Autosomal Dominant Retinitis Pigmentosa Due to Class B Rhodopsin Mutations: An Objective Outcome for Future Treatment Trials


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
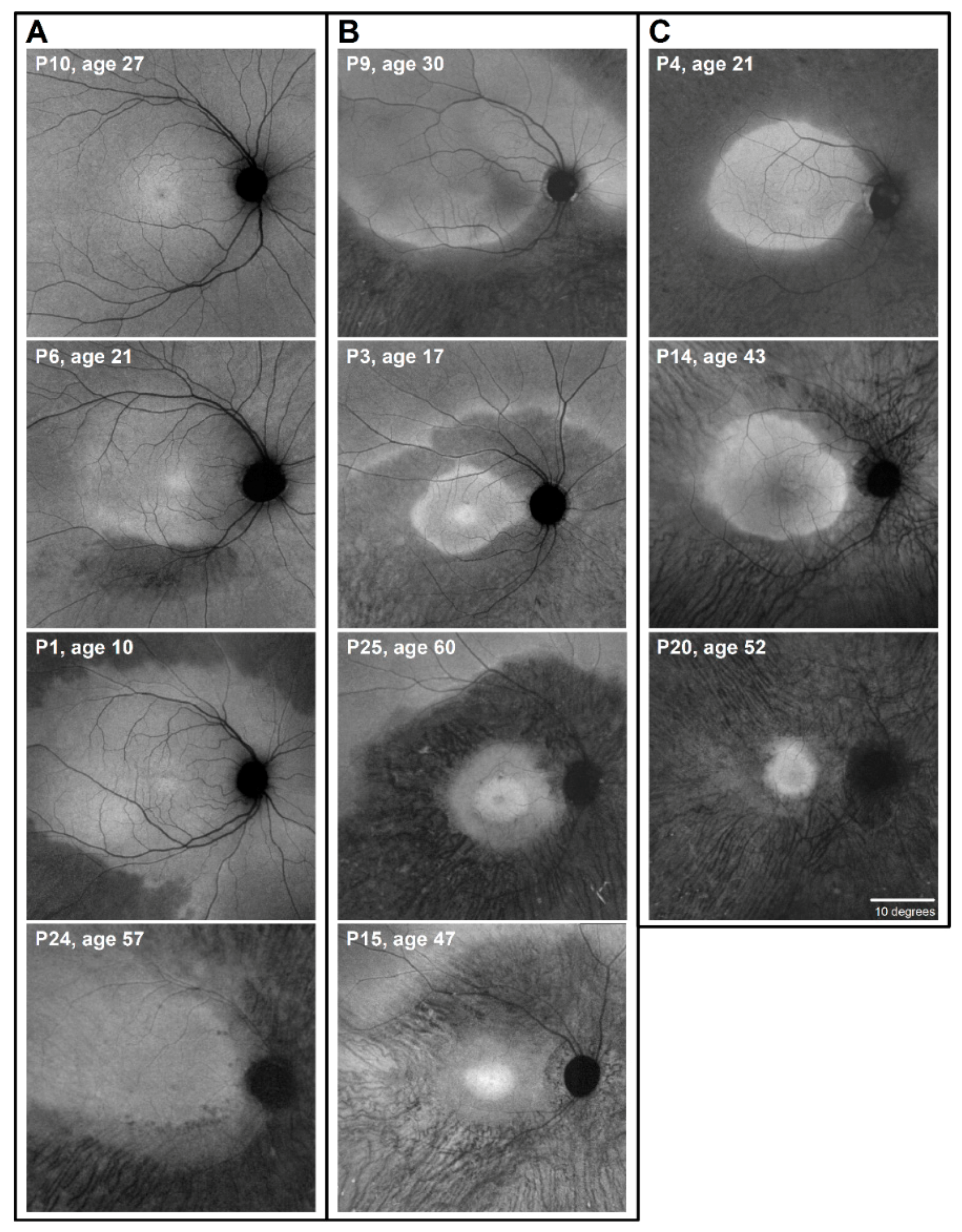
2.1. Autofluorescence (AF) Imaging as an Outcome Measure in RHO-adRP
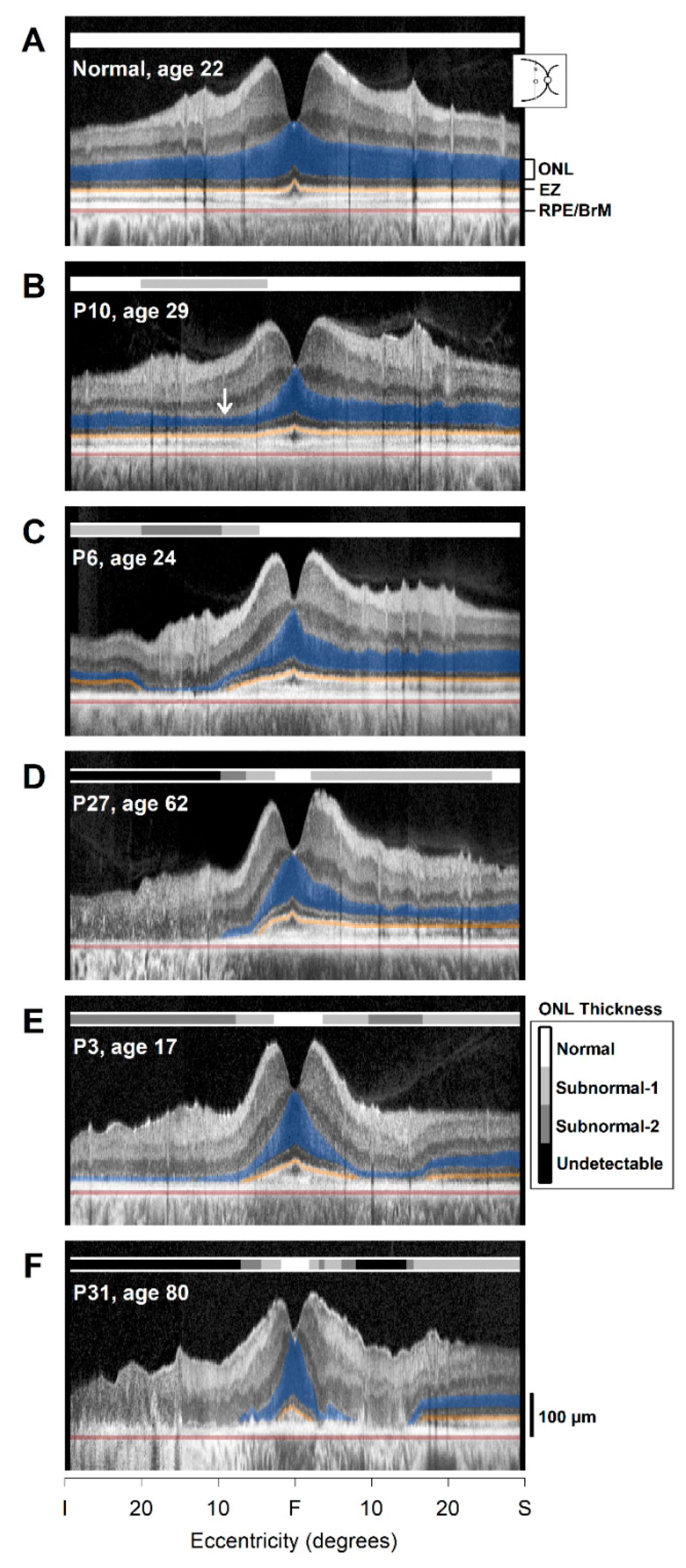
2.2. Photoreceptor Layer Imaging with OCT: Cross-Sectional Observations
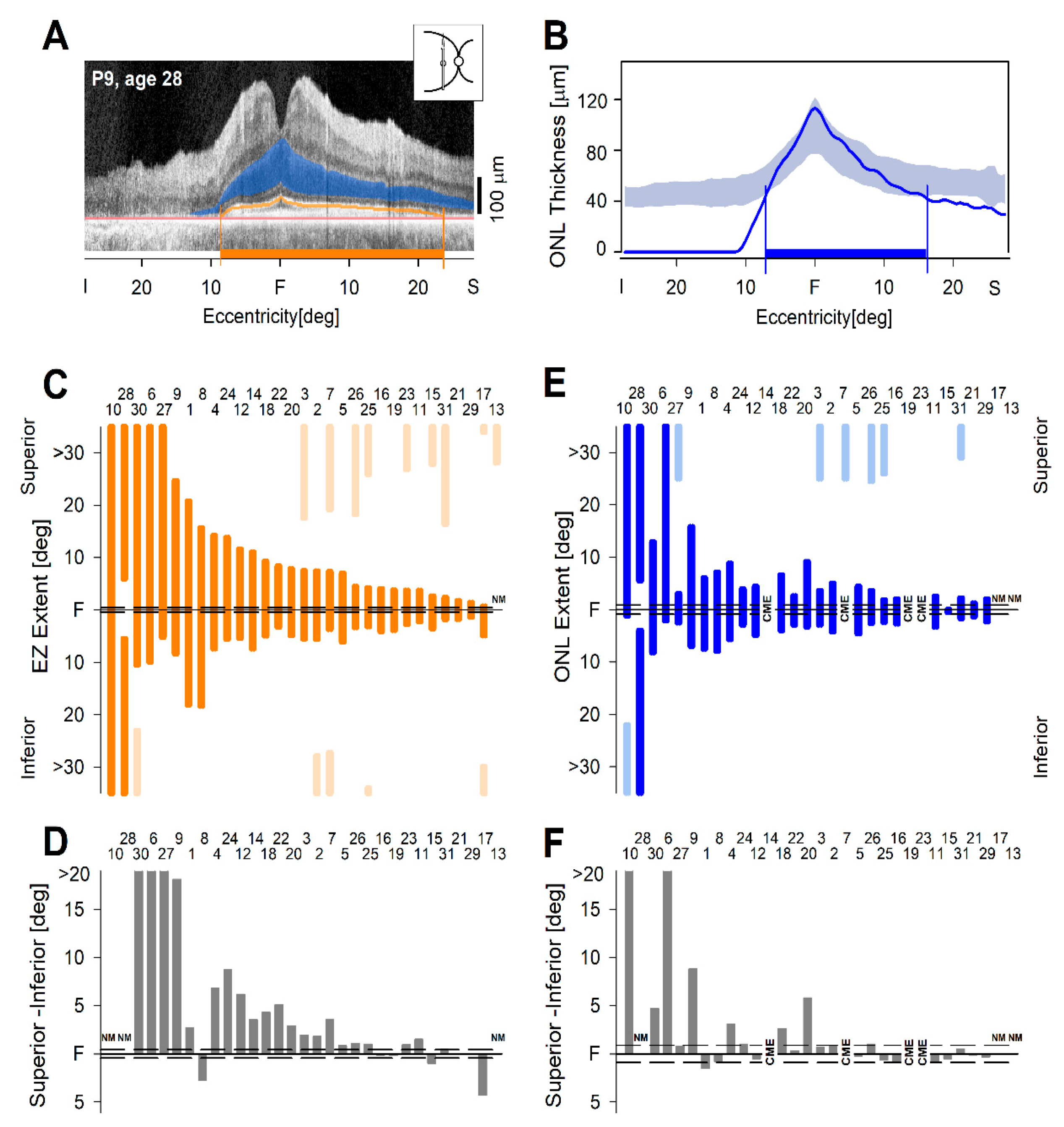
2.3. From Observations to Quantitation of the Cross-Sectional OCT Data
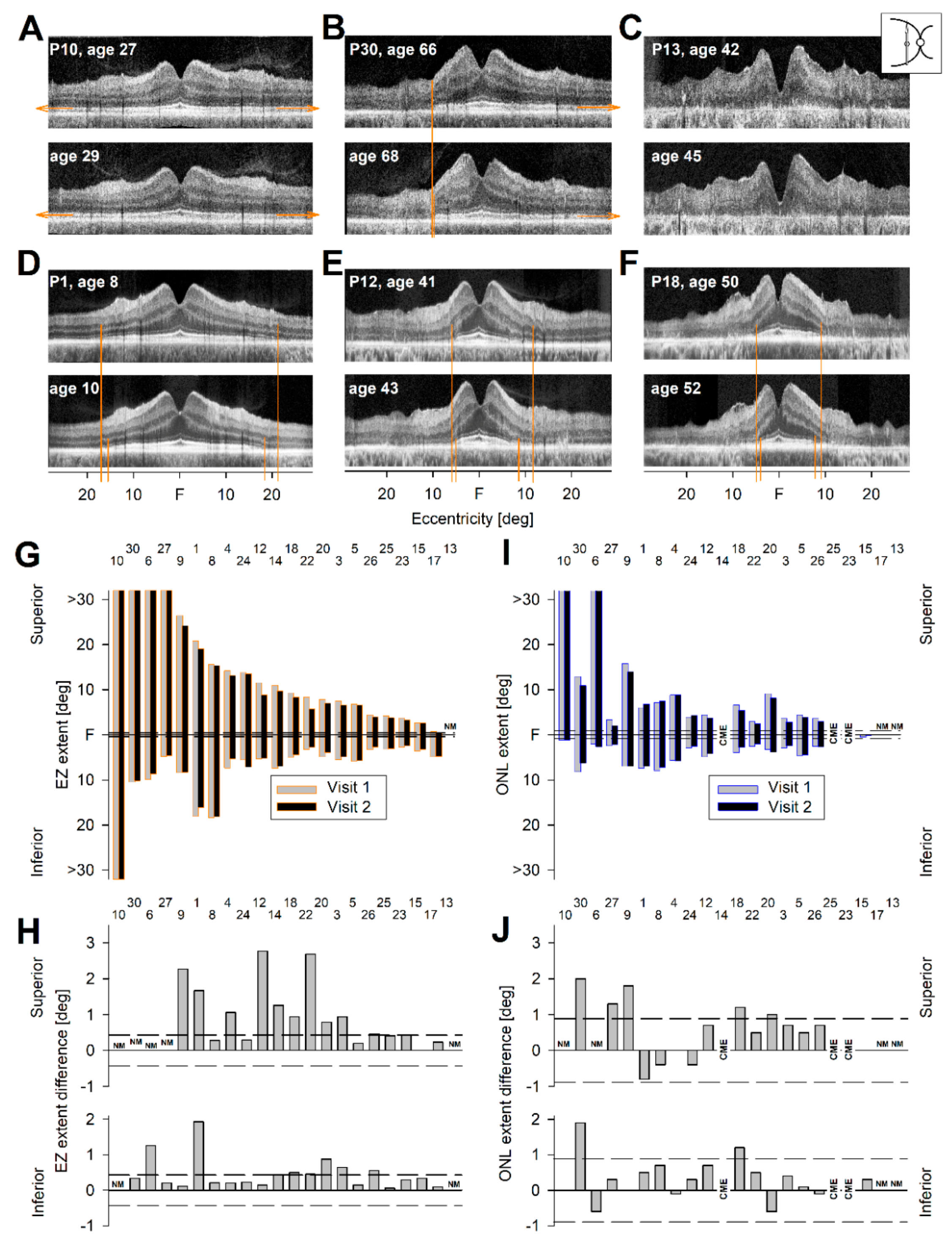
2.4. Longitudinal OCT Data in RHO-adRP
2.5. An Objective Outcome for a Clinical Trial of RHO-adRP
3. Summary and Conclusions
4. Materials and Methods
4.1. Subjects
4.2. Near-Infrared Autofluorescence Imaging
4.3. Optical Coherence Tomography
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bird, A.C. Retinitis pigmentosa—a review. Trans. Ophthalmol. Soc. NZ 1977, 29, 51–58. [Google Scholar]
- Flannery, J.G.; Farber, D.B.; Bird, A.C.; Bok, D. Degenerative changes in a retina affected with autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 1989, 30, 191–211. [Google Scholar]
- Milam, A.H.; Li, Z.Y.; Fariss, R.N. Histopathology of the human retina in retinitis pigmentosa. Prog. Retin. Eye Res. 1998, 17, 175–205. [Google Scholar] [PubMed]
- Bramall, A.N.; Wright, A.F.; Jacobson, S.G.; McInnes, R.R. The genomic, biochemical, and cellular responses of the retina in inherited photoreceptor degenerations and prospects for the treatment of these disorders. Annu. Rev. Neurosci. 2010, 33, 441–472. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.F.; Chakarova, C.F.; Abd El-Aziz, M.M.; Bhattacharya, S.S. Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010, 11, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Hardcastle, A.J.; Sieving, P.A.; Sahel, J.A.; Jacobson, S.G.; Cideciyan, A.V.; Flannery, J.G.; Beltran, W.A.; Aguirre, G.D. Translational retinal research and therapies. Transl. Vis. Sci. Technol. 2018, 7, 8. [Google Scholar] [CrossRef]
- Takahashi, V.K.L.; Takiuti, J.T.; Jauregui, R.; Tsang, S.H. Gene therapy in inherited retinal degenerative diseases, a review. Ophthalmic Genet. 2018, 39, 560–568. [Google Scholar] [CrossRef]
- Miyadera, K. Inherited retinal diseases in dogs: Advances in gene/mutation discovery. Dobutsu Iden Ikushu Kenkyu 2014, 42, 79–89. [Google Scholar]
- Veleri, S.; Lazar, C.H.; Chang, B.; Sieving, P.A.; Banin, E.; Swaroop, A. Biology and therapy of inherited retinal degenerative disease: Insights from mouse models. Dis. Model. Mech. 2015, 8, 109–129. [Google Scholar] [CrossRef]
- Da Cruz, L.; Dorn, J.D.; Humayun, M.S.; Dagnelie, G.; Handa, J.; Barale, P.O.; Sahel, J.A.; Stanga, P.E.; Hafezi, F.; Safran, A.B.; et al. Five-year safety and performance results from the Argus II retinal prosthesis system clinical trial. Ophthalmology 2016, 123, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Simunovic, M.P.; Shen, W.; Lin, J.Y.; Protti, D.A.; Lisowski, L.; Gillies, M.C. Optogenetic approaches to vision restoration. Exp. Eye Res. 2019, 178, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Aleman, T.S.; Cideciyan, A.V.; Sumaroka, A.; Schwartz, S.B.; Windsor, E.A.; Traboulsi, E.I.; Heon, E.; Pittler, S.J.; Milam, A.H.; et al. Identifying photoreceptors in blind eyes caused by RPE65 mutations: Prerequisite for human gene therapy success. Proc. Natl. Acad. Sci. USA 2005, 102, 6177–6182. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Jacobson, S.G. Leber congenital amaurosis (LCA): Potential for improvement of vision. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1680–1695. [Google Scholar] [CrossRef] [PubMed]
- Sumaroka, A.S.; Garafalo, A.V.; Semenov, E.P.; Sheplock, R.; Krishnan, A.K.; Roman, A.J.; Jacobson, S.G.; Cideciyan, A.V. Treatment potential for macular cone vision in Leber congenital amaurosis due to CEP290 or NPHP5 mutations: Predictions from artificial intelligence. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2551–2562. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Heon, E.; Hauswirth, W.W. Improvement and decline in vision with gene therapy in childhood blindness. N. Engl. J. Med. 2015, 372, 1920–1926. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Kumaran, N.; Michaelides, M.; Smith, A.J.; Ali, R.R.; Bainbridge, J.W.B. Retinal gene therapy. Br. Med. Bull. 2018, 126, 13–25. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Han, I.C.; Hochstedler, M.D.; et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019, 25, 225–228. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Hayes, K.C.; Nicholson, B.W.; Weigel-DiFranco, C.; Willett, W. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch. Ophthalmol. 1993, 111, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.R.; Locke, K.G.; Wheaton, D.H.; Fish, G.E.; Spencer, R.; Birch, D.G. A randomized, placebo-controlled clinical trial of docosahexaenoic acid supplementation for X-linked retinitis pigmentosa. Am. J. Ophthalmol. 2004, 137, 704–718. [Google Scholar] [PubMed]
- Sacchetti, M.; Mantelli, F.; Merlo, D.; Lambiase, A. Systematic review of randomized clinical trials on safety and efficacy of pharmacological and nonpharmacological treatments for retinitis pigmentosa. J. Ophthalmol. 2015, 2015, 737053. [Google Scholar] [CrossRef] [PubMed]
- Birch, D.G.; Locke, K.G.; Wen, Y.; Locke, K.I.; Hoffman, D.R.; Hood, D.C. Spectral-domain optical coherence tomography measures of outer segment layer progression in patients with X-linked retinitis pigmentosa. JAMA Ophthalmol. 2013, 131, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Hood, D.C.; Huang, Y.; Banin, E.; Li, Z.Y.; Stone, E.M.; Jacobson, S.G. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc. Natl. Acad. Sci. USA 1998, 95, 7103–7108. [Google Scholar] [CrossRef]
- Jacobson, S.G.; McGuigan, D.B.; Sumaroka, A.; Roman, A.J.; Gruzensky, M.L.; Sheplock, R.; Palma, J.; Schwartz, S.B.; Aleman, T.S.; Cideciyan, A.V. Complexity of the class B phenotype in autosomal dominant retinitis pigmentosa due to rhodopsin mutations. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4847–4858. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef]
- Bird, A.C. Clinical investigation of retinitis pigmentosa. Aust. NZ J. Ophthalmol. 1988, 16, 189–198. [Google Scholar] [CrossRef]
- Heckenlively, J.R.; Rodriguez, J.A.; Daiger, S.P. Autosomal dominant sectoral retinitis pigmentosa two families with transversion mutation in codon 23 of rhodopsin. Arch. Ophthalmol. 1991, 109, 84–91. [Google Scholar] [CrossRef]
- Stone, E.M.; Kimura, A.E.; Nichols, B.E.; Khadivi, P.; Fishman, G.A.; Sheffield, V.C. Regional distribution of retinal degeneration in patients with proline to histidine mutation in codon 23 of the rhodopsin gene. Ophthalmology 1991, 98, 1806–1813. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Kemp, C.M.; Sung, C.H.; Nathans, J. Retinal function and rhodopsin levels in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am. J. Ophthalmol. 1991, 112, 256–271. [Google Scholar] [CrossRef]
- Fishman, G.A.; Stone, E.M.; Gilbert, L.D.; Kenna, P.; Sheffield, V.C. Ocular findings associated with rhodopsin gene codon 58 transversion mutation in autosomal dominant retinitis pigmentosa. Arch. Ophthalmol. 1991, 109, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Matsui, R.; Cideciyan, A.V.; Schwartz, S.B.; Sumaroka, A.; Roman, A.J.; Swider, M.; Huang, W.C.; Sheplock, R.; Jacobson, S.G. Molecular heterogeneity within the clinical diagnosis of pericentral retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6007–6018. [Google Scholar] [CrossRef] [PubMed]
- Grøndahl, J.; Riise, R.; Heiberg, A.; Leren, T.; Christoffersen, T.; Bragadottir, R. Autosomal dominant retinitis pigmentosa in Norway: A 20-year clinical follow-up study with molecular genetic analysis. Two novel rhodopsin mutations: 1003delG and I179F. Acta Ophthalmol. Scand. 2007, 85, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Comander, J.; Weigel-DiFranco, C.; Maher, M.; Place, E.; Wan, A.; Harper, S.; Sandberg, M.A.; Navarro-Gomez, D.; Pierce, E.A. The genetic basis of pericentral retinitis pigmentosa—a form of mild retinitis pigmentosa. Genes (Basel) 2017, 8, 256. [Google Scholar] [CrossRef]
- Takahashi, V.K.L.; Takiuti, J.T.; Carvallho, J.R.L.; Xu, C.L.; Duong, J.K.; Mahajan, V.B.; Tsang, S.H. Fundus autofluorescence and ellipsoid zone (EZ) line width can be an outcome measurement in RHO-associated autosomal dominant retinitis pigmentosa. Graefe’s Arch. Clin. Exp. Ophthalmol. 2019, 257, 725–731. [Google Scholar] [CrossRef]
- Jauregui, R.; Takahashi, V.K.L.; Park, K.S.; Cui, X.; Takiuti, J.T.; Carvaljo, J.R.L.; Tsang, S.H. Multimodal structural disease progression in retinitis pigmentosam according to mode of inheritance. Sci. Rep. 2019, 9, 10712. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Roman, M.I.; Sumaroka, A.; Schwartz, S.B.; Stone, E.M.; Jacobson, S.G. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2007, 24, 1457–1467. [Google Scholar] [CrossRef] [Green Version]
- Cideciyan, A.V.; Jacobson, S.G.; Aleman, T.S.; Gu, D.; Pearce-Kelling, S.E.; Sumaroka, A.; Acland, G.M.; Aguirre, G.D. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2005, 102, 5233–5238. [Google Scholar] [CrossRef]
- Paskowitz, D.M.; LaVail, M.M.; Duncan, J.L. Light and inherited retinal degeneration. Br. J. Ophthalmol. 2006, 90, 1060–1066. [Google Scholar] [CrossRef] [Green Version]
- Curcio, C.A.; Sloan, K.R.; Kalina, R.E.; Hendrickson, A.E. Human photoreceptor topography. J. Comp. Neurol. 1990, 292, 497–523. [Google Scholar] [CrossRef]
- Aleman, T.S.; Cideciyan, A.V.; Sumaroka, A.; Windsor, E.A.; Herrera, W.; White, D.A.; Kaushal, S.; Naidu, A.; Roman, A.J.; Schwartz, S.B.; et al. Retinal laminar architecture in human retinitis pigmentosa caused by rhodopsin gene mutations. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.X.; Locke, K.G.; Ramachandran, R.; Birch, D.G.; Hood, D.C. A comparison of progressive loss of the ellipsoid zone (EZ) band in autosomal dominant and X-linked retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7417–7422. [Google Scholar] [CrossRef] [PubMed]
- Sumaroka, A.; Matsui, R.; Cideciyan, A.V.; McGuigan, D.B.; Sheplock, R.; Schwartz, S.B.; Jacobson, S.G. Outer retinal changes including the ellipsoid zone band in Usher syndrome 1B due to MYO7A mutations. Investig. Ophthalmol. Vis. Sci. 2016, 57, OCT253–OCT261. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Aleman, T.S.; Sumaroka, A.; Cideciyan, A.V.; Roman, A.J.; Windsor, E.A.; Schwartz, S.B.; Rehm, H.L.; Kimberling, W.J. Disease boundaries in the retina of patients with Usher syndrome caused by MYO7A gene mutations. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.S.; Aleman, T.S.; Lillo, C.; Lopes, V.S.; Hughes, L.C.; Stone, E.M.; Jacobson, S.G. Harmonin in the murine retina and the retinal phenotypes of Ush1c-mutant mice and human USH1C. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3881–3889. [Google Scholar] [CrossRef] [PubMed]
- Sujirakul, T.; Lin, M.K.; Duong, J.K.; Wei, Y.; Lopez-Pintado, S.; Tsang, S.H. Multimodal imaging of central retinal disease progression in a 2-year mean follow- up of retinitis pigmentosa. Am. J. Ophthalmol. 2015, 160, 786–798. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Hufnagel, R.B.; Carroll, J.; Sumaroka, A.; Luo, X.; Schwartz, S.B.; Dubra, A.; Land, M.; Michaelides, M.; Gardner, J.C.; et al. Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Hum. Gene Ther. 2013, 24, 993–1006. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumaroka, A.; Cideciyan, A.V.; Charng, J.; Wu, V.; Powers, C.A.; Iyer, B.S.; Lisi, B.; Swider, M.; Jacobson, S.G. Autosomal Dominant Retinitis Pigmentosa Due to Class B Rhodopsin Mutations: An Objective Outcome for Future Treatment Trials. Int. J. Mol. Sci. 2019, 20, 5344. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215344
Sumaroka A, Cideciyan AV, Charng J, Wu V, Powers CA, Iyer BS, Lisi B, Swider M, Jacobson SG. Autosomal Dominant Retinitis Pigmentosa Due to Class B Rhodopsin Mutations: An Objective Outcome for Future Treatment Trials. International Journal of Molecular Sciences. 2019; 20(21):5344. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215344
Chicago/Turabian StyleSumaroka, Alexander, Artur V. Cideciyan, Jason Charng, Vivian Wu, Christian A. Powers, Bhavya S. Iyer, Brianna Lisi, Malgorzata Swider, and Samuel G. Jacobson. 2019. "Autosomal Dominant Retinitis Pigmentosa Due to Class B Rhodopsin Mutations: An Objective Outcome for Future Treatment Trials" International Journal of Molecular Sciences 20, no. 21: 5344. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215344