Depletion of KNL2 Results in Altered Expression of Genes Involved in Regulation of the Cell Cycle, Transcription, and Development in Arabidopsis

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Loss of KNL2 Function Leads to Massive Transcriptional Changes
2.2. Knockout of KNL2 Impairs Expression of Genes Involved in Kinetochore Function
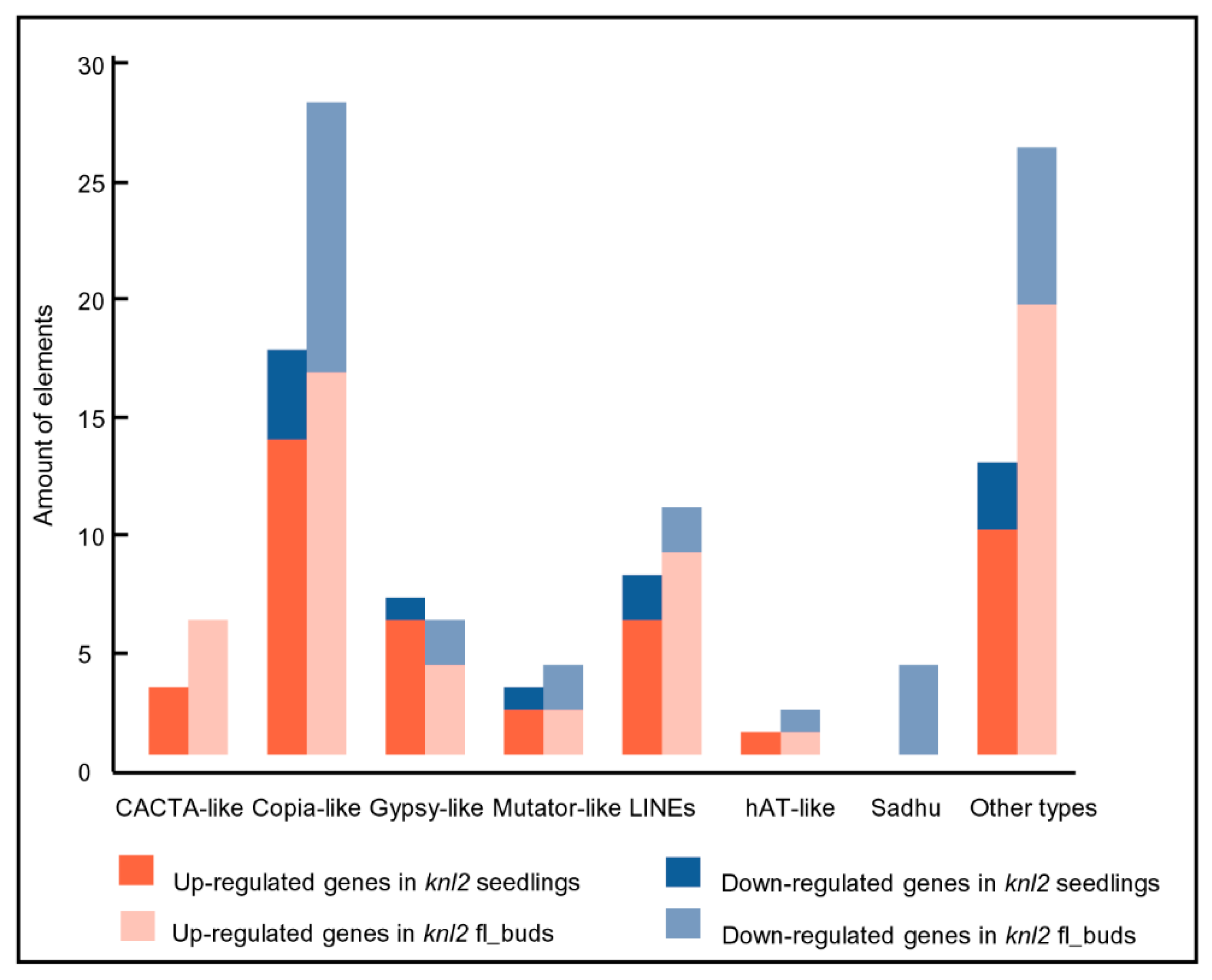
2.3. Reduced DNA Methylation in the knl2 Mutant Might Be Responsible for the Activation of a High Number of Transposons
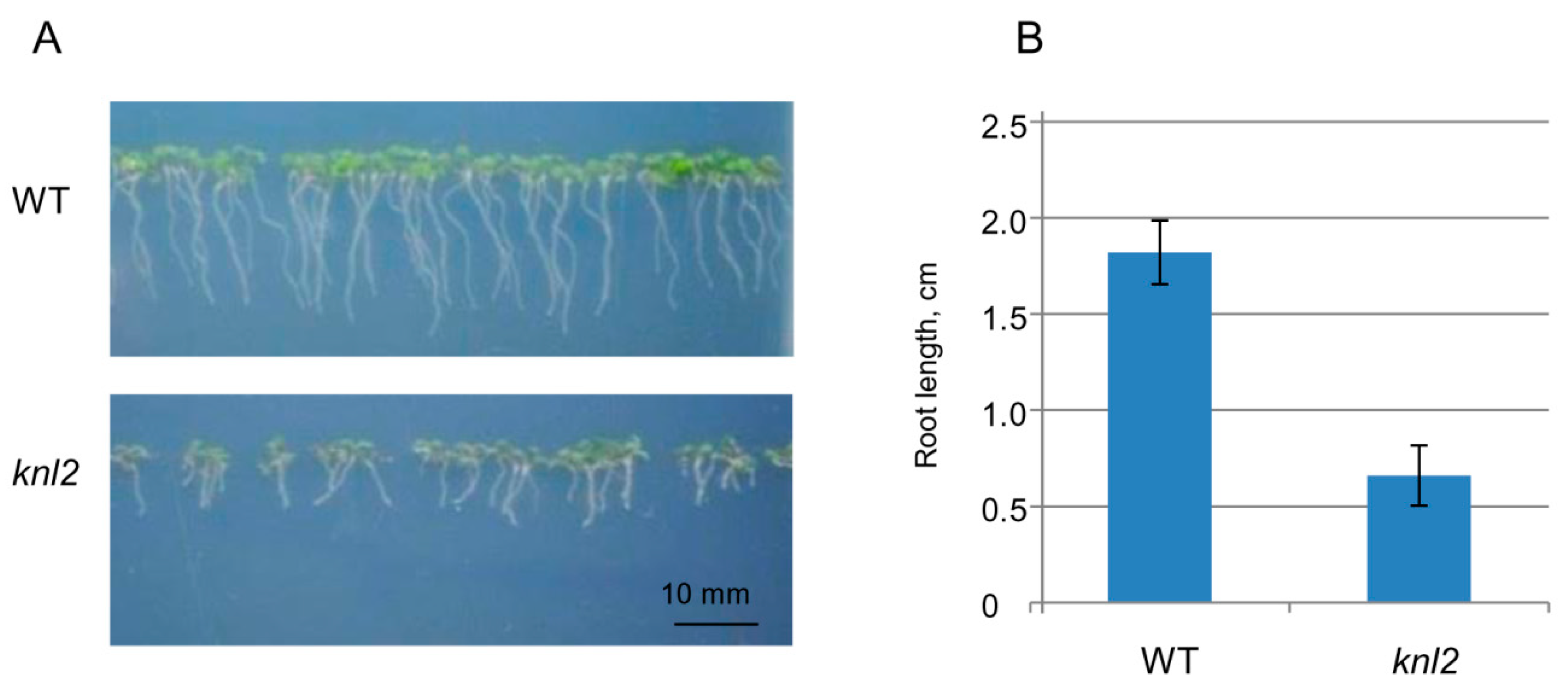
2.4. Altered Expression of the Root-related Genes Explains the Slow Root Growth of knl2
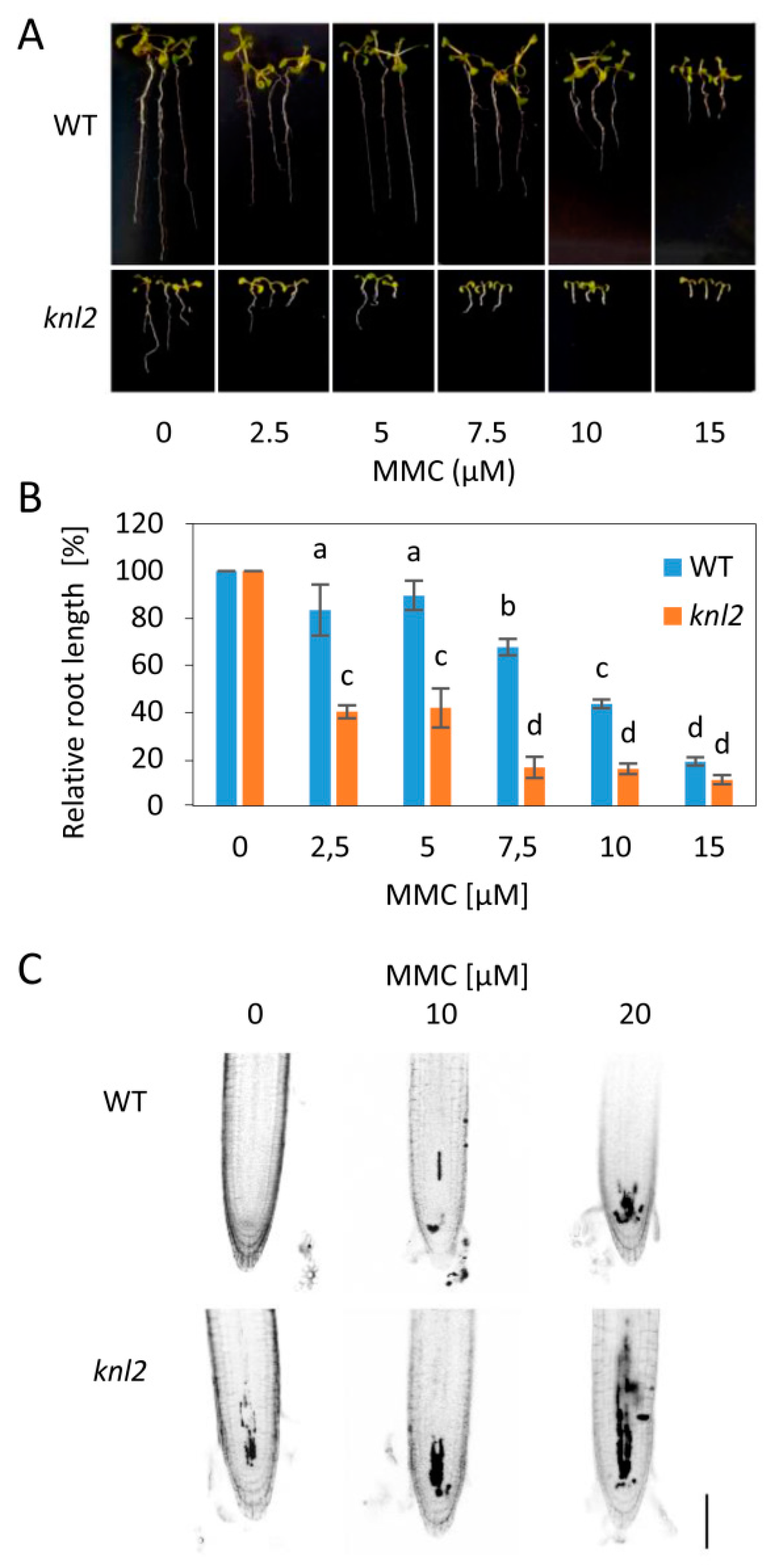
2.5. DNA Damage Repair Genes are Downregulated in the knl2 Plants
2.6. Knockout of KNL2 Results in Deregulated Expression of a High Number of Transcription Factors
2.7. Late Flowering of the knl2 Plants is Determined by the Altered Expression of Flowering Genes
2.8. Knockout of KNL2 Results in Altered Expression of Genes Controlling Seed Development
2.9. Real-time Quantitative PCR Confirms RNA-seq Analysis
2.10. Conclusions
3. Materials and Methods
3.1. Plant Materials and Growth Conditions
3.2. DNA Damage Sensitivity Assays
3.3. RNA Isolation and Illumina Sequencing
3.4. RNA-seq Data Processing
3.5. Analysis of Differentially Expressed Genes (DEGs)
3.6. Gene Expression Validation by Reverse Transcription Quantitative PCR (RT-qPCR)
3.7. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Talbert, P.B.; Henikoff, S. Phylogeny as the basis for naming histones. Trends Genet. 2013, 29, 499–500. [Google Scholar] [CrossRef]
- Silva, M.C.; Jansen, L.E. At the right place at the right time: Novel CENP-A binding proteins shed light on centromere assembly. Chromosoma 2009, 118, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Transcribing centromeres: Noncoding RNAs and kinetochore assembly. Trends Genet. 2018, 34, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Perea-Resa, C.; Blower, M.D. Centromere biology: Transcription goes on stage. Mol. Cell Biol. 2018, 38, e00263-18. [Google Scholar] [CrossRef]
- Bobkov, G.O.M.; Gilbert, N.; Heun, P. Centromere transcription allows CENP-A to transit from chromatin association to stable incorporation. J. Cell Biol. 2018, 217, 1957–1972. [Google Scholar] [CrossRef]
- Bergmann, J.H.; Rodriguez, M.G.; Martins, N.M.; Kimura, H.; Kelly, D.A.; Masumoto, H.; Larionov, V.; Jansen, L.E.; Earnshaw, W.C. Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. EMBO J. 2011, 30, 328–340. [Google Scholar] [CrossRef]
- Kim, I.S.; Lee, M.; Park, K.C.; Jeon, Y.; Park, J.H.; Hwang, E.J.; Jeon, T.I.; Ko, S.; Lee, H.; Baek, S.H.; et al. Roles of Mis18alpha in epigenetic regulation of centromeric chromatin and CENP-A loading. Mol. Cell 2012, 46, 260–273. [Google Scholar] [CrossRef]
- Nardi, I.K.; Zasadzinska, E.; Stellfox, M.E.; Knippler, C.M.; Foltz, D.R. Licensing of centromeric chromatin assembly through the Mis18alpha-Mis18beta heterotetramer. Mol. Cell 2016, 61, 774–787. [Google Scholar] [CrossRef]
- Fujita, Y.; Hayashi, T.; Kiyomitsu, T.; Toyoda, Y.; Kokubu, A.; Obuse, C.; Yanagida, M. Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev. Cell 2007, 12, 17–30. [Google Scholar] [CrossRef]
- Maddox, P.S.; Hyndman, F.; Monen, J.; Oegema, K.; Desai, A. Functional genomics identifies a Myb domain-containing protein family required for assembly of CENP-A chromatin. J. Cell Biol. 2007, 176, 757–763. [Google Scholar] [CrossRef]
- Foltz, D.R.; Jansen, L.E.; Bailey, A.O.; Yates, J.R., 3rd; Bassett, E.A.; Wood, S.; Black, B.E.; Cleveland, D.W. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 2009, 137, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.C.; Kuich, P.H.; Stellfox, M.E.; Ward, J.A.; Bassett, E.A.; Black, B.E.; Foltz, D.R. HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J. Cell Biol. 2011, 194, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Lermontova, I.; Kuhlmann, M.; Friedel, S.; Rutten, T.; Heckmann, S.; Sandmann, M.; Demidov, D.; Schubert, V.; Schubert, I. Arabidopsis KINETOCHORE NULL2 is an upstream component for centromeric histone H3 variant cenH3 deposition at centromeres. Plant Cell 2013, 25, 3389–3404. [Google Scholar] [CrossRef]
- Sandmann, M.; Talbert, P.; Demidov, D.; Kuhlmann, M.; Rutten, T.; Conrad, U.; Lermontova, I. Targeting of arabidopsis KNL2 to centromeres depends on the conserved CENPC-k motif in Its C terminus. Plant Cell 2017, 29, 144–155. [Google Scholar] [CrossRef]
- Zhang, D.; Martyniuk, C.J.; Trudeau, V.L. SANTA domain: A novel conserved protein module in Eukaryota with potential involvement in chromatin regulation. Bioinformatics 2006, 22, 2459–2462. [Google Scholar] [CrossRef]
- Kral, L. Possible identification of CENP-C in fish and the presence of the CENP-C motif in M18BP1 of vertebrates. F1000Research 2015, 4, 474. [Google Scholar] [CrossRef]
- French, B.T.; Westhorpe, F.G.; Limouse, C.; Straight, A.F. Xenopus laevis M18BP1 directly binds existing CENP-A nucleosomes to promote centromeric chromatin assembly. Dev. Cell 2017, 42, 190e10–199e10. [Google Scholar] [CrossRef]
- Hori, T.; Shang, W.H.; Hara, M.; Ariyoshi, M.; Arimura, Y.; Fujita, R.; Kurumizaka, H.; Fukagawa, T. Association of M18BP1/KNL2 with CENP-A nucleosome is essential for centromere formation in non-mammalian vertebrates. Dev. Cell 2017, 42, 181e3–189e3. [Google Scholar] [CrossRef]
- Liebelt, F.; Jansen, N.S.; Kumar, S.; Gracheva, E.; Claessens, L.A.; Verlaan-de Vries, M.; Willemstein, E.; Vertegaal, A.C.O. The poly-SUMO2/3 protease SENP6 enables assembly of the constitutive centromere-associated network by group deSUMOylation. Nat. Commun. 2019, 10, 3987. [Google Scholar] [CrossRef]
- Silva, M.C.; Bodor, D.L.; Stellfox, M.E.; Martins, N.M.; Hochegger, H.; Foltz, D.R.; Jansen, L.E. Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev. Cell 2012, 22, 52–63. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO: TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, H.; Lloyd, C.W. Arabidopsis mutants and the network of microtubule-associated functions. Mol. Plant 2008, 1, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Wang, L.; Zhang, L.; Ren, D.; Ren, R.; Copenhaver, G.P.; Ma, H.; Wang, Y. Arabidopsis cell division cycle 20.1 is required for normal meiotic spindle assembly and chromosome segregation. Plant Cell 2015, 27, 3367–3382. [Google Scholar] [CrossRef] [PubMed]
- Kevei, Z.; Baloban, M.; Da Ines, O.; Tiricz, H.; Kroll, A.; Regulski, K.; Mergaert, P.; Kondorosi, E. Conserved CDC20 cell cycle functions are carried out by two of the five isoforms in Arabidopsis thaliana. PLoS ONE 2011, 6, e20618. [Google Scholar] [CrossRef]
- Bucher, E.; Reinders, J.; Mirouze, M. Epigenetic control of transposon transcription and mobility in Arabidopsis. Curr. Opin. Plant Biol. 2012, 15, 503–510. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef]
- Deniz, O.; Frost, J.M.; Branco, M.R. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019, 20, 417–431. [Google Scholar] [CrossRef]
- Rangwala, S.H.; Richards, E.J. Differential epigenetic regulation within an Arabidopsis retroposon family. Genetics 2007, 176, 151–160. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, C.; Lin, Q.; Liu, A.; Wang, T.; Feng, X.; Liu, J.; Han, H.; Ma, Y.; Bonea, D.; et al. A tetratricopeptide repeat domain-containing protein SSR1 located in mitochondria is involved in root development and auxin polar transport in Arabidopsis. Plant J. Cell Mol. Biol. 2015, 83, 582–599. [Google Scholar] [CrossRef]
- Munoz-Bertomeu, J.; Cascales-Minana, B.; Mulet, J.M.; Baroja-Fernandez, E.; Pozueta-Romero, J.; Kuhn, J.M.; Segura, J.; Ros, R. Plastidial glyceraldehyde-3-phosphate dehydrogenase deficiency leads to altered root development and affects the sugar and amino acid balance in Arabidopsis. Plant Physiol. 2009, 151, 541–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garay-Arroyo, A.; Ortiz-Moreno, E.; de la Paz Sanchez, M.; Murphy, A.S.; Garcia-Ponce, B.; Marsch-Martinez, N.; de Folter, S.; Corvera-Poire, A.; Jaimes-Miranda, F.; Pacheco-Escobedo, M.A.; et al. The MADS transcription factor XAL2/AGL14 modulates auxin transport during Arabidopsis root development by regulating PIN expression. EMBO J. 2013, 32, 2884–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapia-Lopez, R.; Garcia-Ponce, B.; Dubrovsky, J.G.; Garay-Arroyo, A.; Perez-Ruiz, R.V.; Kim, S.H.; Acevedo, F.; Pelaz, S.; Alvarez-Buylla, E.R. An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol. 2008, 146, 1182–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, K.; Nagata, N.; Yee, K.M.; Braybrook, S.A.; Pelletier, J.; Fujioka, S.; Yoshida, S.; Fischer, R.L.; Goldberg, R.B.; Harada, J.J. TANMEI/EMB2757 encodes a WD repeat protein required for embryo development in Arabidopsis. Plant Physiol. 2005, 139, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.; Liu, J.X.; Guo, H.; Yin, Y.; Howell, S.H. Regulation and processing of a plant peptide hormone, AtRALF23, in Arabidopsis. Plant J. Cell Mol. Biol. 2009, 59, 930–939. [Google Scholar] [CrossRef]
- Manova, V.; Gruszka, D. DNA damage and repair in plants—From models to crops. Front. Plant Sci. 2015, 6, 885. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Chen, C.; Markmann-Mulisch, U.; Timofejeva, L.; Schmelzer, E.; Ma, H.; Reiss, B. The Arabidopsis AtRAD51 gene is dispensable for vegetative development but required for meiosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10596–10601. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wang, H.; Xu, Y.; Brinkman, K.L.; Ishiyama, H.; Wong, S.T.; Xu, B. The kinetochore protein Bub1 participates in the DNA damage response. DNA Repair 2012, 11, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Zeitlin, S.G.; Baker, N.M.; Chapados, B.R.; Soutoglou, E.; Wang, J.Y.; Berns, M.W.; Cleveland, D.W. Double-strand DNA breaks recruit the centromeric histone CENP-A. Proc. Natl. Acad. Sci. USA 2009, 106, 15762–15767. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [Green Version]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY transcription factors: Molecular regulation and stress responses in plants. Front. Plant Sci. 2016, 7, 760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Ma, S.; Ye, N.; Jiang, M.; Cao, J.; Zhang, J. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Finatto, T.; Viana, V.E.; Woyann, L.G.; Busanello, C.; da Maia, L.C.; de Oliveira, A.C. Can WRKY transcription factors help plants to overcome environmental challenges? Genet. Mol. Biol. 2018, 41, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Rizhsky, L.; Liang, H.; Shuman, J.; Shulaev, V.; Mittler, R. Enhanced tolerance to environmental stress in transgenic plants expressing the transcriptional coactivator multiprotein bridging factor 1c. Plant Physiol. 2005, 139, 1313–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Nolan, T.M.; Ye, H.; Zhang, M.; Tong, H.; Xin, P.; Chu, J.; Chu, C.; Li, Z.; Yin, Y. Arabidopsis WRKY46, WRKY54, and WRKY70 transcription factors are involved in brassinosteroid-regulated plant growth and drought responses. Plant Cell 2017, 29, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.J.; Yan, J.Y.; Xu, X.Y.; Yu, D.Q.; Li, G.X.; Zhang, S.Q.; Zheng, S.J. Transcription factor WRKY46 regulates osmotic stress responses and stomatal movement independently in Arabidopsis. Plant J. Cell Mol. Biol. 2014, 79, 13–27. [Google Scholar] [CrossRef]
- Sheikh, A.H.; Eschen-Lippold, L.; Pecher, P.; Hoehenwarter, W.; Sinha, A.K.; Scheel, D.; Lee, J. Regulation of WRKY46 transcription factor function by mitogen-activated protein kinases in arabidopsis THALIANA. Front. Plant Sci. 2016, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Krishnaswamy, S.; Verma, S.; Rahman, M.H.; Kav, N.N. Functional characterization of four APETALA2-family genes (RAP2.6, RAP2.6L, DREB19 and DREB26) in Arabidopsis. Plant Mol. Biol. 2011, 75, 107–127. [Google Scholar] [CrossRef]
- Matias-Hernandez, L.; Aguilar-Jaramillo, A.E.; Marin-Gonzalez, E.; Suarez-Lopez, P.; Pelaz, S. RAV genes: Regulation of floral induction and beyond. Ann. Bot. 2014, 114, 1459–1470. [Google Scholar] [CrossRef]
- Higginson, T.; Li, S.F.; Parish, R.W. AtMYB103 regulates tapetum and trichome development in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 2003, 35, 177–192. [Google Scholar] [CrossRef]
- Adamczyk, B.J.; Fernandez, D.E. MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol. 2009, 149, 1713–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verelst, W.; Twell, D.; de Folter, S.; Immink, R.; Saedler, H.; Munster, T. MADS-complexes regulate transcriptome dynamics during pollen maturation. Genome Biol. 2007, 8, R249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smaczniak, C.; Immink, R.G.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Ruiz, R.V.; Garcia-Ponce, B.; Marsch-Martinez, N.; Ugartechea-Chirino, Y.; Villajuana-Bonequi, M.; de Folter, S.; Azpeitia, E.; Davila-Velderrain, J.; Cruz-Sanchez, D.; Garay-Arroyo, A.; et al. XAANTAL2 (AGL14) Is an important component of the complex gene regulatory network that underlies Arabidopsis shoot apical meristem transitions. Mol. Plant 2015, 8, 796–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, D.R.; Bowman, J.L.; Meyerowitz, E.M. Early flower development in Arabidopsis. Plant Cell 1990, 2, 755–767. [Google Scholar]
- Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 24 November 2010).
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bouché, F.; Lobet, G.; Tocquin, P.; Périlleux, C. FLOR-ID: An interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res. 2016, 44, D1167–D1171. [Google Scholar] [CrossRef] [Green Version]
- Meinke, D.; Muralla, R.; Sweeney, C.; Dickerman, A. Identifying essential genes in Arabidopsis thaliana. Trends Plant Sci. 2008, 13, 483–491. [Google Scholar] [CrossRef]
- Arvidsson, S.; Kwasniewski, M.; Riano-Pachon, D.M.; Mueller-Roeber, B. QuantPrime—A flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinform. 2008, 9, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czechowski, T.; Stitt, M.; Altmann, T.; Udvardi, M.K.; Scheible, W.R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005, 139, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO ID | Biological Process | p-Value | FDR Rate, % | Gene Count |
|---|---|---|---|---|
| GO:0051315 | Attachment of mitotic spindle microtubules to kinetochore | 0.002143 | 2.39 | 2 |
| GO:0007049 | Cell cycle | 0.003143 | 2.94 | 41 |
| GO:0000075 | Cell cycle checkpoint | 0.004300 | 3.40 | 5 |
| GO:0022402 | Cell cycle process | 0.001001 | 0.92 | 30 |
| GO:0007059 | Chromosome segregation | 0.007086 | 4.99 | 11 |
| GO:0051304 | Chromosome separation | 0.001526 | 1.15 | 6 |
| GO:0051103 | DNA ligation involved in DNA repair | 6.37 × 10−5 | 0.06 | 4 |
| GO:0006310 | DNA recombination | 4.72 × 10−6 | 0.00 | 20 |
| GO:0006281 | DNA repair | 0.000745 | 0.79 | 26 |
| GO:0006302 | Double-strand break repair via | 1.79 × 10−5 | 0.02 | 15 |
| homologous recombination | ||||
| GO:0006303 | Double-strand break repair via | 0.000127 | 0.15 | 5 |
| nonhomologous end joining | ||||
| GO:0035825 | Homologous recombination | 0.001124 | 0.93 | 8 |
| GO:0007127 | Meiosis I | 0.001483 | 1.10 | 9 |
| GO:0051307 | Meiotic chromosome separation | 0.001680 | 1.31 | 4 |
| GO:0051310 | Metaphase plate congression | 0.002143 | 2.40 | 2 |
| GO:0007080 | Mitotic metaphase plate congression | 0.002143 | 2.44 | 2 |
| GO:0006312 | Mitotic recombination | 0.000820 | 0.82 | 5 |
| GO:0045786 | Negative regulation of cell cycle | 0.005582 | 4.02 | 7 |
| GO:0000726 | Non-recombinational repair | 0.000127 | 0.15 | 5 |
| GO:0007131 | Reciprocal meiotic recombination | 0.001124 | 0.94 | 8 |
| GO:0071156 | Regulation of cell cycle arrest | 0.006230 | 4.82 | 2 |
| GO ID | Biological Process | p-Value | FDR Rate, % | Gene Count |
|---|---|---|---|---|
| GO:0007049 | Cell cycle | 3.13 × 10−9 | 0.00 | 83 |
| GO:0048468 | Cell development | 3.30 × 10−8 | 0.00 | 56 |
| GO:0030154 | Cell differentiation | 2.36 × 10−5 | 0.08 | 95 |
| GO:0051301 | Cell division | 5.97 × 10−7 | 0.00 | 54 |
| GO:0016049 | Cell growth | 3.06 × 10−6 | 0.05 | 62 |
| GO:0032989 | Cellular component morphogenesis | 2.59 × 10−8 | 0.00 | 72 |
| GO:0016043 | Cellular component organization | 2.03 × 10−8 | 0.00 | 261 |
| GO:0070192 | Chromosome organization | 0.000320 | 0.52 | 11 |
| involved in meiotic cell cycle | ||||
| GO:0007059 | Chromosome segregation | 0.001374 | 1.31 | 17 |
| GO:0000910 | Cytokinesis | 3.63 × 10−5 | 0.15 | 21 |
| GO:0007010 | Cytoskeleton organization | 6.02 × 10−6 | 0.04 | 33 |
| GO:0061640 | Cytoskeleton-dependent cytokinesis | 0.000676 | 0.81 | 17 |
| GO:0000086 | G2/M transition of mitotic cell cycle | 0.000205 | 0.41 | 7 |
| GO:0048229 | Gametophyte development | 3.69 × 10−9 | 0.00 | 70 |
| GO:0045143 | Homologous chromosome segregation | 5.80 × 10−5 | 0.18 | 10 |
| GO:0035825 | Homologous recombination | 2.30 × 10−5 | 0.09 | 13 |
| GO:0007127 | Meiosis I | 2.23 × 10−5 | 0,09 | 15 |
| GO:0061982 | Meiosis I cell cycle process | 7.78 × 10−6 | 0.08 | 16 |
| GO:0051321 | Meiotic cell cycle | 4.41 × 10−5 | 0.15 | 27 |
| GO:0045132 | Meiotic chromosome segregation | 1.62 × 10−5 | 0.10 | 14 |
| GO:0051307 | Meiotic chromosome separation | 0.001212 | 1.24 | 5 |
| GO:0140013 | Meiotic nuclear division | 1.12 × 10−5 | 0.07 | 20 |
| GO:0000226 | Microtubule cytoskeleton organization | 0.000596 | 0.78 | 17 |
| GO:0007018 | Microtubule-based movement | 0.000235 | 0.44 | 10 |
| GO:0000278 | Mitotic cell cycle | 5.20 × 10−5 | 0.19 | 36 |
| GO:0000281 | Mitotic cytokinesis | 0.001316 | 1.29 | 16 |
| GO:0006312 | Mitotic recombination | 0.001079 | 1,20 | 6 |
| GO:0098813 | Nuclear chromosome segregation | 0.000706 | 0.85 | 16 |
| GO:0000280 | Nuclear division | 0.000107 | 0.20 | 24 |
| GO:0007131 | Reciprocal meiotic recombination | 2.30 × 10−5 | 0.09 | 13 |
| GO:0051726 | Regulation of cell cycle | 0.000251 | 0.45 | 32 |
| GO:0007129 | Synapsis | 7.30 × 10−5 | 0.19 | 9 |
| GO ID | Biological Process | p-Value | FDR Rate, % | Gene Count |
|---|---|---|---|---|
| GO:0048653 | Anther development | 0.000711 | 0.84 | 13 |
| GO:0048589 | Developmental growth | 0.004788 | 4.32 | 54 |
| GO:0060560 | Developmental growth involved | 2.06 × 10−5 | 0.10 | 51 |
| in morphogenesis | ||||
| GO:0044703 | Multi-organism reproductive process | 0.002391 | 2.32 | 26 |
| GO:0009555 | Pollen development | 4.10 × 10−11 | 0.00 | 61 |
| GO:0010584 | Pollen exine formation | 2.12 × 10−5 | 0.09 | 10 |
| GO:0009846 | Pollen germination | 0.000968 | 1.15 | 13 |
| GO:0048868 | Pollen tube development | 1.57 × 10−6 | 0.00 | 36 |
| GO:0009860 | Pollen tube growth | 7.09 × 10−9 | 0.00 | 34 |
| GO:0009856 | Pollination | 2.44 × 10−5 | 0.08 | 44 |
| GO:0010769 | Regulation of cell morphogenesis involved | 0.001673 | 1.84 | 9 |
| in differentiation | ||||
| GO:0080092 | Regulation of pollen tube growth | 0.000708 | 0.85 | 9 |
| GO:0000003 | Reproduction | 3.55 × 10−4 | 0.50 | 189 |
| GO:0048443 | Stamen development | 0.002527 | 2.46 | 15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boudichevskaia, A.; Houben, A.; Fiebig, A.; Prochazkova, K.; Pecinka, A.; Lermontova, I. Depletion of KNL2 Results in Altered Expression of Genes Involved in Regulation of the Cell Cycle, Transcription, and Development in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 5726. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225726
Boudichevskaia A, Houben A, Fiebig A, Prochazkova K, Pecinka A, Lermontova I. Depletion of KNL2 Results in Altered Expression of Genes Involved in Regulation of the Cell Cycle, Transcription, and Development in Arabidopsis. International Journal of Molecular Sciences. 2019; 20(22):5726. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225726
Chicago/Turabian StyleBoudichevskaia, Anastassia, Andreas Houben, Anne Fiebig, Klara Prochazkova, Ales Pecinka, and Inna Lermontova. 2019. "Depletion of KNL2 Results in Altered Expression of Genes Involved in Regulation of the Cell Cycle, Transcription, and Development in Arabidopsis" International Journal of Molecular Sciences 20, no. 22: 5726. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225726