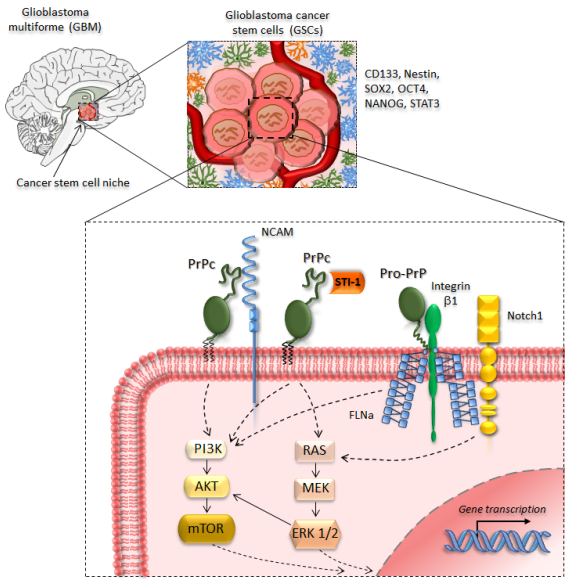
3. PrPc in Glioblastoma and Glioma Cancer Stem Cells
4. Targeting PrPc in Cancer Stem Cells
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATG | Autophagy |
| BSE | Bovine spongiform encephalopathy |
| cAMP | Cyclic adenosine monophosphate |
| CAV-1 | Caveolin-1 |
| CJD | Creutzfeldt–Jakob disease |
| CNS | Central nervous system |
| CSCs | Cancer stem cells |
| CWD | Chronic wasting disease |
| EMT | Epithelial-mesenchymal transition |
| ERK | Extracellular-signal-regulated kinase |
| FFI | Fatal familial insomnia |
| FLNA | Filamin A |
| GP78 | Ubiquitin-protein E3 ligase glycoprotein 78 |
| GSCs | Glioma stem-like cells |
| GSS | Gerstmann–Sträussler–Sheinker syndrome |
| HIF-1α | Hypoxia-inducible factor-1α |
| HOP | Hsp70/Hsp90 organizing protein |
| HSPA1L | Heat-shock 70 kDa protein-1-like |
| LR/37/67 | 37/67 kDa non-integrin laminin receptor |
| MAPK | Mitogen-activated protein kinase |
| MT1-MMP | Membrane type 1-matrix metalloproteinase |
| MVBs | Multivesicular bodies |
| NCAM | Neural cell adhesion molecule |
| NF2 | Neurofibromatosis type 2 |
| mTOR | Mammalian target of rapamycin |
| NSCs | Neural stem cells |
| PDAC | Pancreatic ductal adenocarcinoma |
| PI3K | Phosphoinositide 3-kinase |
| PKA | Protein kinase A |
| PrPc | Cellular prion protein |
| PrPsc | Scrapie prion protein |
| ROS | Reactive oxygen species |
| STI-1 | Stress-inducible protein |
| SVZ | Subependymal ventricular zone |
| TIMP | Tissue inhibitor of metalloproteinase |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
References
- Oesch, B.; Westaway, D.; Wälchli, M.; McKinley, M.P.; Kent, S.B.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Scott, M.R.; DeArmond, S.J.; Cohen, F.E. Prion protein biology. Cell 1998, 93, 337–348. [Google Scholar] [CrossRef]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Fornai, F.; Ferrucci, M.; Gesi, M.; Bandettini di Poggio, A.; Giorgi, F.S.; Biagioni, F.; Paparelli, A. A hypothesis on prion disorders: Are infectious, inherited, and sporadic causes so distinct? Brain Res. Bull. 2006, 69, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Lindquist, S. Conversion of PrP to a self perpetuating PrPSc-like conformation in the cytosol. Science 2002, 298, 1785–1788. [Google Scholar] [CrossRef] [PubMed]
- Heiseke, A.; Aguib, Y.; Riemer, C.; Baier, M.; Schätzl, H.M. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J. Neurochem. 2009, 109, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.K.; Moon, M.H.; Lee, Y.J.; Seol, J.W.; Park, S.Y. Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol. Aging 2013, 34, 146–156. [Google Scholar] [CrossRef]
- Natale, G.; Pompili, E.; Biagioni, F.; Paparelli, S.; Lenzi, P.; Fornai, F. Histochemical approaches to assess cell-to-cell transmission of misfolded proteins in neurodegenerative diseases. Eur. J. Histochem. 2013, 57, e5. [Google Scholar] [CrossRef] [Green Version]
- Ryskalin, L.; Busceti, C.L.; Limanaqi, F.; Biagioni, F.; Gambardella, S.; Fornai, F. A Focus on the Beneficial Effects of Alpha Synuclein and a Re-Appraisal of Synucleinopathies. Curr. Protein Pept. Sci. 2018, 19, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Mitteregger, G.; Vosko, M.; Krebs, B.; Xiang, W.; Kohlmannsperger, V.; Nölting, S.; Hamann, G.F.; Kretzschmar, H.A. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007, 17, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Biasini, E.; Turnbaugh, J.A.; Unterberger, U.; Harris, D.A. Prion protein at the crossroads of physiology and disease. Trends Neurosci. 2012, 35, 92–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roffé, M.; Beraldo, F.H.; Bester, R.; Nunziante, M.; Bach, C.; Mancini, G.; Gilch, S.; Vorberg, I.; Castilho, B.A.; Martins, V.R.; et al. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. USA 2010, 107, 13147–13152. [Google Scholar] [CrossRef] [Green Version]
- Wulf, M.A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 34. [Google Scholar] [CrossRef]
- Zhang, C.C.; Steele, A.D.; Lindquist, S.; Lodish, H.F. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc. Natl. Acad. Sci. USA 2006, 103, 2184–2189. [Google Scholar] [CrossRef] [Green Version]
- Santos, T.G.; Silva, I.R.; Costa-Silva, B.; Lepique, A.P.; Martins, V.R.; Lopes, M.H. Enhanced neural progenitor/stem cells self-renewal via the interaction of stress-inducible protein 1 with the prion protein. Stem Cells 2011, 29, 1126–1136. [Google Scholar] [CrossRef]
- Martin-Lannerée, S.; Halliez, S.; Hirsch, T.Z.; Hernandez-Rapp, J.; Passet, B.; Tomkiewicz, C.; Villa-Diaz, A.; Torres, J.M.; Launay, J.M.; Béringue, V.; et al. The Cellular Prion Protein Controls Notch Signaling in Neural Stem/Progenitor Cells. Stem Cells 2017, 35, 754–765. [Google Scholar] [CrossRef]
- Meslin, F.; Hamai, A.; Gao, P.; Jalil, A.; Cahuzac, N.; Chouaib, S.; Mehrpour, M. Silencing of prion protein sensitizes breast adriamycin-resistant carcinoma cells to TRAIL-mediated cell death. Cancer Res. 2007, 67, 10910–10919. [Google Scholar] [CrossRef]
- Du, L.; Rao, G.; Wang, H.; Li, B.; Tian, W.; Cui, J.; He, L.; Laffin, B.; Tian, X.; Hao, C.; et al. CD44-positive cancer stem cells expressing cellular prion protein contribute to metastatic capacity in colorectal cancer. Cancer Res. 2013, 73, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, A.; Bajetto, A.; Thellung, S.; Begani, G.; Villa, V.; Nizzari, M.; Pattarozzi, A.; Solari, A.; Gatti, M.; Pagano, A.; et al. Cellular prion protein controls stem cell-like properties of human glioblastoma tumor-initiating cells. Oncotarget 2016, 7, 38638–38657. [Google Scholar] [CrossRef] [PubMed]
- Iglesia, R.P.; Prado, M.B.; Cruz, L.; Martins, V.R.; Santos, T.G.; Lopes, M.H. Engagement of cellular prion protein with the co-chaperone Hsp70/90 organizing protein regulates the proliferation of glioblastoma stem-like cells. Stem Cell Res. Ther. 2017, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Comincini, S.; Ferrara, V.; Arias, A.; Malovini, A.; Azzalin, A.; Ferretti, L.; Benericetti, E.; Cardarelli, M.; Gerosa, M.; Passarin, M.G.; et al. Diagnostic value of PRND gene expression profiles in astrocytomas: Relationship to tumor grades of malignancy. Oncol. Rep. 2007, 17, 989–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Pan, Y.; Zhang, D.; Guo, C.; Shi, Y.; Wang, J.; Chen, Y.; Wang, X.; Liu, J.; Guo, X.; et al. Cellular prion protein promotes proliferation and G1/S transition of human gastric cancer cells SGC7901 and AGS. FASEB J. 2007, 21, 2247–2256. [Google Scholar] [CrossRef]
- Li, C.; Yu, S.; Nakamura, F.; Yin, S.; Xu, J.; Petrolla, A.A.; Singh, N.; Tartakoff, A.; Abbott, D.W.; Xin, W.; et al. Binding of pro-prion to filamin A disrupts cytoskeleton and correlates with poor prognosis in pancreatic cancer. J. Clin. Investig. 2009, 119, 2725–2736. [Google Scholar] [CrossRef]
- McEwan, J.F.; Windsor, M.L.; Cullis-Hill, S.D. Antibodies to prion protein inhibit human colon cancer cell growth. Tumour Biol. 2009, 30, 141–147. [Google Scholar] [CrossRef]
- Yun, C.W.; Yun, S.; Lee, J.H.; Han, Y.S.; Yoon, Y.M.; An, D.; Lee, S.H. Silencing Prion Protein in HT29 Human Colorectal Cancer Cells Enhances Anticancer Response to Fucoidan. Anticancer Res. 2016, 36, 4449–4458. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yu, S.; Huang, D.; Cui, M.; Hu, H.; Zhang, L.; Wang, W.; Parameswaran, N.; Jackson, M.; Osborne, B.; et al. Cellular Prion Protein Mediates Pancreatic Cancer Cell Survival and Invasion through Association with and Enhanced Signaling of Notch1. Am. J. Pathol. 2016, 186, 2945–2956. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Yun, C.W.; Lee, S.H. Cellular prion protein enhances drug resistance of colorectal cancer cells via regulation of a survival signal pathway. Biomol. Ther. 2018, 26, 313–321. [Google Scholar] [CrossRef]
- Lee, J.H.; Yun, C.W.; Han, Y.S.; Kim, S.; Jeong, D.; Kwon, H.Y.; Kim, H.; Baek, M.J.; Lee, S.H. Melatonin and 5-fluorouracil co-suppress colon cancer stem cells by regulating cellular prion protein-Oct4 axis. J. Pineal Res. 2018, 65, e12519. [Google Scholar] [CrossRef] [PubMed]
- Le Corre, D.; Ghazi, A.; Balogoun, R.; Pilati, C.; Aparicio, T.; Martin-Lannerée, S.; Marisa, L.; Djouadi, F.; Poindessous, V.; Crozet, C.; et al. The cellular prion protein controls the mesenchymal-like molecular subtype and predicts disease outcome in colorectal cancer. EBioMedicine 2019, 46, 94–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.R.; Arantes, C.P.; Muras, A.G.; Nomizo, R.; Brentani, R.R.; Martins, V.R. Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J. Neurochem. 2007, 103, 2164–2176. [Google Scholar] [CrossRef] [PubMed]
- Arantes, C.; Nomizo, R.; Lopes, M.H.; Hajj, G.N.; Lima, F.R.; Martins, V.R. Prion protein and its ligand stress inducible protein 1 regulate astrocyte development. Glia 2009, 57, 1439–1449. [Google Scholar] [CrossRef]
- Hartmann, C.A.; Martins, V.R.; Lima, F.R.S. High levels of cellular prion protein improve astrocyte development. FEBS Lett. 2013, 587, 238–244. [Google Scholar] [CrossRef]
- Büeler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef]
- Moore, R.C.; Lee, I.Y.; Silverman, G.L.; Harrison, P.M.; Strome, R.; Heinrich, C.; Karunaratne, A.; Pasternak, S.H.; Chishti, M.A.; Liang, Y.; et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J. Mol. Biol. 1999, 292, 797–817. [Google Scholar] [CrossRef]
- Watts, J.C.; Drisaldi, B.; Ng, V.; Yang, J.; Strome, B.; Horne, P.; Sy, M.S.; Yoong, L.; Young, R.; Mastrangelo, P.; et al. The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J. 2007, 26, 4038–4050. [Google Scholar] [CrossRef]
- Prestori, F.; Rossi, P.; Bearzatto, B.; Lainé, J.; Necchi, D.; Diwakar, S.; Schiffmann, S.N.; Axelrad, H.; D’Angelo, E. Altered neuron excitability and synaptic plasticity in the cerebellar granular layer of juvenile prion protein knock-out mice with impaired motor control. J. Neurosci. 2008, 28, 7091–7103. [Google Scholar] [CrossRef]
- Senatore, A.; Colleoni, S.; Verderio, C.; Restelli, E.; Morini, R.; Condliffe, S.B.; Bertani, I.; Mantovani, S.; Canovi, M.; Micotti, E.; et al. Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC α(2)δ-1 Subunit. Neuron 2012, 74, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Mangé, A.; Dong, L.; Lehmann, S.; Schachner, M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell Neurosci. 2003, 22, 227–233. [Google Scholar] [CrossRef]
- Chiarini, L.B.; Freitas, A.R.; Zanata, S.M.; Brentani, R.R.; Martins, V.R.; Linden, R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002, 21, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.; Cabral, A.L.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.H.; Hajj, G.N.; Muras, A.G.; Mancini, G.L.; Castro, R.M.; Ribeiro, K.C.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [PubMed]
- Spielhaupter, C.; Schätzl, H.M. PrPC directly interacts with proteins involved in signaling pathways. J. Biol. Chem. 2001, 276, 44604–44612. [Google Scholar] [CrossRef] [PubMed]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Forlenza, O.V.; Cabral, A.L.; Veiga, S.S.; Juliano, M.A.; Roesler, R.; Walz, R.; Minetti, A.; et al. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. Mol. Brain Res. 2000, 76, 85–92. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Machado, C.F.; Roffe, M.; Hajj, G.N.; Lee, K.S.; Magalhães, A.C.; Caetano, F.A.; Mancini, G.L.; et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin γ1 chain. FASEB J. 2011, 25, 265–279. [Google Scholar] [CrossRef]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225. [Google Scholar] [CrossRef]
- Mohanty, S.T.; Cairney, C.J.; Chantry, A.D.; Madan, S.; Fernandes, J.A.; Howe, S.J.; Moore, H.D.; Thompson, M.J.; Chen, B.; Thrasher, A.; et al. A small molecule modulator of prion protein increases human mesenchymal stem cell lifespan, ex vivo expansion, and engraftment to bone marrow in NOD/SCID mice. Stem Cells 2012, 30, 1134–1143. [Google Scholar] [CrossRef]
- Lee, Y.J.; Baskakov, I.V. Treatment with normal prion protein delays differentiation and helps to maintain high proliferation activity in human embryonic stem cells. J. Neurochem. 2010, 114, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Martellucci, S.; Santacroce, C.; Santilli, F.; Piccoli, L.; Delle Monache, S.; Angelucci, A.; Misasi, R.; Sorice, M.; Mattei, V. Cellular and Molecular Mechanisms Mediated by recPrP(C) Involved in the Neuronal Differentiation Process of Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 345. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Emsley, J.G.; Ozdinler, P.H.; Lindquist, S.; Macklis, J.D. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 3416–3421. [Google Scholar] [CrossRef] [PubMed]
- Prodromidou, K.; Papastefanaki, F.; Sklaviadis, T.; Matsas, R. Functional cross-talk between the cellular prion protein and the neural cell adhesion molecule is critical for neuronal differentiation of neural stem/precursor cells. Stem Cells 2014, 32, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Baskakov, I.V. The cellular form of the prion protein guides the differentiation of human embryonic stem cells into neuron-, oligodendrocyte-, and astrocyte-committed lineages. Prion 2014, 8, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. Biomed. Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef]
- Mange, A.; Beranger, F.; Poec’h, K.; Onodera, T.; Frobert, Y.; Lehmann, S. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol. Cell 2004, 96, 125–132. [Google Scholar] [CrossRef]
- Watt, N.T.; Taylor, D.R.; Gillott, A.; Thomas, D.A.; Perera, W.S.S.; Hooper, N.M. Reactive oxygen species-mediated β-cleavage of the prion protein in the cellular response to oxidative stress. J. Biol. Chem. 2005, 280, 35914–35921. [Google Scholar] [CrossRef]
- Haigh, C.L.; McGlade, A.R.; Collins, S.J. MEK1 transduces the prion protein N2 fragment antioxidant effects. Cell Mol. Life Sci. 2015, 72, 1613–1629. [Google Scholar] [CrossRef]
- Février, B.; Laude, H.; Raposo, G.; Vilette, D. Exosomes: Carriers of prions? Med. Sci. 2005, 21, 132–133. [Google Scholar]
- Hajj, G.N.; Arantes, C.P.; Dias, M.V.; Roffé, M.; Costa-Silva, B.; Lopes, M.H.; Porto-Carreiro, I.; Rabachini, T.; Lima, F.R.; Beraldo, F.H.; et al. The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell Mol. Life Sci. 2013, 70, 3211–3227. [Google Scholar] [CrossRef] [PubMed]
- Abdulrahman, B.A.; Abdelaziz, D.H.; Schatzl, H.M. Autophagy regulates exosomal release of prions in neuronal cells. J. Biol. Chem. 2018, 293, 8956–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, M.V.; Teixeira, B.L.; Rodrigues, B.R.; Sinigaglia-Coimbra, R.; Porto-Carreiro, I.; Roffé, M.; Hajj, G.N.; Martins, V.R. PRNP/prion protein regulates the secretion of exosomes modulating CAV1/caveolin-1-suppressed autophagy. Autophagy 2016, 12, 2113–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.C.; Lin, C.H.; Shih, N.C.; Liu, H.L.; Wang, W.C.; Lin, K.Y.; Liu, Z.Y.; Tseng, Y.J.; Chang, H.K.; Lin, Y.C.; et al. Cellular prion protein transcriptionally regulated by NFIL3 enhances lung cancer cell lamellipodium formation and migration through JNK signaling. Oncogene 2019. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, L.; Ryan, Y.; Hilton, D.A.; Lyons-Rimmer, J.; Dave, F.; Maze, E.A. Cellular prion protein (PrPC) in the development of Merlin-deficient tumours. Oncogene 2017, 36, 6132–6142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, M.H.; Santos, T.G.; Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Cunha, I.W.; Wasilewska-Sampaio, A.P.; Costa-Silva, B.; Marchi, F.A.; Bleggi-Torres, L.F.; Sanematsu, P.I.; et al. Disruption of prion protein-HOP engagement impairs glioblastoma growth and cognitive decline and improves overall survival. Oncogene 2015, 34, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Tao, L.; Xu, J.; Li, Q.; Yu, J.; Jin, Y.; Chen, Q.; Xu, Z.; Zou, Q.; Liu, X. CD44/cellular prion protein interact in multidrug resistant breast cancer cells and correlate with responses to neoadjuvant chemotherapy in breast cancer patients. Mol. Carcinog. 2014, 53, 686–697. [Google Scholar] [CrossRef]
- de Lacerda, T.C.S.; Costa-Silva, B.; Giudice, F.S.; Dias, M.V.S.; de Oliveira, G.P.; Teixeira, B.L. Prion protein binding to HOP modulates the migration and invasion of colorectal cancer cells. Clin. Exp. Metastasis. 2016, 33, 441–451. [Google Scholar] [CrossRef]
- Chieng, C.K.-L.; Say, Y.-H. Cellular prion protein contributes to LS 174T colon cancer cell carcinogenesis by increasing invasiveness and resistance against doxorubicin-induced apoptosis. Tumour Biol. J. Int. Soc. Oncodeve Biol. Med. 2015, 36, 8107–8120. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Kakeya, T.; Yamazaki, T.; Takekida, K.; Nakamura, N.; Matsuda, H.; Takatori, K.; Tanimura, A.; Tanamoto, K.; Sawada, J. G1-dependent prion protein expression in human glioblastoma cell line T98G. Biol. Pharm. Bull. 2002, 25, 728–733. [Google Scholar] [CrossRef]
- Erlich, R.B.; Kahn, S.A.; Lima, F.R.; Muras, A.G.; Martins, R.A.; Linden, R.; Chiarini, L.B.; Martins, V.R.; Moura Neto, V. STI1 promotes glioma proliferation through MAPK and PI3K pathways. Glia 2007, 55, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, A.; Sbalchiero, E.; Barbieri, G.; Palumbo, S.; Muzzini, C.; Comincini, S. The doppel (Dpl) protein influences in vitro migration capability in astrocytoma-derived cells. Cell Oncol. 2008, 30, 491–501. [Google Scholar] [PubMed]
- Fonseca, A.C.; Romão, L.; Amaral, R.F.; Assad Kahn, S.; Lobo, D.; Martins, S.; Marcondes de Souza, J.; Moura-Neto, V.; Lima, F.R. Microglial stress inducible protein 1 promotes proliferation and migration in human glioblastoma cells. Neuroscience 2012, 200, 130–141. [Google Scholar] [CrossRef]
- Aguib, Y.; Heiseke, A.; Gilch, S.; Riemer, C.; Baier, M.; Schätzl, H.M.; Ertmer, A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009, 5, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Arcella, F.; Biagioni, M.; Oliva, A.; Bucci, D.; Frati, A.; Esposito, V.; Cantore, G.; Giangaspero, F.; Fornai, F. Rapamycin inhibits the growth of glioblastoma. Brain Res. 2013, 1495, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Biagioni, F.; Lenzi, P.; Gambardella, S.; Ferese, R.; Calierno, M.T.; Falleni, A.; Grimaldi, A.; Frati, A.; Esposito, V.; et al. Rapamycin promotes differentiation increasing βIII-tubulin, NeuN, and NeuroD while suppressing nestin expression in glioblastoma cells. Oncotarget 2017, 8, 29574–29599. [Google Scholar] [CrossRef]
- Cui, J.; He, W.; Yi, B.; Zhao, H.; Lu, K.; Ruan, H.; Ma, D. mTOR pathway is involved in ADP-evoked astrocyte activation and ATP release in the spinal dorsal horn in a rat neuropathic pain model. Neuroscience 2014, 275, 395–403. [Google Scholar] [CrossRef]
- Goldshmit, Y.; Kanner, S.; Zacs, M.; Frisca, F.; Pinto, A.R.; Currie, P.D.; Pinkas-Kramarski, R. Rapamycin increases neuronal survival, reduces inflammation and astrocyte proliferation after spinal cord injury. Mol. Cell. Neurosci. 2015, 68, 82–91. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [Green Version]
- Rodolfo, C.; Di Bartolomeo, S.; Cecconi, F. Autophagy in stem and progenitor cells. Cell. Mol. Life Sci. 2016, 73, 475–496. [Google Scholar] [CrossRef]
- Ryskalin, L.; Limanaqi, F.; Biagioni, F.; Frati, A.; Esposito, V.; Calierno, M.T.; Lenzi, P.; Fornai, F. The emerging role of m-TOR up-regulation in brain Astrocytoma. Histol. Histopathol. 2017, 32, 413–431. [Google Scholar] [PubMed]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Li, T.; Ma, H.; Yang, Y.; Zhang, C.; Hai, L.; Liu, P.; Yuan, F.; Li, J.; Yi, L.; et al. Autophagy suppresses self-renewal ability and tumorigenicity of glioma-initiating cells and promotes Notch1 degradation. Cell Death Dis. 2018, 9, 1063. [Google Scholar] [CrossRef]
- Barbieri, G.; Palumbo, S.; Gabrusiewicz, K.; Azzalin, A.; Marchesi, N.; Spedito, A.; Biggiogera, M.; Sbalchiero, E.; Mazzini, G.; Miracco, C.; et al. Silencing of cellular prion protein (PrPC) expression by DNA-antisense oligonucleotides induces autophagy-dependent cell death in glioma cells. Autophagy 2011, 7, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Li, C.C.; Eaton, S.A.; Young, P.E.; Lee, M.; Shuttleworth, R.; Humphreys, D.T.; Grau, G.E.; Combes, V.; Bebawy, M.; Gong, J.; et al. Glioma microvesicles carry selectively packaged coding and non-coding RNAs which alter gene expression in recipient cells. RNA Biol. 2013, 10, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Sun, J.; Lan, Q. Glioblastoma microvesicles promote endothelial cell proliferation through Akt/beta-catenin pathway. Int. J. Clin. Exp. Pathol. 2014, 7, 4857–4866. [Google Scholar]
- Li, C.; Yu, S.; Nakamura, F.; Pentikäinen, O.T.; Singh, N.; Yin, S.; Xin, W.; Sy, M.S. Pro-prion binds filamin A, facilitating its interaction with integrin beta1, and contributes to melanomagenesis. J. Biol. Chem. 2010, 285, 30328–30339. [Google Scholar] [CrossRef]
- Jiang, B.; Liu, J.; Lee, M.H. Targeting a Designer TIMP-1 to the Cell Surface for Effective MT1-MMP Inhibition: A Potential Role for the Prion Protein in Renal Carcinoma Therapy. Molecules 2019, 24, 255. [Google Scholar] [CrossRef]
- Wiegmans, A.P.; Saunus, J.M.; Ham, S.; Lobb, R.; Kutasovic, J.R.; Dalley, A.J.; Miranda, M.; Atkinson, C.; Foliaki, S.T.; Ferguson, K.; et al. Secreted cellular prion protein binds doxorubicin and correlates with anthracycline resistance in breast cancer. JCI Insight 2019, 4, e124092. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.; Mishima, Y. Increased in vitro radio-sensitivity of malignant melanoma induced by the in vivo administration of chlorpromazine. British J. Dermatol. 1972, 86, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Yde, C.W.; Clausen, M.P.; Bennetzen, M.V.; Lykkesfeldt, A.E.; Mouritsen, O.G.; Guerra, B. The antipsychotic drug chlorpromazine enhances the cytotoxic effect of tamoxifen in tamoxifen-sensitive and tamoxifen-resistant human breast cancer cells. Anti-Cancer Drugs 2009, 20, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Zhang, W.; Langford, C.; Suto, M.J.; Griguer, C.E. Repositioning chlorpromazine for treating chemoresistant glioma through the inhibition of cytochrome c oxidase bearing the COX4-1 regulatory subunit. Oncotarget 2017, 8, 37568–37583. [Google Scholar] [CrossRef]
- Luo, G.; Wang, W.; Wu, Q.; Lu, Y.; Su, T.; Gu, N.; Li, K.; Wang, J.; Du, R.; Zhao, X.; et al. MGr1-Antigen/37 kDa laminin receptor precursor promotes cellular prion protein induced multi-drug-resistance of gastric cancer. Oncotarget 2017, 8, 71630–71641. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [Green Version]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell. Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, Y.S.; Yoon, Y.M.; Yun, C.W.; Yun, S.P.; Kim, S.M.; Kwon, H.Y.; Jeong, D.; Baek, M.J.; Lee, H.J.; et al. Role of HSPA1L as a cellular prion protein stabilizer in tumor progression via HIF-1α/GP78 axis. Oncogene 2017, 36, 6555–6567. [Google Scholar] [CrossRef]
- Yi, L.; Zhou, X.; Li, T.; Liu, P.; Hai, L.; Tong, L.; Ma, H.; Tao, Z.; Xie, Y.; Zhang, C.; et al. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J. Exp. Clin. Cancer Res. 2019, 38, 339. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryskalin, L.; Busceti, C.L.; Biagioni, F.; Limanaqi, F.; Familiari, P.; Frati, A.; Fornai, F. Prion Protein in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5107. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205107
Ryskalin L, Busceti CL, Biagioni F, Limanaqi F, Familiari P, Frati A, Fornai F. Prion Protein in Glioblastoma Multiforme. International Journal of Molecular Sciences. 2019; 20(20):5107. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205107
Chicago/Turabian StyleRyskalin, Larisa, Carla L. Busceti, Francesca Biagioni, Fiona Limanaqi, Pietro Familiari, Alessandro Frati, and Francesco Fornai. 2019. "Prion Protein in Glioblastoma Multiforme" International Journal of Molecular Sciences 20, no. 20: 5107. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205107