A Newly Established Murine Cell Line as a Model for Hepatocellular Cancer in Non-Alcoholic Steatohepatitis

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
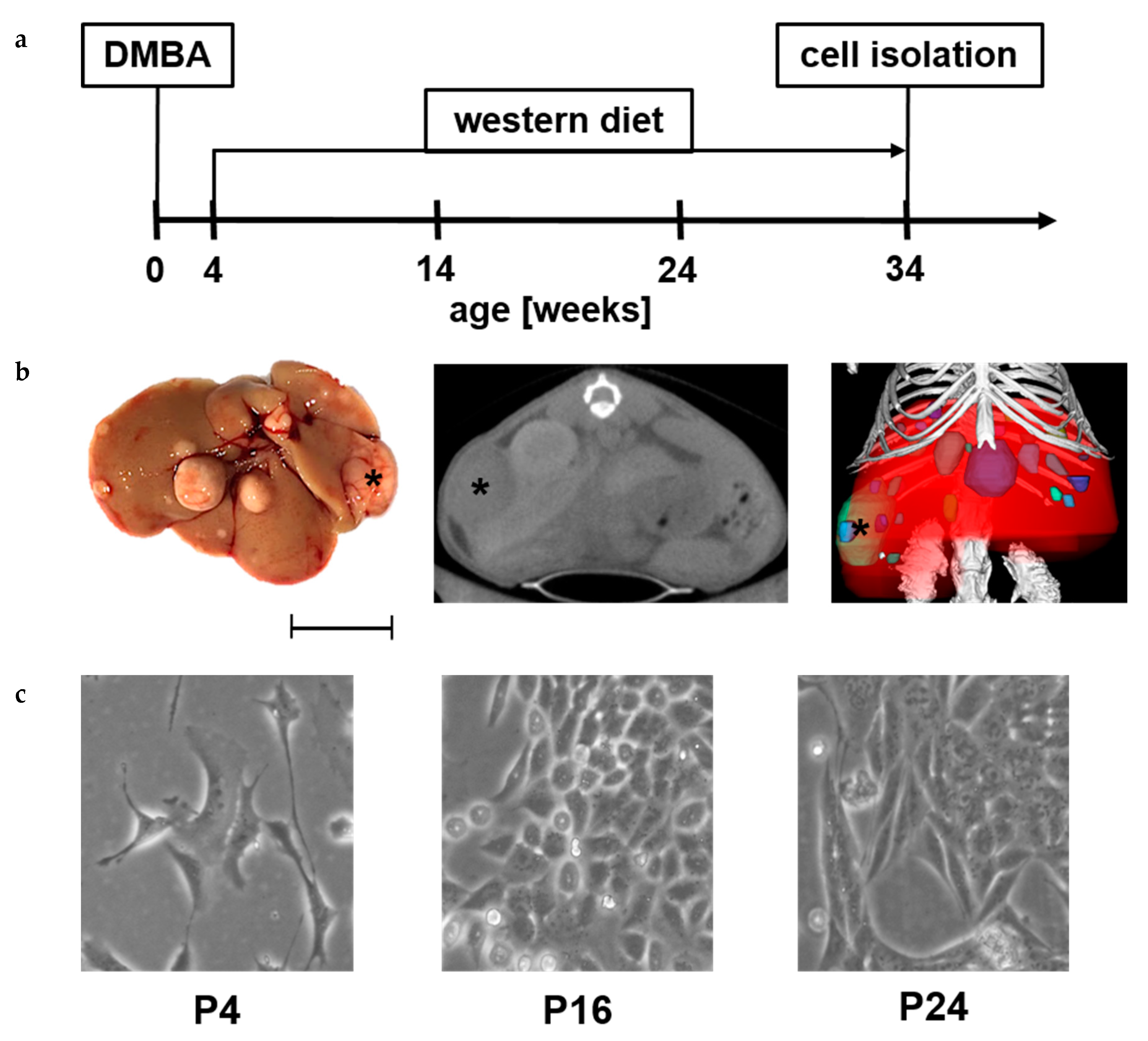
2.1. Isolation of N-HCC25 Cell Line
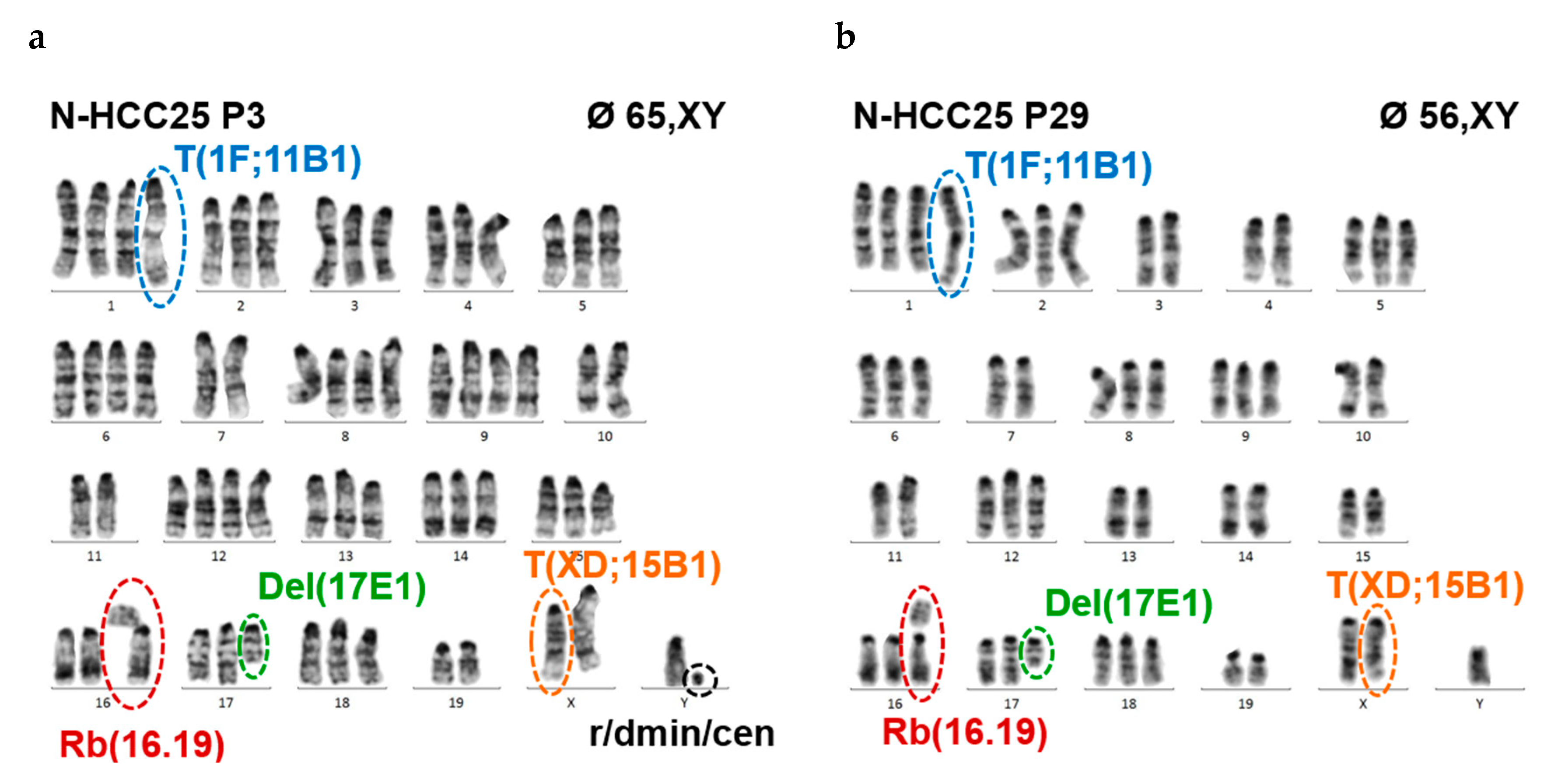
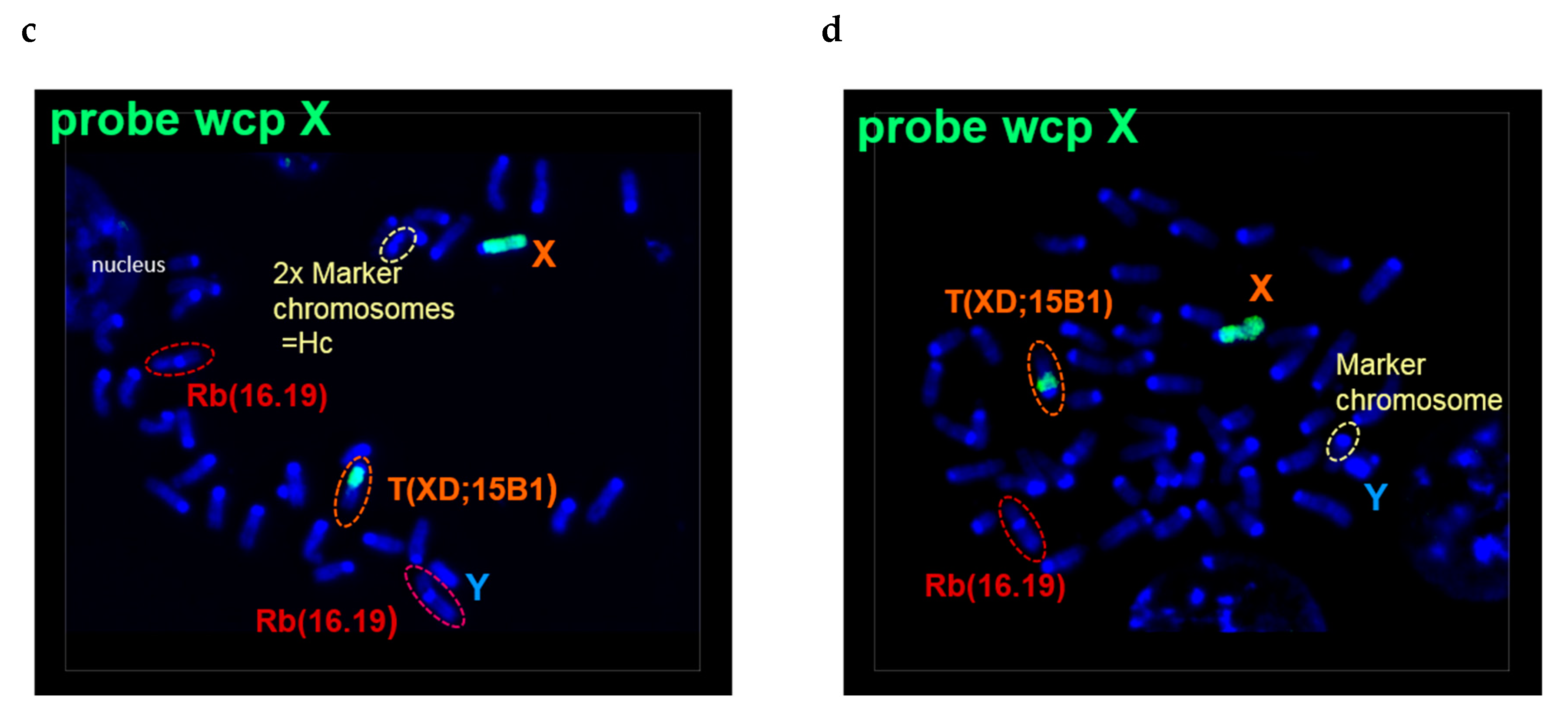
2.2. N-HCC25 Cells Exhibit Chromosomal Aberrations Associated with HCC Pathology
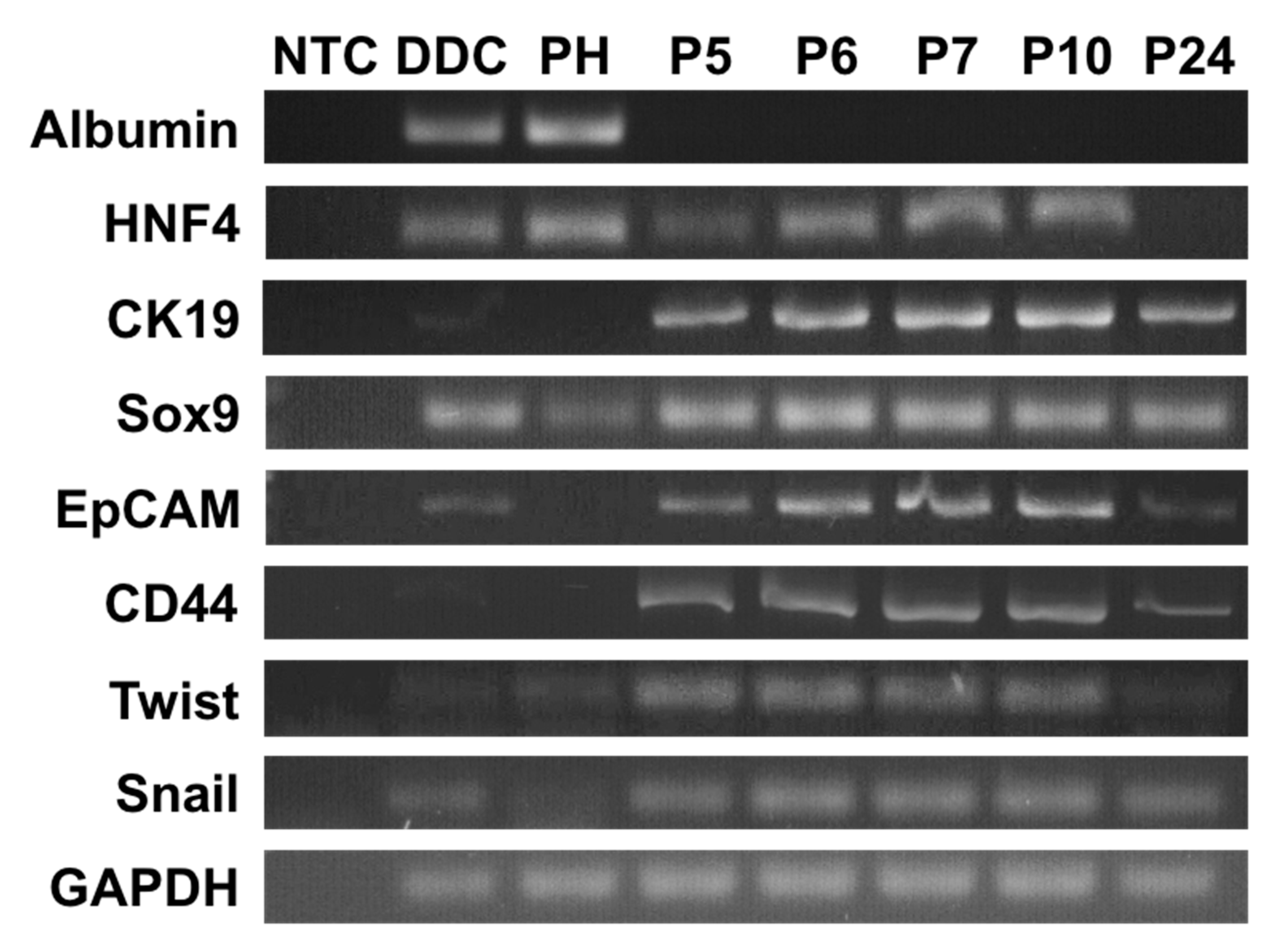
2.3. N-HCC25 Cells Derive from Hepatocytes and Express Cancer-Related Markers
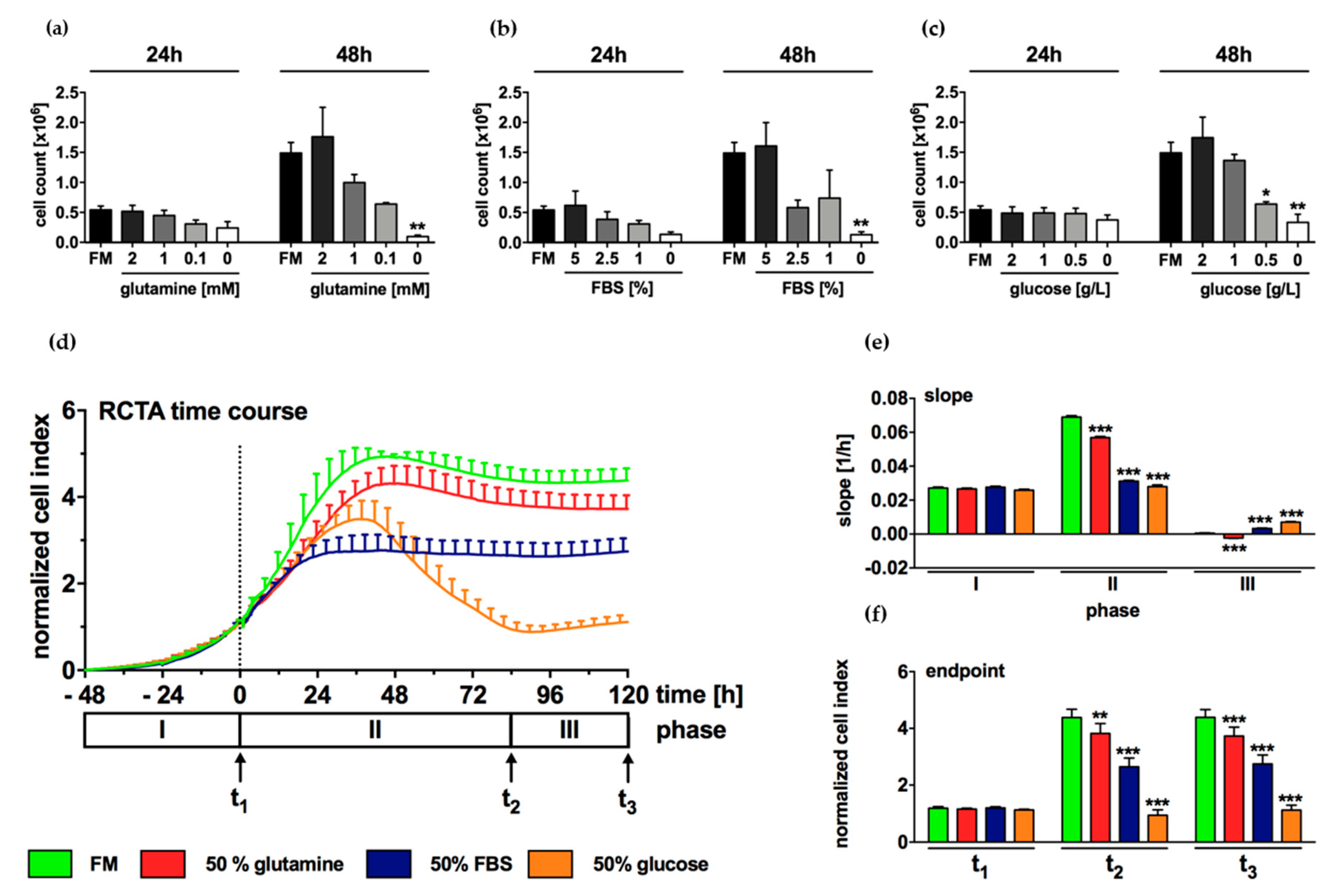
2.4. Growth of N-HCC25 Cells Mostly Depends on Sufficient Levels of Glucose and FBS
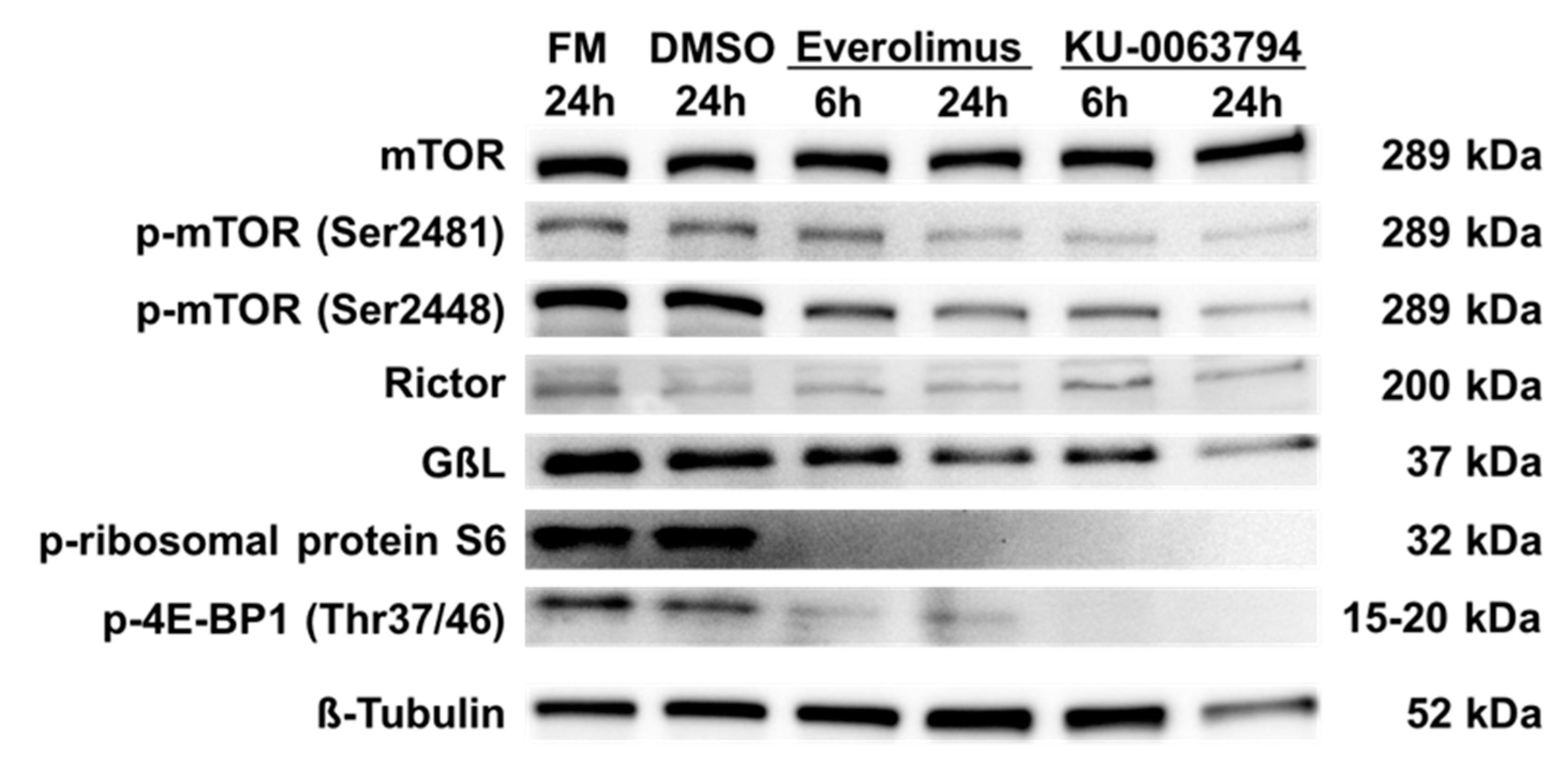
2.5. N-HCC25 Cells Exhibit mTOR Activity, which Can Be Blocked by Specific Inhibitors
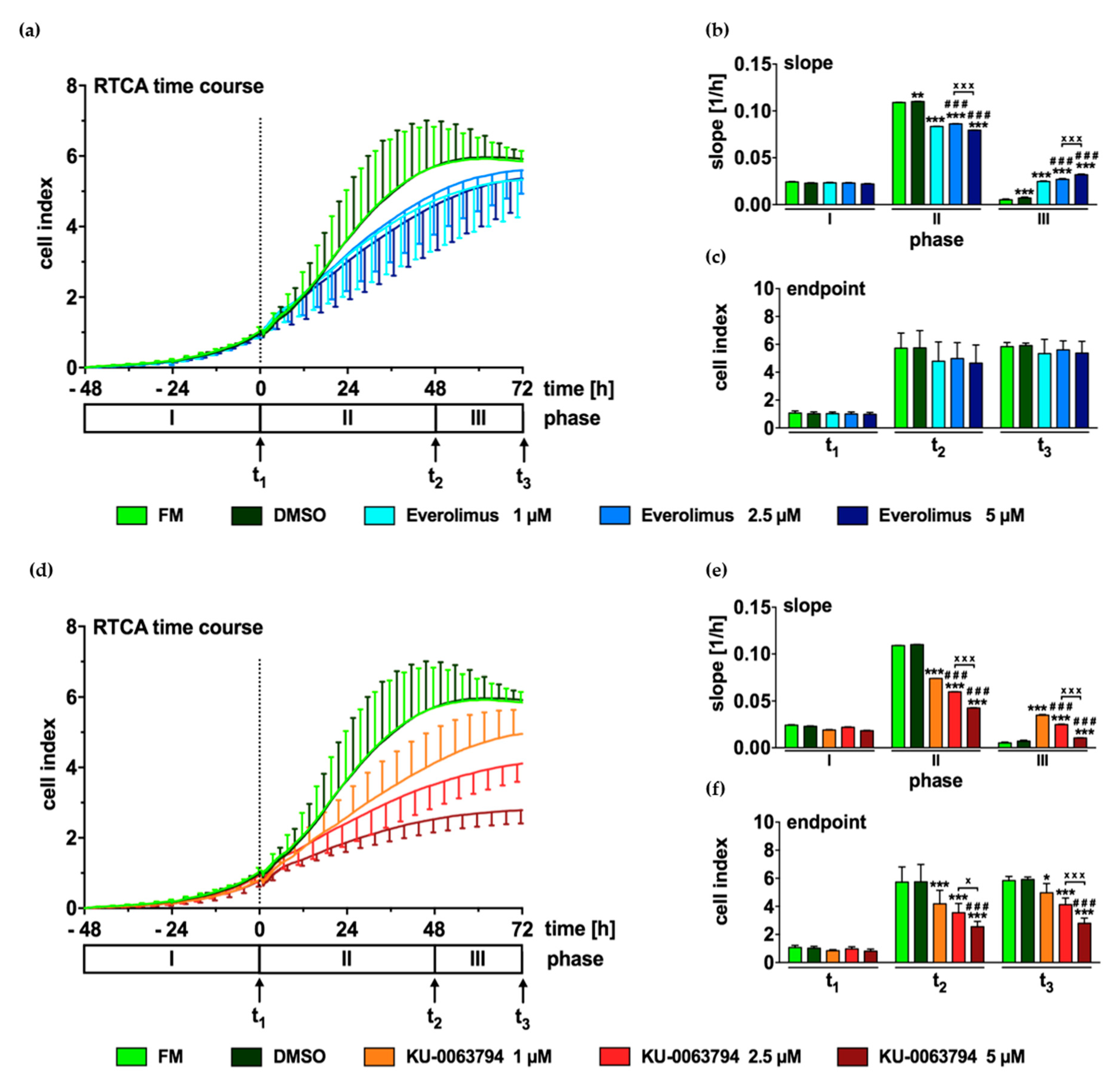
2.6. mTOR Inhibition Leads to Decreased Proliferation of N-HCC25 Cells
2.7. mTOR Pathway Inhibition Drives N-HCC25 Cells Into G1/G0 Cell Cycle Arrest
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Isolation of Primary Mouse Hepatocytes
4.3. Cell Culture and Stimulation
4.4. Cytogenetic Analysis
4.5. PCR
4.6. Western Blot
4.7. Starvation Experiments
4.8. RTCA (Real-Time Cell Analysis)
4.9. Flow Cytometry
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| AKT | protein kinase B |
| BSA | bovine serum albumin |
| CI | cell index |
| CSCs | cancer stem cells |
| DDC | liver samples from DDC (diethyl1,4-dihydro-2,4,6-trimethyl- 3,5-pyridinedicarboxylate) treated mice |
| DMBA | 7,12-Dimethylbenz[a]anthracene |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | dimethyl-sulfoxide |
| EMT | epithelial-mesenchymal transition |
| EpCAM | Epithelial cell adhesion molecule |
| EpICD | intracellular domain of EpCAM |
| FBS | fetal bovine serum |
| FISH | fluorescence in situ hybridization |
| FM | full medium control |
| G1/G0 | G1/G0-phase of cell cycle |
| G2/M | G2/M- phase of cell cycle |
| GAPDH | glycerinaldehyde- 3- phosphate- dehydrogenase |
| gDNA | genomic DNA |
| GLUT1 | glucose transporter 1 |
| GßL | G protein β-subunit-like protein, also known as mLST8 |
| HBV | hepatitis B virus |
| Hc | pericentric heterochromatin |
| HCC | Hepatocellular cancer |
| HCV | hepatitis C virus |
| HNF4 | hepatocyte nuclear factor 4 |
| HRP | horseradish peroxidase |
| LST8 | mammalian lethal with SEC13 protein 8 |
| mTOR | mechanistic (previously: mammalian) target of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| N-HCC25 | newly established cell line isolated from murine NASH-HCC model |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NTC | No template control |
| p-4E-BP1 | phosphorylated eukaryotic translation initiation factor 4E binding protein 1 |
| p-mTOR (Ser2448) | phospho-mTOR (Serin 2448) |
| p-mTOR (Ser2481) | phospho-mTOR (Serin 2481) |
| p-rp S6 | phospho-ribosomal protein S6 |
| P4 | passage of cells number 4 |
| PBS | phosphate buffered saline |
| PCR | polymerase chain reaction |
| PH | primary hepatocytes |
| PI | propidium iodide |
| PI3K | Phophoinositide-3-kinase |
| Rb | Robertsonian Fusion |
| Rictor | rapamycin insensitive companion of mTOR |
| RIPA | Radioimmunoprecipitation assay buffer |
| RNA | ribonucleic acid |
| RTCA | real-time cell analyser |
| S | S-phase of cell cycle |
| SD | standard deviation |
| SEM | standard error of the mean |
| SIRT | sirtuin |
| Snail | snail familiy zinc finger 1 |
| Sox9 | SRY (sex determing region) -box 9 |
| T | unbalanced translocation |
| t1–3 | timepoint |
| TACE | transarterial chemoembolization |
| TBS-T | tween supplemented tris-buffered saline |
| TICs | tumour initiating cells |
| Twist | twist basic helix-loop-helix transcription factor 1 |
| wcp | whole-chromosome-painting |
| µCT | micro comupter-tomography |
References
- Lamarca, A.; Mendiola, M.; Barriuso, J. Hepatocellular carcinoma: Exploring the impact of ethnicity on molecular biology. Crit. Rev. Oncol. Hematol. 2016, 105, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.E.; de Lope, C.R.; Bruix, J. Current strategy for staging and treatment: The BCLC update and future prospects. Semin. Liver Dis. 2010, 30, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Imamura, H.; Matsuyama, Y.; Tanaka, E.; Ohkubo, T.; Hasegawa, K.; Miyagawa, S.; Sugawara, Y.; Minagawa, M.; Takayama, T.; Kawasaki, S.; et al. Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. J. Hepatol. 2003, 38, 200–207. [Google Scholar] [CrossRef]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Bruix, J.; Gores, G.J.; Mazzaferro, V. Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut 2014, 63, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Colvin, H.; Mizushima, T.; Eguchi, H.; Takiguchi, S.; Doki, Y.; Mori, M. Gastroenterological surgery in Japan: The past, the present and the future. Ann. Gastroenterol. Surg. 2017, 1, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Lin, Y.; Zhang, X.; Chen, Y.X.; Zeng, X.; Yue, H.Y.; Hou, J.L.; Deng, X.; Zhang, J.P.; Han, Z.G.; et al. Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4alpha gene. Hepatology 2008, 48, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.R.; Rosso, N.; Bedogni, G.; Tiribelli, C.; Bellentani, S. Global epidemiology of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: What we need in the future. Liver Int. Off. J. Int. Assoc. Study Liver 2018, 38 (Suppl. 1), 47–51. [Google Scholar] [CrossRef]
- Puchades Renau, L.; Berenguer, M. Introduction to hepatitis C virus infection: Overview and history of hepatitis C virus therapies. Hemodial. Int. 2018, 22 (Suppl. 1), S8–S21. [Google Scholar] [CrossRef]
- Sun, D.; Zhu, L.; Yao, D.; Chen, L.; Fu, L.; Ouyang, L. Recent progress in potential anti-hepatitis B virus agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2018, 147, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Paik, Y.H.; Lindquist, J.N.; Lemasters, J.J.; Brenner, D.A. Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology 2004, 126, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Krebs, A.; Bauer, M.; Hahn, E.G. Hepatitis C and liver fibrosis. Cell Death Differ. 2003, 10 (Suppl. 1), S59–S67. [Google Scholar] [CrossRef]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Jou, J.; Choi, S.S.; Diehl, A.M. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.J.; Leon, P.; Ryan, J.C. Beyond insulin resistance: Innate immunity in nonalcoholic steatohepatitis. Hepatology 2008, 48, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Mummadi, R.R.; Kasturi, K.S.; Chennareddygari, S.; Sood, G.K. Effect of bariatric surgery on nonalcoholic fatty liver disease: Systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2008, 6, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Oseini, A.M.; Sanyal, A.J. Therapies in non-alcoholic steatohepatitis (NASH). Liver Int. 2017, 37 (Suppl. 1), 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyault, S.; Rickman, D.S.; de Reynies, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Herault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, J.K.; Choi, Y.K.; Kang, Y.N.; Jang, B.K.; Kang, K.J.; Jeon, Y.H.; Lee, H.W.; Jeon, J.H.; Koo, S.H.; Jeong, W.I.; et al. Retinoic acid-related orphan receptor alpha reprograms glucose metabolism in glutamine-deficient hepatoma cells. Hepatology 2015, 61, 953–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, M.; Tardito, S.; Pillozzi, S.; Arcangeli, A.; Armento, A.; Uggeri, J.; Missale, G.; Bianchi, M.G.; Barilli, A.; Dall’Asta, V.; et al. Glutamine depletion by crisantaspase hinders the growth of human hepatocellular carcinoma xenografts. Br. J. Cancer 2014, 111, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Wang, M.D.; Zhou, X.; Qin, C.J.; Fu, G.B.; Tang, L.; Wu, H.; Huang, S.; Zhao, L.H.; Zeng, M.; et al. Blocking preferential glucose uptake sensitizes liver tumor-initiating cells to glucose restriction and sorafenib treatment. Cancer Lett. 2017, 388, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amann, T.; Hellerbrand, C. GLUT1 as a therapeutic target in hepatocellular carcinoma. Expert Opin. Ther. Targets 2009, 13, 1411–1427. [Google Scholar] [CrossRef] [PubMed]
- Sahin, F.; Kannangai, R.; Adegbola, O.; Wang, J.; Su, G.; Torbenson, M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin. Cancer Res. 2004, 10, 8421–8425. [Google Scholar] [CrossRef] [PubMed]
- Sieghart, W.; Fuereder, T.; Schmid, K.; Cejka, D.; Werzowa, J.; Wrba, F.; Wang, X.; Gruber, D.; Rasoul-Rockenschaub, S.; Peck-Radosavljevic, M.; et al. Mammalian target of rapamycin pathway activity in hepatocellular carcinomas of patients undergoing liver transplantation. Transplantation 2007, 83, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008, 135, 1972–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, P.; Klupp, J.; Langrehr, J.M. mTOR inhibitors: An overview. Liver Transpl. 2001, 7, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.M.; Perry, C.M. Everolimus. Drugs 2004, 64, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the mTOR pathway in hepatocellular carcinoma: Current state and future trends. J. Hepatol. 2014, 60, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.X.; Kudo, M.; Assenat, E.; Cattan, S.; Kang, Y.K.; Lim, H.Y.; Poon, R.T.; Blanc, J.F.; Vogel, A.; Chen, C.L.; et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: The EVOLVE-1 randomized clinical trial. JAMA 2014, 312, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Evert, M.; Calvisi, D.F.; Evert, K.; De Murtas, V.; Gasparetti, G.; Mattu, S.; Destefanis, G.; Ladu, S.; Zimmermann, A.; Delogu, S.; et al. V-AKT murine thymoma viral oncogene homolog/mammalian target of rapamycin activation induces a module of metabolic changes contributing to growth in insulin-induced hepatocarcinogenesis. Hepatology 2012, 55, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Grabinski, N.; Ewald, F.; Hofmann, B.T.; Staufer, K.; Schumacher, U.; Nashan, B.; Jucker, M. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol. Cancer 2012, 11, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirstein, M.M.; Boukouris, A.E.; Pothiraju, D.; Buitrago-Molina, L.E.; Marhenke, S.; Schutt, J.; Orlik, J.; Kuhnel, F.; Hegermann, J.; Manns, M.P.; et al. Activity of the mTOR inhibitor RAD001, the dual mTOR and PI3-kinase inhibitor BEZ235 and the PI3-kinase inhibitor BKM120 in hepatocellular carcinoma. Liver Int. 2013, 33, 780–793. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Shimomura, M.; Kobayashi, K.; Kojima, S.; Nakatsura, T. Growth inhibition by NVP-BEZ235, a dual PI3K/mTOR inhibitor, in hepatocellular carcinoma cell lines. Oncol. Rep. 2011, 26, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.E.; Mercer, C.A.; Carnevalli, L.S.; Park, J.; Andersen, J.B.; Conner, E.A.; Tanaka, K.; Matsutani, T.; Iwanami, A.; Aronow, B.J.; et al. mTOR inhibitors synergize on regression, reversal of gene expression, and autophagy in hepatocellular carcinoma. Sci. Transl. Med. 2012, 4, 139ra184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Martinez, J.M.; Moran, J.; Clarke, R.G.; Gray, A.; Cosulich, S.C.; Chresta, C.M.; Alessi, D.R. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J. 2009, 421, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yongxi, T.; Haijun, H.; Jiaping, Z.; Guoliang, S.; Hongying, P. Autophagy inhibition sensitizes KU-0063794-mediated anti-HepG2 hepatocellular carcinoma cell activity in vitro and in vivo. Biochem. Biophys. Res. Commun. 2015, 465, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Poitou, C.; Veyrie, N.; Bouillot, J.L.; Basdevant, A.; Paradis, V.; Tordjman, J.; Clement, K. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology 2012, 56, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.J.; Conley, P.B.; Chen, L.; Sladek, F.M.; Darnell, J.E., Jr.; Crabtree, G.R. A transcriptional hierarchy involved in mammalian cell-type specification. Nature 1992, 355, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.F.; Ding, J.; Yin, C.; Zhong, W.; Wu, K.; Zeng, X.; Yang, W.; Chen, Y.X.; Zhang, J.P.; Zhang, X.; et al. Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res. 2010, 70, 7640–7651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.; You, H.; Dang, H.; LeBlanc, F.; Galicia, V.; Lu, S.C.; Stiles, B.; Rountree, C.B. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology 2010, 52, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, S.; Kon, A.; Kudo, D.; Nakazawa, H.; Kakizaki, I.; Sasaki, M.; Endo, M.; Takagaki, K. A hyaluronan synthase suppressor, 4-methylumbelliferone, inhibits liver metastasis of melanoma cells. FEBS Lett. 2005, 579, 2722–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walesky, C.; Edwards, G.; Borude, P.; Gunewardena, S.; O’Neil, M.; Yoo, B.; Apte, U. Hepatocyte nuclear factor 4 alpha deletion promotes diethylnitrosamine-induced hepatocellular carcinoma in rodents. Hepatology 2013, 57, 2480–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Choi, G.H.; Na, D.C.; Ahn, E.Y.; Kim, G.I.; Lee, J.E.; Cho, J.Y.; Yoo, J.E.; Choi, J.S.; Park, Y.N. Human hepatocellular carcinomas with “Stemness”-related marker expression: Keratin 19 expression and a poor prognosis. Hepatology 2011, 54, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xiong, L.; Sun, T.; Peng, R.; Zou, L.; Zhu, H.; Zhang, J.; Li, H.; Zhao, J. Expression features of SOX9 associate with tumor progression and poor prognosis of hepatocellular carcinoma. Diagn. Pathol. 2012, 7, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mima, K.; Okabe, H.; Ishimoto, T.; Hayashi, H.; Nakagawa, S.; Kuroki, H.; Watanabe, M.; Beppu, T.; Tamada, M.; Nagano, O.; et al. CD44s regulates the TGF-beta-mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res. 2012, 72, 3414–3423. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.J.; Li, C.J.; Wu, M.Y.; Chu, P.Y. Overexpression of epithelial cell adhesion molecule as a predictor of poor outcome in patients with hepatocellular carcinoma. Exp. Ther. Med. 2018, 16, 4810–4816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uenishi, T.; Kubo, S.; Yamamoto, T.; Shuto, T.; Ogawa, M.; Tanaka, H.; Tanaka, S.; Kaneda, K.; Hirohashi, K. Cytokeratin 19 expression in hepatocellular carcinoma predicts early postoperative recurrence. Cancer Sci. 2003, 94, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.Y.; Xi, T.; Lau, W.Y.; Dong, H.; Zhu, Z.; Shen, F.; Wu, M.C.; Cong, W.M. Hepatocellular carcinoma expressing cholangiocyte phenotype is a novel subtype with highly aggressive behavior. Ann. Surg. Oncol. 2011, 18, 2210–2217. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Baeuerle, P.A.; Gires, O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009, 69, 5627–5629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettger, W.J.; McKeehan, W.L. Mechanisms of cellular nutrition. Physiol. Rev. 1986, 66, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tang, Y.; Xie, Z.J.; Lu, J.N.; Deng, J.H.; Huang, X.W.; Chuan Hooi, S.; He, M.; Lu, G.D. AKT activation was not essential for hepatocellular carcinoma cell survival under glucose deprivation. Anti-Cancer Drugs 2017, 28, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardito, S.; Chiu, M.; Uggeri, J.; Zerbini, A.; Da Ros, F.; Dall’Asta, V.; Missale, G.; Bussolati, O. L-Asparaginase and inhibitors of glutamine synthetase disclose glutamine addiction of beta-catenin-mutated human hepatocellular carcinoma cells. Curr. Cancer Drug Targets 2011, 11, 929–943. [Google Scholar] [PubMed]
- Levy, P.L.; Duponchel, S.; Eischeid, H.; Molle, J.; Michelet, M.; Diserens, G.; Vermathen, M.; Vermathen, P.; Dufour, J.F.; Dienes, H.P.; et al. Hepatitis C virus infection triggers a tumor-like glutamine metabolism. Hepatology 2017, 65, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Andersen, J.B.; Thorgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Baba, H.A.; Wohlschlaeger, J.; Cicinnati, V.R.; Hilgard, P.; Lang, H.; Sotiropoulos, G.C.; Takeda, A.; Beckebaum, S.; Schmitz, K.J. Phosphorylation of p70S6 kinase predicts overall survival in patients with clear margin-resected hepatocellular carcinoma. Liver Int. 2009, 29, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med. Oncol. 2010, 27, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, R.E.; Wu, J. Mammalian target of rapamycin inhibition in hepatocellular carcinoma. World J. Hepatol. 2014, 6, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Scheller, T.; Hellerbrand, C.; Moser, C.; Schmidt, K.; Kroemer, A.; Brunner, S.M.; Schlitt, H.J.; Geissler, E.K.; Lang, S.A. mTOR inhibition improves fibroblast growth factor receptor targeting in hepatocellular carcinoma. Br. J. Cancer 2015, 112, 841–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., III; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Horsch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M.; et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): A randomised, placebo-controlled, phase 3 study. Lancet 2011, 378, 2005–2012. [Google Scholar] [CrossRef]
- Fischer, L.; Klempnauer, J.; Beckebaum, S.; Metselaar, H.J.; Neuhaus, P.; Schemmer, P.; Settmacher, U.; Heyne, N.; Clavien, P.A.; Muehlbacher, F.; et al. A randomized, controlled study to assess the conversion from calcineurin-inhibitors to everolimus after liver transplantation--PROTECT. Am. J. Transpl. 2012, 12, 1855–1865. [Google Scholar] [CrossRef] [PubMed]
- De Simone, P.; Nevens, F.; De Carlis, L.; Metselaar, H.J.; Beckebaum, S.; Saliba, F.; Jonas, S.; Sudan, D.; Fung, J.; Fischer, L.; et al. Everolimus with reduced tacrolimus improves renal function in de novo liver transplant recipients: A randomized controlled trial. Am. J. Transpl. 2012, 12, 3008–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roat, E.; De Biasi, S.; Bertoncelli, L.; Rompianesi, G.; Nasi, M.; Gibellini, L.; Pinti, M.; Del Giovane, C.; Zanella, A.; Di Benedetto, F.; et al. Immunological advantages of everolimus versus cyclosporin A in liver-transplanted recipients, as revealed by polychromatic flow cytometry. Cytometry A 2012, 81, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Cholongitas, E.; Antoniadis, N.; Goulis, I.; Fouzas, I.; Vasiliadis, T.; Akriviadis, E.; Papanikolaou, V. Renal function improvement in liver transplant recipients after early everolimus conversion: A clinical practice cohort study in Spain. Liver Transpl. 2016, 22, 132–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, E.T.; Cao, C.; Niermann, K.; Mu, Y.; Zeng, F.; Hallahan, D.E.; Lu, B. Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene 2005, 24, 5414–5422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Tourneau, C.; Faivre, S.; Serova, M.; Raymond, E. mTORC1 inhibitors: Is temsirolimus in renal cancer telling us how they really work? Br. J. Cancer 2008, 99, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Petrulionis, M.; Lin, S.; Gao, C.; Galli, U.; Richter, S.; Winkler, S.; Houben, P.; Schultze, D.; Hatano, E.; et al. Therapeutic potential and adverse events of everolimus for treatment of hepatocellular carcinoma-systematic review and meta-analysis. Cancer Med. 2013, 2, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Kim, K.H.; Kim, O.H.; Lee, S.K.; Hong, H.E.; Choi, B.J.; Jeong, W.; Kim, S.J. Everolimus Plus Ku0063794 Regimen Promotes Anticancer Effects against Hepatocellular Carcinoma Cells through the Paradoxical Inhibition of Autophagy. Cancer Res. Treat. 2018, 50, 1023–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Autophagy in health and disease. 1. Regulation and significance of autophagy: An overview. Am. J. Physiol. Cell Physiol. 2010, 298, C776–C785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesse, D.; Jaschke, A.; Kanzleiter, T.; Witte, N.; Augustin, R.; Hommel, A.; Puschel, G.P.; Petzke, K.J.; Joost, H.G.; Schupp, M.; et al. GTPase ARFRP1 is essential for normal hepatic glycogen storage and insulin-like growth factor 1 secretion. Mol. Cell. Biol. 2012, 32, 4363–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Aneuploidy (4×) | Loss of Additional Chromosomes (2x) | ||
|---|---|---|---|---|
| P3 | P29 | P3 | P29 | |
| 2 | 95% | 4% | - | 8% |
| 3 | 60% | - | 10% | 50% |
| 4 | 30% | 4% | 10% | 85% |
| 5 | 75% | - | - | 15% |
| 6 | 45% | 54% | - | 4% |
| 7 | 50% | 4% | 20% | 88% |
| 8 | 70% | 38% | - | - |
| 9 | 55% | 4% | 5% | 31% |
| 10 | 35% | - | 15% | 81% |
| 11 | 25% | - | - | - |
| 12 | 65% | 19% | - | 8% |
| 13 | 20% | - | 5% | 38% |
| 14 | 30% | - | - | 58% |
| 15 | 15% | 8% | - | 8% |
| 16 | 25% | 19% | - | 11% |
| 17 | 5% | - | - | 8% |
| 18 | - | - | 50% | 46% |
| 19 | 15% | 15% | 15% | 4% |
| ploidy grade | #chromosomes | range | ||
| P3 | hypo-tetraploid | 65 | 60–69 | |
| P29 | hypo-triploid | 56 | 50–59 | |
| Kind of Aberration | P3 | P29 |
|---|---|---|
| T (1F;11B1) | 96% | 100% |
| Rb 16.19 | 73% | 86% |
| Del(17E1) | 92% | 100% |
| T (XD;15B1) | 82% | 93% |
| loss of Y | 44% | 55% |
| r/dmin | 48% | 61% |
| Primer | Sequence | Annealing Temperature | Cycles | |
|---|---|---|---|---|
| Albumin | for rev | TCCTGGGCACGTTCTTGTAT TGCTTTCTGGGTGTAGCGAA | 58.5 °C | 30 |
| HNF4 | for rev | AAGGTGCCAACCTCAATTCATC CACATTGTCGGCTAAACCTGC | 60 °C | 30 |
| CK19 | for rev | GTCCTACAGATTGACAATG CACGCTCTGGATCTGTGACAG | 57 °C | 30 |
| Sox9 | for rev | GTGAAGAACGGACAAGCGGA GATTGCCCAGAGTGCTCGC | 60 °C | 40 |
| EpCAM | for rev | CGGCTCAGAGAGACTGTGTC GATCCAGTAGGTCCTCACGC | 57.5 °C | 30 |
| CD44 | for rev | CAGAGGCGACTAGATCCCTC GAGTCACAGTGCGGGAACTC | 59 °C | 30 |
| Twist | for rev | GCCGGAGACCTAGATGTCATTG CCACGCCCTGATTCTTGTGA | 60 °C | 40 |
| Snail | for rev | TCTGCACGACCTGTGGAAAG GTTGGAGCGGTCAGCAAAAG | 60 °C | 40 |
| GAPDH | for rev | AGGTCGGTGTGAACGGATTTG TGTAGACCATGTAGTTGAGGTCA | 60 °C | 40 |
| Antibody | Target Size | Order Number | Dilution | Blocking Solution |
|---|---|---|---|---|
| mTOR | 289 kDa | CST 2983 | 1:500 | milk powder |
| p-mTOR (Ser2481) | 289 kDa | CST 2974 | 1:1000 | BSA |
| p-mTOR (Ser2448) | 289 kDa | CST 5536 | 1:500 | BSA |
| Rictor | 200 kDa | CST 2114 | 1:2500 | BSA |
| GßL | 37 kDa | CST 3274 | 1:1000 | BSA |
| p-ribosomal protein S6 | 32 kDa | SC 293144 | 1:200 | BSA |
| p-4E-BP1 | 15–20 kDa | CST 2855 | 1:1000 | BSA |
| ß-Tubulin | 52 kDa | PA5 16863 | 1:1000 | milk powder |
| Goat-anti-mouse | - | CST 7076 | 1:1000 | - |
| Goat-anti-rabbit | - | CST 7074 | 1:2500 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kroh, A.; Walter, J.; Schüler, H.; Nolting, J.; Eickhoff, R.; Heise, D.; Neumann, U.P.; Cramer, T.; Ulmer, T.F.; Fragoulis, A. A Newly Established Murine Cell Line as a Model for Hepatocellular Cancer in Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2019, 20, 5658. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225658
Kroh A, Walter J, Schüler H, Nolting J, Eickhoff R, Heise D, Neumann UP, Cramer T, Ulmer TF, Fragoulis A. A Newly Established Murine Cell Line as a Model for Hepatocellular Cancer in Non-Alcoholic Steatohepatitis. International Journal of Molecular Sciences. 2019; 20(22):5658. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225658
Chicago/Turabian StyleKroh, Andreas, Jeanette Walter, Herdit Schüler, Jochen Nolting, Roman Eickhoff, Daniel Heise, Ulf Peter Neumann, Thorsten Cramer, Tom Florian Ulmer, and Athanassios Fragoulis. 2019. "A Newly Established Murine Cell Line as a Model for Hepatocellular Cancer in Non-Alcoholic Steatohepatitis" International Journal of Molecular Sciences 20, no. 22: 5658. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225658