mTOR Links Tumor Immunity and Bone Metabolism: What are the Clinical Implications?

 , and
, and 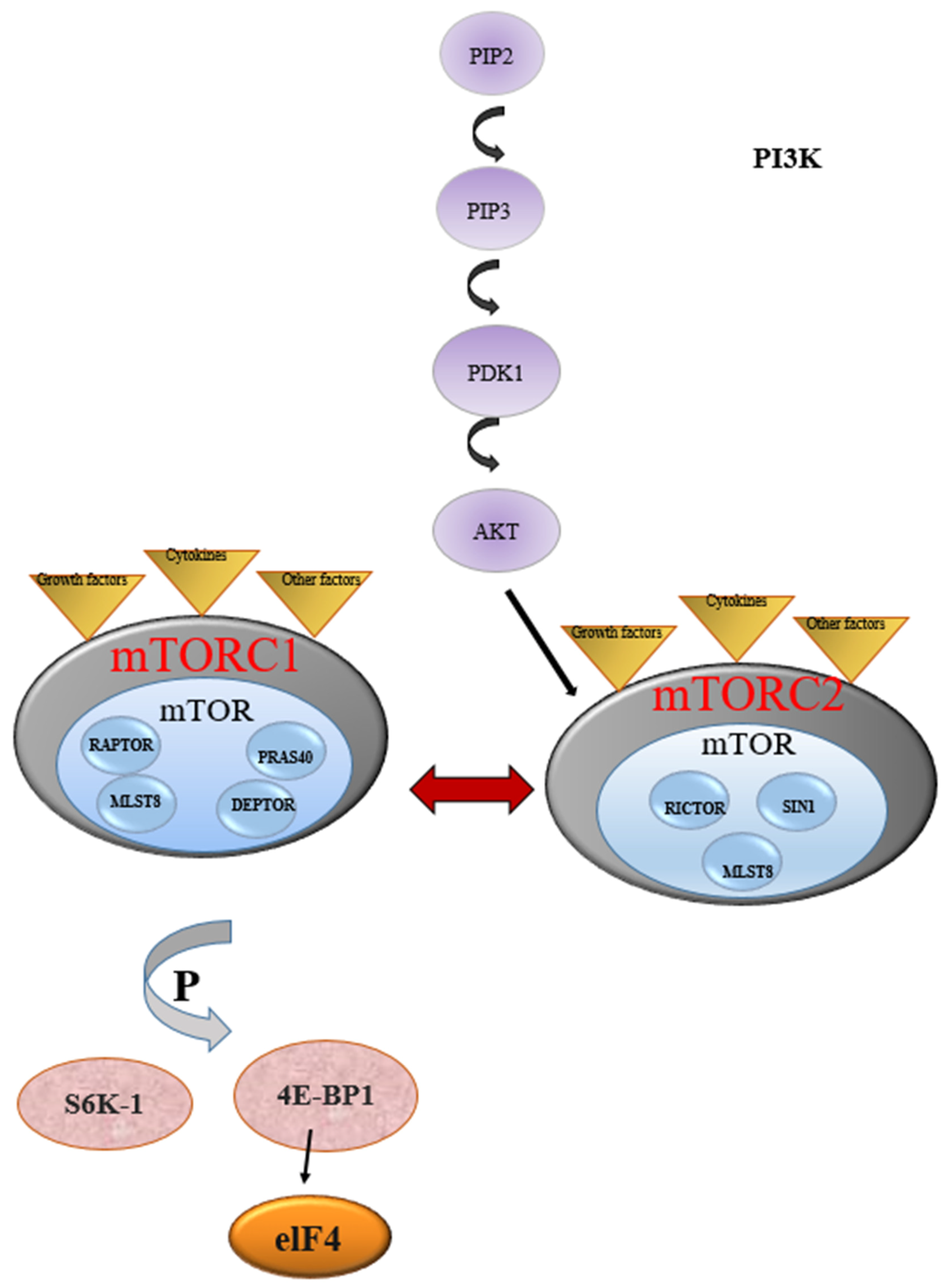{kind=link}
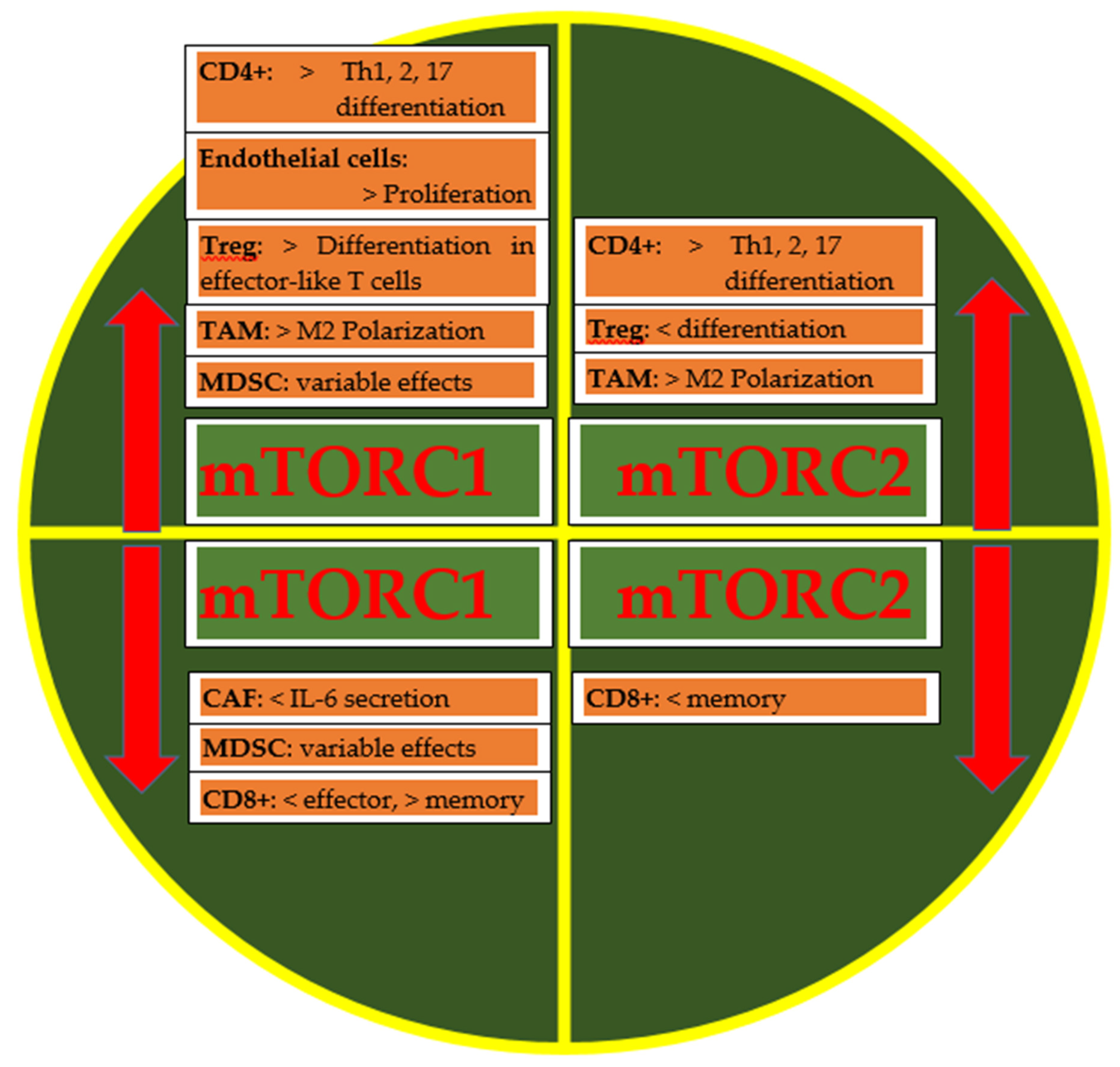{kind=link}
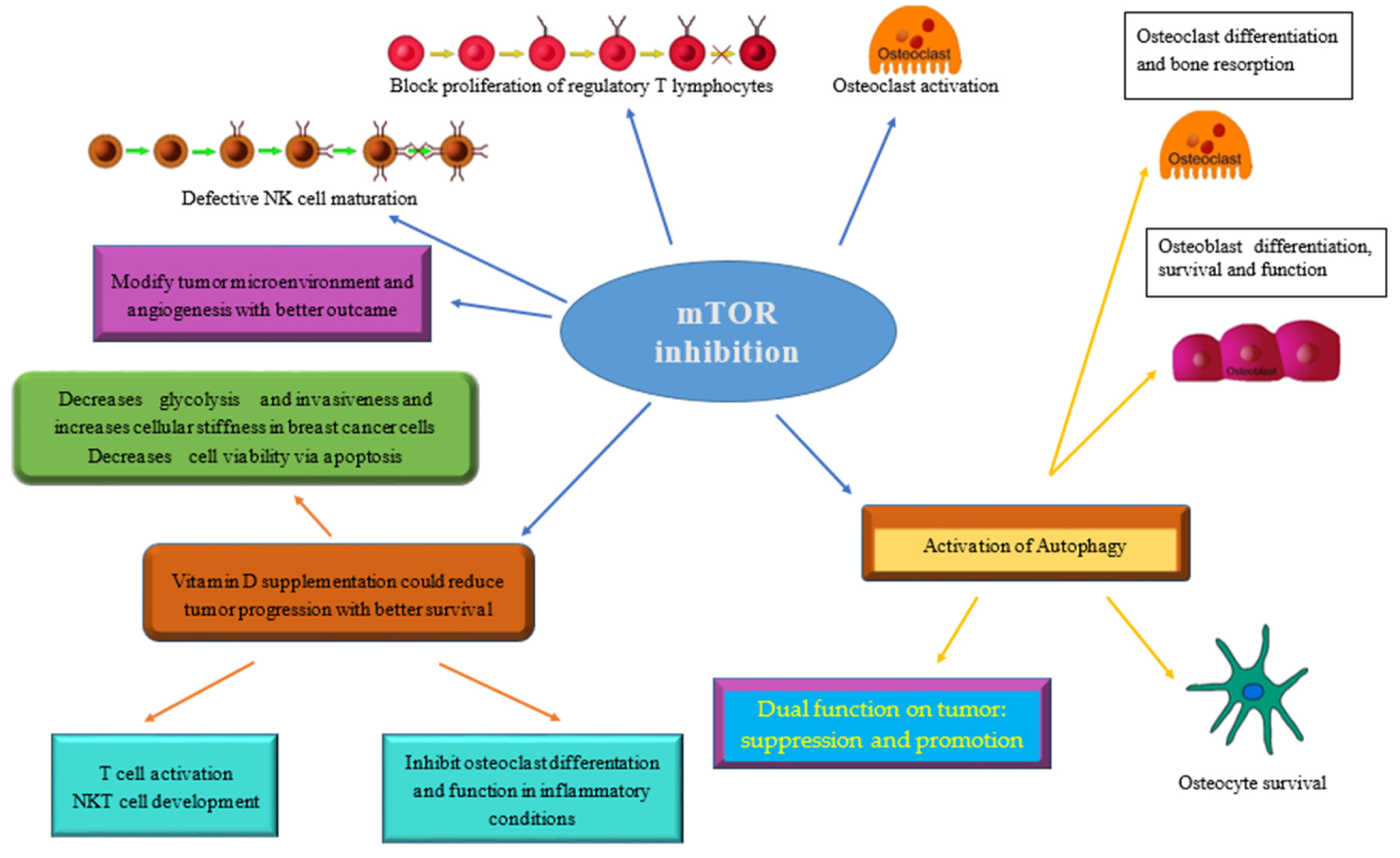{kind=link}
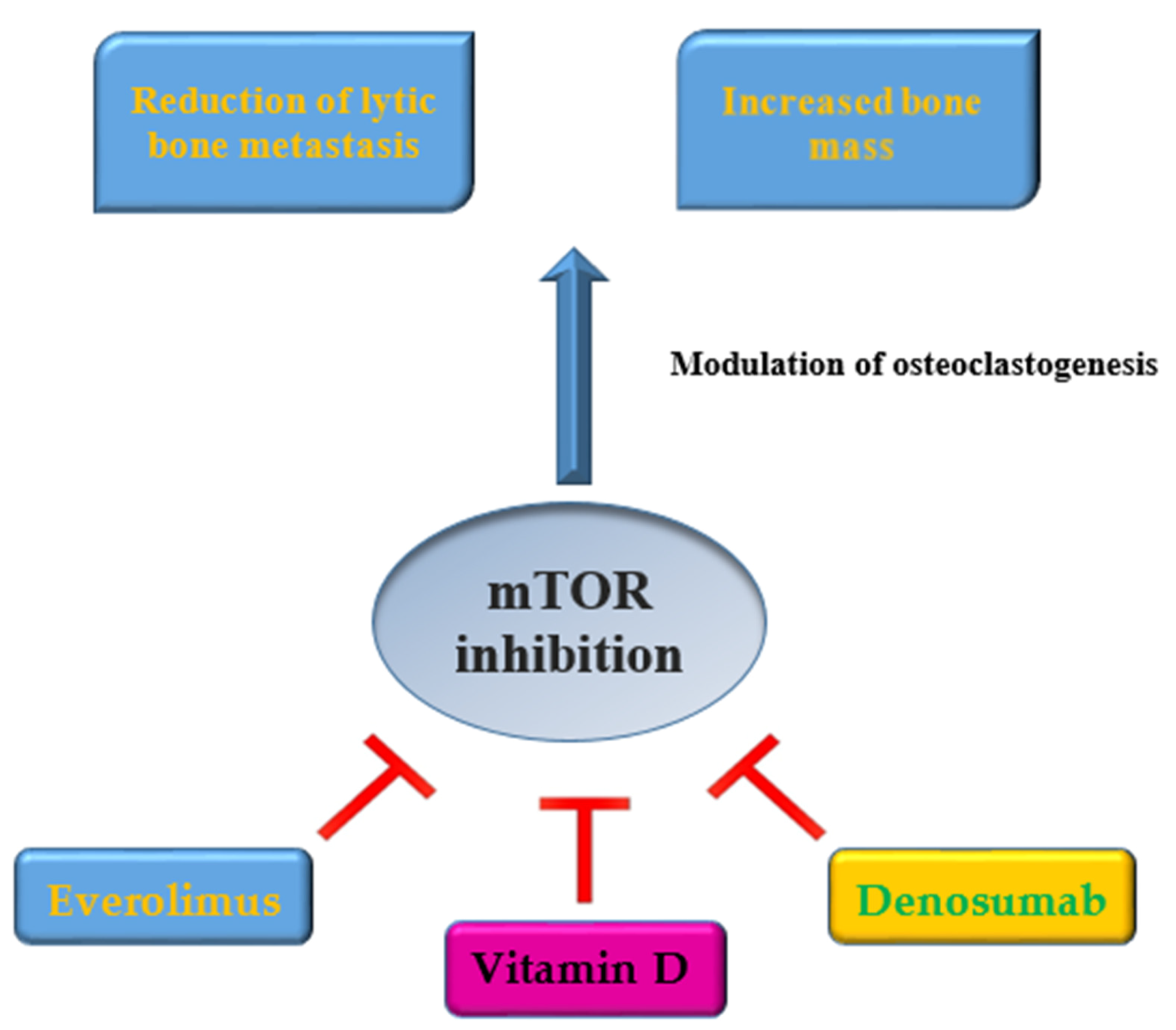{kind=link}
Abstract
:1. Introduction
2. Bone and the Immune System
3. mTOR and the Immune System
4. mTOR and Bone Metabolism
5. Vitamin D: Between mTOR and the Immune System
6. Clinical Implications
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PI3K | phosphoinositide 3-kinase |
| AKT | protein kinase B |
| mTOR | mammalian target of rapamycin |
| TME | umor microenvironment |
| IL-10 | Interleukin-10 |
| TGF-β | transforming growth factor |
| CTLA-4 | cytotoxic T-Lymphocyte protein 4 |
| PD-1 | Programmed Death 1 |
| TOR1 | target of rapamycin 1 |
| mTORC1/2 | mTOR complex ½ |
| Raptor | regulatory-associated protein of mTOR |
| mLST8 | mammalian lethal with sec-13 protein 8 |
| DEPTOR | DEP domain-containing mTOR-interacting protein |
| S6K1 | ribosomal protein S6 kinase B1 |
| RPS6 | ribosomal protein S6 |
| PRAS40 | Proline-rich-AKT-substrate 40kDa |
| Rictor | rapamycin-insensitive companion of mTOR |
| mSin1 | mammalian stress-activated map kinase-interacting protein 1 |
| Protor1/2 | protein observed with Rictor 1 and 2 |
| M-CSF | macrophage colony-stimulating factor |
| 4E-BP1 | 4E-binding protein 1 |
| TSC1 | tuberic complex sclerosis complex-1 |
| eIF4E | starting factor of eukaryotic translation 4E |
| CSLCs | cancer stem cells |
| ADCC | antibody-dependent cellular cytotoxicity |
| OPG | Osteoprotegerin |
| M-CSF | macrophage colony-stimulating factor |
| VD | Vitamin D |
| 25-VD | 25-hydroxy-vitamin D |
| VDBP | vitamin D-binding protein |
| Protor1/2 | protein observed with Rictor 1 and 2 |
| TSC1/2 | tuberous sclerosis 1 and 2 |
| AMPK | AMP-activated protein kinase |
| PTEN | phosphatase and tensin homologue |
| TSC | tuberous sclerosis complex |
| p-mTOR | phosphorylated mTOR |
| p-4EBP1 | phosphorylated 4EBP1 |
| eIF4E | eukaryotic translation initiation factor 4E |
| PIK3CA | phosphoinositide 3-kinase catalytic subunit alpha |
| PIK3CB | phosphoinositide 3-kinase catalytic subunit beta isoform |
| RPS6KA3 | ribosomal protein S6 kinase A3 |
| IGF1R | insulin-like growth factor 1 receptor |
| VEGF | vascular endothelial growth factor |
| p-Akt | phosphorylated Akt |
| S6K1 | ribosomal protein S6 kinase B1 |
| RPS6 | ribosomal protein S6 |
| ncRNAs | non-coding RNAs |
| lncRNA | long non-coding RNA |
| PIK3CD | phosphoinositide 3-kinase catalytic subunit delta |
| PIK3R3 | phosphoinositide-3-kinase regulatory subunit 3 |
References
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010, 1, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Dai, H.B.; Qiu, Z.L. mTOR signaling in osteosarcoma: Oncogenesis and therapeutic aspects. Oncol. Rep. 2016, 36, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H. mTOR signaling in immune cells and its implications for cancer immunotherapy. Cancer Lett. 2017, 408, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Uche, U.U.; Piccirillo, A.R.; Kataoka, S.; Grebinoski, S.J.; D’Cruz, L.M.; Kane, L.P. PIK3IP1/TrIP restricts activation of T cells through inhibition of PI3K/Akt. J. Exp. Med. 2018, 215, 3165–3179. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Ayuk, S.M.; Abrahamse, H. mTOR Signaling Pathway in Cancer Targets Photodynamic Therapy In Vitro. Cells 2019, 8, 431. [Google Scholar] [CrossRef]
- Teachey, D.T.; Grupp, S.A.; Brown, V.I. Mammalian target of rapamycin inhibitors and their potential role in therapy in leukaemia and other haematological malignancies. Br. J. Haematol. 2009, 145, 569–580. [Google Scholar] [CrossRef]
- Khan, K.H.; Yap, T.A.; Yan, L.; Cunningham, D. Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin. J. Cancer 2013, 32, 253–265. [Google Scholar] [CrossRef]
- Deng, L.; Chen, L.; Zhao, L.; Xu, Y.; Peng, X.; Wang, X.; Ding, L.; Jin, J.; Teng, H.; Wang, Y.; et al. Ubiquitination of Rheb governs growth factor-induced mTORC1 activation. Cell Res. 2019, 29, 136–150. [Google Scholar] [CrossRef]
- Sharif, T.; Martell, E.; Dai, C.; Singh, S.K.; Gujar, S. Regulation of the proline regulatory axis and autophagy modulates stemness in TP73/p73 deficient cancer stem-like cells. Autophagy 2019, 15, 934–936. [Google Scholar] [CrossRef]
- Massimini, M.; Palmieri, C.; De Maria, R.; Romanucci, M.; Malatesta, D.; De Martinis, M.; Maniscalco, L.; Ciccarelli, A.; Ginaldi, L.; Buracco, P.; et al. 17-AAG and Apoptosis, Autophagy and Mitophagy in Canine Osteosarcoma Cell Lines. Vet. Pathol. 2017, 54, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Chagin, A.S. Effectors of mTOR-autophagy pathway: Targeting cancer, affecting the skeleton. Cur. Opin. Pharmacol. 2016, 28, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef] [PubMed]
- Ginaldi, L.; De Martinis, M. Osteoimmunology and beyond. Curr. Med. Chem. 2016, 23, 3754–3774. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Massi, D.; Teng, M.W.L.; Mandala, M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef]
- Xue, G.; Zippelius, A.; Wicki, A.; Mandalà, M.; Tang, F.; Massi, D.; Hemmings, B.A. Integrated Akt/PKB signaling in immunomodulation and its potential role in cancer immunotherapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int J. Mol. Sci. 2018, 19, 2453. [Google Scholar] [CrossRef]
- Mingari, M.C.; Gerosa, F.; Carra, G.; Accolla, R.S.; Moretta, A.; Zubler, R.H.; Waldmann, T.A.; Moretta, L. Human interleukin-2 promotes proliferation of activated B cells via surface receptors similar to those of activated T cells. Nature 1984, 6, 601. [Google Scholar] [CrossRef]
- Chester, C.; Fritsch, K.; Kohrt, H.E. Natural Killer Cell Immunomodulation: Targeting Activating, Inhibitory, and Co-stimulatory Receptor Signaling for Cancer Immunotherapy. Front. Immunol. 2015, 6, 601. [Google Scholar] [CrossRef]
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br. J. Cancer 2015, 112, 1501–1509. [Google Scholar] [CrossRef]
- Yang, C.; Tsaih, S.W.; Lemke, A.; Flister, M.J.; Thakar, M.S.; Malarkannan, S. mTORC1 and mTORC2 differentially promote natural killer cell development. eLife 2018, 7, e35619. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zou, Q.; Su, B. Regulation of T cell immunity by cellular metabolism. Front. Med. 2018, 12, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle 2015, 14, 3331–3339. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Saqcena, M.; Chatterjee, A.; Garcia, A.; Frias, M.A.; Foster, D.A. Reciprocal regulation of AMP-activated protein kinase and phospholipase D. J. Biol. Chem. 2015, 290, 6986–6993. [Google Scholar] [CrossRef]
- Ginaldi, L.; De Martinis, M.; Ciccarelli, F.; Saitta, S.; Imbesi, S.; Mannucci, C.; Gangemi, S. Increased levels of interleukin 31 (IL-31) in osteoporosis. BMC Immunol. 2015, 16, 60. [Google Scholar] [CrossRef]
- Ginaldi, L.; De Martinis, M.; Saitta, S.; Sirufo, M.M.; Mannucci, C.; Casciaro, M.; Ciccarelli, F.; Gangemi, S. Interleukin-33 serum levels in postmenopausal women with osteoporosis. Sci. Rep. 2019, 9, 3786. [Google Scholar] [CrossRef]
- Irelli, A.; Cocciolone, V.; Cannita, K.; Zugaro, L.; Di Staso, M.; Lanfiuti Baldi, P.; Paradisi, S.; Sidoni, T.; Ricevuto, E.; Ficorella, C. Bone targeted therapy for preventing skeletal-related events in metastatic breast cancer. Bone 2016, 87, 169–175. [Google Scholar] [CrossRef]
- Glantschnig, H.; Fisher, J.E.; Wesolowski, G.; Rodan, G.A.; Reszka, A.A. M-CSF, TNFalpha and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ. 2003, 10, 1165–1177. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Feige, U.; Sarosi, I.; Bolon, B.; Tafuri, A.; Morony, S.; Capparelli, C.; Li, J.; Elliott, R.; McCabe, S.; et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 1999, 402, 304–309. [Google Scholar] [CrossRef]
- Van Dam, P.A.; Verhoeven, Y.; Trinh, X.B.; Wouters, A.; Lardon, F.; Prenen, H.; Smits, E.; Baldewijns, M.; Lammens, M. RANK/RANKL signaling inhibition may improve the effectiveness of checkpoint blockade in cancer treatment. Crit. Rev. Oncol. Hematol. 2019, 133, 85–91. [Google Scholar] [CrossRef]
- Ciccarelli, F.; De Martinis, M.; Ginaldi, L. Glucocorticoids in Patients with Rheumatic Diseases: Friends or Enemies of Bone? Curr. Med. Chem. 2015, 22, 596–603. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, M.; Sirufo, M.M.; Ginaldi, L. Osteoporosis: Current and emerging therapies targeted to immunological checkpoints. Curr. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.; Clézardin, P. Bone-Targeted Therapies in Cancer-Induced Bone Disease. Calcif. Tissue Int. 2018, 102, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.M.; Li, L.; Indran, I.R.; Chew, N.; Yong, E.L. TRAF6 Mediates Suppression of Osteoclastogenesis and Prevention of Ovariectomy-Induced Bone Loss by a Novel Prenylflavonoid. J. Bone Miner. Res. 2017, 32, 846–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.M.; Zhao, L.; Liu, T.T.; Jiao, P.L.; Zhao, D.D.; Shih, M.S.; Tao, B.; Sun, L.H.; Zhao, H.Y.; Liu, J.M. Rictor/mTORC2 loss in osteoblasts impairs bone mass and strength. Bone 2016, 90, 50–58. [Google Scholar] [CrossRef]
- Bakker, A.D.; Jaspers, R.T. IL-6 and IGF-1 Signaling Within and Between Muscle and Bone: How Important is the mTOR Pathway for Bone Metabolism? Curr. Osteoporos. Rep. 2015, 13, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Bakker, A.D.; Gakes, T.; Hogervorst, J.M.; de Wit, G.M.; Klein-Nulend, J.; Jaspers, R.T. Mechanical Stimulation and IGF-1 Enhance mRNA Translation Rate in Osteoblasts Via Activation of the AKT-mTOR Pathway. J. Cell Physiol. 2016, 231, 1283–1290. [Google Scholar] [CrossRef]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [Green Version]
- Cusato, J.; Genova, C.; Tomasello, C.; Carrega, P.; Ottonello, S.; Pietra, G.; Mingari, M.C.; Cossu, I.; Rijavec, E.; Leggieri, A.; et al. Influence of Vitamin D in Advanced Non-Small Cell Lung Cancer Patients Treated with Nivolumab. Cancers (Basel) 2019, 11, 125. [Google Scholar] [CrossRef] [Green Version]
- Lisse, T.S.; Liu, T.; Irmler, M.; Beckers, J.; Chen, H.; Adams, J.S.; Hewison, M. Gene targeting by the vitamin D response element binding protein reveals a role for vitamin D in osteoblast mTOR signaling. FASEB J. 2011, 25, 937–947. [Google Scholar] [CrossRef] [Green Version]
- Lisse, T.S.; Hewison, M. Vitamin D: A new player in the world of mTOR signaling. Cell Cycle 2011, 10, 1888–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, G.; Ren, H.; Qiu, T.; Zhang, Z.; Zhao, W.; Yu, X.; Huang, J.; Tang, J.; Liang, D.; Yao, Z.; et al. Mammalian target of rapamycin as a therapeutic target in osteoporosis. J. Cell Physiol. 2018, 233, 3929–3944. [Google Scholar] [CrossRef] [PubMed]
- Stucci, L.S.; D’Oronzo, S.; Tucci, M.; Macerollo, A.; Ribero, S.; Spagnolo, F.; Marra, E.; Picasso, V.; Orgiano, L.; Marconcini, R.; et al. Italian Melanoma Intergroup (IMI). Vitamin D in melanoma: Controversies and potential role in combination with immune check-point inhibitors. Cancer Treat. Rev. 2018, 69, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Bersanelli, M.; Leonetti, A.; Buti, S. The link between calcitriol and anticancer immunotherapy: Vitamin D as the possible balance between inflammation and autoimmunity in the immune-checkpoint blockade. Immunotherapy 2017, 9, 1127–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Essen, M.R.; Kongsbak, M.; Schjerling, P.; Olgaard, K.; Odum, N.; Geisler, C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat. Immunol. 2010, 11, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Cantorna, M.T. The vitamin D receptor is required for iNKT cell development. Proc. Natl. Acad. Sci. USA 2008, 105, 5207–5212. [Google Scholar] [CrossRef] [Green Version]
- Zaza, G.; Granata, S.; Caletti, C.; Signorini, L.; Stallone, G.; Lupo, A. mTOR Inhibition Role in Cellular Mechanisms. Transplantation 2018, 102 (2S Suppl. 1), S3–S16. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Francipane, M.G.; Lagasse, E. Selective targeting of human colon cancer stem-like cells by the mTOR inhibitor Torin-1. Oncotarget 2013, 4, 1948–1962. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Su, Z.; Fei, H.; Liu, X.; Pan, Q. The novel mTOR inhibitor Torin-2 induces autophagy and downregulates the expression of UHRF1 to suppress hepatocarcinoma cell growth. Oncol. Rep. 2015, 34, 1708–1716. [Google Scholar] [CrossRef]
- Naruse, T.; Yanamoto, S.; Okuyama, K.; Yamashita, K.; Omori, K.; Nakao, Y.; Yamada, S.I.; Umeda, M. Therapeutic implication of mTORC2 in oral squamous cell carcinoma. Oral Oncol. 2017, 65, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Hau, A.M.; Nakasaki, M.; Nakashima, K.; Krish, G.; Hansel, D.E. Differential mTOR pathway profiles in bladder cancer cell line subtypes to predict sensitivity to mTOR inhibition. Urol. Oncol. 2017, 35, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, S.M.; Boufraqech, M.; Zhang, L.; Mehta, A.; Kapur, P.; Zhang, Y.; Li, Z.; Shen, M.; Kebebew, E. Torin2 targets dysregulated pathways in anaplastic thyroid cancer and inhibits tumor growth and metastasis. Oncotarget 2015, 6, 18038–18049. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xu, C.; Kirubakaran, S.; Zhang, X.; Hur, W.; Liu, Y.; Kwiatkowski, N.P.; Wang, J.; Westover, K.D.; Gao, P.; et al. Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer Res. 2013, 73, 2574–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alameen, A.A.; Simioni, C.; Martelli, A.M.; Zauli, G.; Ultimo, S.; McCubrey, J.A.; Gonelli, A.; Marisi, G.; Ulivi, P.; Capitani, S.; et al. Healthy CD4+ T lymphocytes are not affected by targeted therapies against the PI3K/Akt/mTOR pathway in T-cell acute lymphoblastic leukemia. Oncotarget 2016, 7, 55690–55703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Frias, M.A.; Chatterjee, A.; Yellen, P.; Foster, D.A. The Enigma of Rapamycin Dosage. Mol. Cancer Ther. 2016, 15, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Templeton, A.J.; Dutoit, V.; Cathomas, R.; Rothermundt, C.; Bartschi, D.; Droge, C.; Gautschi, O.; Borner, M.; Fechter, E.; Stenner, F.; et al. Swiss Group for Clinical Cancer. Phase 2 trial of sinle-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08). Eur. Urol. 2013, 64, 150–158. [Google Scholar] [CrossRef]
- Karkeni, E.; Morin, S.O.; BouTayeh, B.; Goubard, A.; Josselin, E.; Castellano, R.; Fauriat, C.; Guittard, G.; Olive, D.; Nunès, J.A. Vitamin D Controls Tumor Growth and CD8+ T Cell Infiltration in Breast Cancer. Front. Immunol. 2019, 10, 1307. [Google Scholar] [CrossRef] [Green Version]
- Gnant, M.; Baselga, J.; Rugo, H.S.; Noguchi, S.; Burris, H.A.; Piccart, M.; Hortobagyi, G.N.; Eakle, J.; Mukai, H.; Iwata, H.; et al. Effect of everolimus on bone marker levels and progressive disease in bone in BOLERO-2. J. Natl. Cancer Inst. 2013, 105, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Hadji, P.; Stoetzer, O.; Decker, T.; Kurbacher, C.M.; Marmé, F.; Schneeweiss, A.; Mundhenke, C.; Distelrath, A.; Fasching, P.A.; Lux, M.P.; et al. The impact of mammalian target of rapamycin inhibition on bone health in postmenopausal women with hormone receptor-positive advanced breast cancer receiving everolimus plus exemestane in the phase IIIb 4EVER trial. J. Bone Oncol. 2018, 14, 100199. [Google Scholar] [CrossRef]
- Bochen, F.; Balensiefer, B.; Körner, S.; Bittenbring, J.T.; Neumann, F.; Koch, A.; Bumm, K.; Marx, A.; Wemmert, S.; Papaspyrou, G.; et al. Vitamin D deficiency in head and neck cancer patients—Prevalence, prognostic value and impact on immune function. Oncoimmunology 2018, 7, e1476817. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irelli, A.; Sirufo, M.M.; Scipioni, T.; De Pietro, F.; Pancotti, A.; Ginaldi, L.; De Martinis, M. mTOR Links Tumor Immunity and Bone Metabolism: What are the Clinical Implications? Int. J. Mol. Sci. 2019, 20, 5841. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235841
Irelli A, Sirufo MM, Scipioni T, De Pietro F, Pancotti A, Ginaldi L, De Martinis M. mTOR Links Tumor Immunity and Bone Metabolism: What are the Clinical Implications? International Journal of Molecular Sciences. 2019; 20(23):5841. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235841
Chicago/Turabian StyleIrelli, Azzurra, Maria Maddalena Sirufo, Teresa Scipioni, Francesca De Pietro, Amedeo Pancotti, Lia Ginaldi, and Massimo De Martinis. 2019. "mTOR Links Tumor Immunity and Bone Metabolism: What are the Clinical Implications?" International Journal of Molecular Sciences 20, no. 23: 5841. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235841