Advances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy


Human Genetics Department, Unit of Research and Development, Nacional Health Institute Doctor Ricardo Jorge, 4000-055 Porto, Portugal
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(23), 5897; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235897
Submission received: 11 October 2019
/
Revised: 21 November 2019
/
Accepted: 22 November 2019
/
Published: 24 November 2019
(This article belongs to the Section Molecular Biophysics)
Abstract
:Sphingolipidoses are inherited genetic diseases characterized by the accumulation of glycosphingolipids. Sphingolipidoses (SP), which usually involve the loss of sphingolipid hydrolase function, are of lysosomal origin, and represent an important group of rare diseases among lysosomal storage disorders. Initial treatments consisted of enzyme replacement therapy, but, in recent decades, various therapeutic approaches have been developed. However, these commonly used treatments for SP fail to be fully effective and do not penetrate the blood–brain barrier. New approaches, such as genome editing, have great potential for both the treatment and study of sphingolipidoses. Here, we review the most recent advances in the treatment and modelling of SP through the application of CRISPR-Cas9 genome editing. CRISPR-Cas9 is currently the most widely used method for genome editing. This technique is versatile; it can be used for altering the regulation of genes involved in sphingolipid degradation and synthesis pathways, interrogating gene function, generating knock out models, or knocking in mutations. CRISPR-Cas9 genome editing is being used as an approach to disease treatment, but more frequently it is utilized to create models of disease. New CRISPR-Cas9-based tools of gene editing with diminished off-targeting effects are evolving and seem to be more promising for the correction of individual mutations. Emerging Prime results and CRISPR-Cas9 difficulties are also discussed.
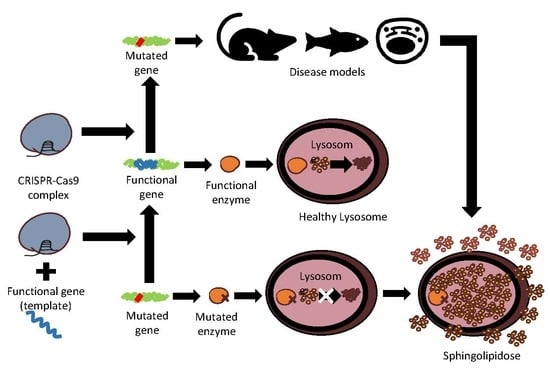
1. Lysosomal Storage Diseases and Sphingolipidoses
The present study was based on high-quality, peer-reviewed publications available at NCBI Pubmed until October 2019. The following work aims to provide a review covering the application of CRISPR-Cas9, with a focus on sphingolipidoses.
Lysosomal storage diseases (LSDs) are an important subdivision of inherited metabolic diseases. As a group, these monogenic diseases are estimated to affect 1 in 7500 newborns [1].
Generally, LSDs are multisystemic and degenerative. Patients present a continuum of disease severity, which are usually classified by the type of disorder and the age of onset of clinical signs. Different types of defects may underlie LSDs, including, but not limited to, defects in lysosomal hydrolases, lysosomal membrane proteins, activator proteins, or transport proteins. Nevertheless, they all present a common feature, which is the storage of macromolecules in the lysosomes. When one of the several lysosomal hydrolases is dysfunctional, or has lost activity, due to mutations or incorrect protein folding, the related substrate accumulates inside the cell. Depending on the type of storage materials, LSDs are classically divided in subgroups that reflect the analogous affected lysosomal pathway. Such is the case, for example, of the sphingolipidoses, mucopolysaccharidoses, and glycoproteinoses. Additionally, there are diseases involving integral membrane proteins, disorders of lysosome-related organelles, and disorders involving lipofuscin [2].
Sphingolipidoses are a subgroup of LSDs usually caused by defective sphingolipid hydrolases that act upon the sphingolipid degradative pathway. Most sphingolipidoses present autosomal recessive inheritance. In some population groups, a few mutations account for a large proportion of the mutant variants. Nevertheless, a myriad of mutations can cause these disorders. Mutated genes coding for sphingolipid hydrolases or deregulation of lysosomal-related proteins are at the basis of sphingolipidoses. In these diseases, cells are unable to degrade specific lipids (Figure 1), which then accumulate inside the cell until reaching toxic or nonfunctional levels, leading to multisystemic pathologies [3,4,5].
These rare disorders have been associated with other frequent diseases, most likely because the toxic effects on the cells makes them prone to the development of other, more common diseases [6,7]. For instance, causal mutations of Gaucher disease are considered to constitute a risk for Parkinson’s disease. Therefore, diseases involving mutant glycosphingolipids represent a significant multidimensional problem. Moreover, recent results in Niemann–Pick A/B demonstrate a tight link between cellular sphingolipid metabolism and immunity, which supports the hypothesis of a central role of sphingolipids in processes beyond those of the lysosome [8]. Recently, a transcription factor EB (TFEB), which is responsible for the transcription coordination of most lysosomal genes, has been identified, this transcription controller provides a new tool to manipulate and study lysosomal diseases [9]. The discovery of TFEB led to the search for a motif associated with genes with lysosomal function, and the coordinated expression resulted in the identification of a new mechanism, the coordinated lysosomal expression and regulation (CLEAR) [10].
Although the LSDs are rare, research in the field has been abundant; LSDs have been the stage for the development of new therapies and have provided knowledge that extends beyond LSDs.
1.1. Gaucher Disease
The most common sphingolipidosis is Gaucher disease (GD). It is caused by mutations in the GBA1 gene that encodes lysosomal acid-β-glucocerebrosidase (GCase), the enzyme responsible for the degradation of glucosylceramide (GlcCer). The Gaucher′s macrophage is the classic pathologic hallmark of the disease, presenting a lysosomal compartment enlarged by the storage of undegraded glycolipid molecules. These cells have a pivotal role in the development of the disease since they are capable of inducing local tissue reactions [1].
Depending on the level severity, deterioration, and type of impairment, GD can be categorized in three main groups, type I, II, and III. The most frequent form of GD is type I, also known as nonneuropathic GD as it spares the nervous system. Thus, it is considered to be the less aggressive form of GD. It can be subdivided according to the age of onset and may have clinical onset at any time during the human life cycle [6,11]. Types II and III are characterized by the presence of primary neurologic disease affecting the central nervous system (CNS). Type II is the most severe form of GD—it has severe and rapid degeneration leading to death before 2–4 years of age [12,13]. Type III usually has a later onset with slower progression. In spite of the three conventional GD classifications, GD presents an important continuum of clinical forms. The symptoms of Gaucher are extensive, affecting many organs systems, such as skeletal, cardiovascular, and neurological. In some cases, GD variants have been associated with other neurological diseases such as Parkinson’s disease and peripheral neuropathy [6,11].
GD can be caused by GBA1 mutations or, less frequently, by mutations in the gene coding for the activator protein saposin C. The application of new sequencing approaches leads to a continuously growing number of GBA1 mutations. Nevertheless some mutations have been reported as more frequent in specific population groups [12,13,14].
The degree of endoplasmic reticulum retention (ER) and proteasomal degradation has recently been proposed as an important factor influencing the outcome of GCase mutants. Different mutations in GBA1, which impact the ability of the protein to fold correctly, present variable levels of ER retention and undergo ER-associated degradation (ERAD) in the proteasome [15].
It is known that GD patients are at a higher risk to develop Parkinson′s disease (PD) in comparison to the non-GD population. Thus, the mutated GCase seems to be a predisposing factor for the development of PD. Mutant GCase contributes to the accumulation and aggregation of α-synuclein, one of the main causes of PD, which may be a reason for pathological interaction [16]. The presence of mutant GCase is also thought to lead to the enhancement of neuronal vulnerability to neurodegenerative processes, triggered by increased alpha-synuclein expression [17] and by ERAD, which is also known to be involved in the process leading to Parkinson′s disease [15].
1.2. Fabry Disease
Fabry disease (FD) is a sphingolipidosis caused by a mutated alpha-galactosidase A, encoded by the GLA gene. FD follows an X-inheritance pattern, but methylation skewing can also lead to female presentation of the disease. When the enzyme is nonfunctional or not present, its substrate, globotriaosylceramide (Gb3), accumulates in the lysosomes [18,19].
The classic form of FD is clinically heterogeneous, with different symptoms along the patient’s life, and usually has more severe clinical expression in males than in females. The lifespan is decreased, often due to renal failure, heart diseases, and strokes. However, there are also reports of late-onset Fabry disease in males, involving only a single organ system (cardiac or renal) due to missense mutations that lead to sufficient residual enzyme activity that prevent symptoms in childhood and early adulthood [20,21].
1.3. GM2-Gangliosidoses
Tay–Sachs Disease (TSD) is one of the most common GM2-gangliosidoses, the substrate is the main glycolipid of neuronal cell plasma membranes. GM2-gangliosidoses are a group of autosomal recessive LSDs related to deficiency in lysosomal hexosaminidases (Hex). Deficient enzyme activity leads to abnormal accumulation of GM2 ganglioside and impaired neuronal cellular activity [22]. Defects in the gene-coding ganglioside GM2 activator protein (GM2A) also lead to TSD. The Hex enzymes are all dimers encoded by the same two genes, HEXA (encodes α subunit) and HEXB (encodes β subunit). HexA enzyme is composed of subunits α and β, and HexB by two β subunits [23]. In the case of TSD, HexA enzyme is mutated whereas in Sandhoff disease (SD), it is HexB that is mutated.
SD is classified in three main age-related forms: (1) infantile, with progressive neurologic impairment, hypotonia, bilateral cherry-red spots in the retina, and seizures; (2) juvenile, with dementia, cerebellar ataxia, mental retardation, and spinal muscular atrophy; and (3) adult, with spinocerebellar degeneration or motor neuron disorders [24].
TSD is also classified by age of onset. The infantile form is characterized by mental and motor developmental delay, hypotension, cherry-red spots, and dysphagia; the juvenile form is characterized by ataxia and spasm progression; and the later-onset form is characterized by gradual decline in motor, cerebral, and spinocerebellar function [25].
Variant B1, particularly prevalent in populations of Northern Iberian descent, particularly in individuals of Portuguese ancestry, is due to a higher frequency of a specific HEXA mutation. This atypical juvenile variant is characterized by differential activity towards sulphated and nonsulphated substrates. The deficiency in this enzyme has altered kinetic properties due to active site disturbance. In most cases, this variant is caused by at least one c.533 G > A allele [26,27].
1.4. Niemann–Pick Disease
Niemann–Pick diseases (NPD) are autosomal recessive inherited diseases subdivided into hydrolase deficient entities or trafficking-impaired disorders. In acid sphingomyelinase-deficient NPD (ASM-deficient NPD), underlying defects are due to SMPD1 gene mutations (type A, B, and intermediate forms), whereas in NP type C (NPC), which is currently described as a cholesterol trafficking defect, is due to NPC1 or NPC2 gene mutations [28,29]. NPC was historically classified as a sphingolipidosis, but it is not caused by a deficiency in sphingolipid catabolism. Rather, it is caused by a cholesterol trafficking defect.
The ASM is a lysosomal enzyme which degrades sphingomyelin. When the enzyme is not present or is nonfunctional, sphingomyelin accumulates in different organs. Type A NPD leads to hepatosplenomegaly and severe CNS problems in infancy, with a frequent lifespan of only 2–3 years after birth. Type B NPD causes hepatosplenomegaly and lung problems, but typically does not impact the CNS. Frequently, NPD diseases cause lipid storage and foam cell infiltration in tissues, pulmonary insufficiency, and central nervous system issues [30,31].
1.5. Krabbe Disease
Like most LSDs, Krabbe Disease (KD) is an autosomal recessive disease. Its underlying causal deficiency is in the lysosomal enzyme β-galactocerebrosidase (Galc), which results from a mutated GALC gene that encodes that enzyme. When deficiency of such enzyme is present, galactosylceramide, the major natural substrate, accumulates in lysosomes. This sphingolipid contains sphingosine, galactose, and fatty acid, and is almost exclusively localized in the myelin sheath [32]. Besides galactosylceramide, Galc has other substrates: galactosylsphingosine or psychosine, monogalactosyldiglyceride, and the precursor of seminolipid. Additionally, KD can also be associated with the mutated activator protein, saposin A [32].
This disease damages the white matter of the brain; it is also known as globoid cell leukodystrophy. KD has been subdivided into four age/symptom-based types. The most common subtype, which presents faster progression, is early infantile type (birth–5 months) the symptoms include irritability, regression of psychomotor development, feeding difficulties, and, as the disease progresses, hypertonicity, seizures, loss of vision and hearing, and early death. At the other end of the spectrum is the adult type (>16 years) with slow development of progressive spastic paraparesis or gait abnormalities [33,34].
1.6. GM1-Gangliosidosis and Morquio B Syndrome
The deficient activity of the lysosomal hydrolase β-galactosidase (β-gal), coded by GLB1, can result in two distinguishable clinical entities, GM1-gangliosidosis and Morquio B syndrome. GM1-gangliosidosis is caused by lysosomal accumulation of ganglioside GM1 and its derivative GA1.
GM1-gangliosidosis is a neurodegenerative disease with GM1-ganglioside accumulation impairing neuronal functioning, whereas Morquio B is a progressive condition that mainly affects the skeleton and has an accumulation of keratan sulfate, which is excreted in the urine.
GM1-gangliosidosis is characterized by progressive neurologic dysfunction, presenting different degrees of severity depending on the subtype, which is subdivided according to the age of onset into Type I (infantile), Type II (late infantile/juvenile), and Type III (adult) [35].
Allelic mutations of the GLB1 gene cause heterogeneous clinical phenotypes, such as GM1-gangliosidosis and Morquio B syndrome. An important structural model of the protein was created and is convenient for modelling the mutant variants in order to better understand the structural bases of the diseases [36]. The existence of intermediate phenotypes that are clinically difficult to distinguish demonstrates the need to further investigate the disease. The in silico analysis and three-dimensional modelling of mutated GLB1 proteins are most helpful, but still not sufficient to fully differentiate between the two entities or establish a genotype/phenotype correlation [37].
1.7. Current Treatments for Sphingolipidoses
Currently, the most common treatments for sphingolipidoses are enzyme replacement therapy (ERT), substrate reduction therapy (SRT), and pharmacological chaperones (PC). ERT is currently the most widely used treatment. However, ERT has a variety of limitations. One major difficulty is that the replacement enzymes are not capable of crossing the blood–brain barrier. Additionally, all of the aforementioned therapeutic approaches are transitory and require periodically administration of the therapeutic agent. Furthermore, in the case of ERT, there is potential for an immunological reaction to the replacement enzyme. SRT is capable of targeting more than one sphingolipidosis as it utilizes inhibitors to block glycosphingolipid synthesis. However, this method can lead to partial depletion of glycosphingolipids from the plasma membrane, which can result in severe side effects due to off-targeting of the inhibitors (even targeting nonlysosomal enzymes) or other unknown interactions. Unlike most ERTs, some of the molecules used for SRT are able to cross the blood–brain barrier. PC treatment can only be successfully used in variants of diseases that are caused by enzyme misfolding, as chaperones assist in protein folding processes. As in the case of STR, this method is also prone to off-targeting [4,5].
ERT was the first therapy applied to sphingolipidoses and it resulted from a series of observations from various scientists; in terms of therapeutic advances, it was most important and provided the stepping stone for many other ERTs that are now available [38]. Concomitantly, it also brought upon these rare diseases a great interest from social, scientific, medical, and pharmaceutical points-of-view.
2. CRISPR-Cas9 and Prime Editing Background
CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats) is a programmable tool for site-specific gene editing with low cost, easy application, and high efficiency when compared to other well-known gene editing tools like TALEN (transcription activator-like effector nuclease) and ZFN (zinc finger nuclease [39].
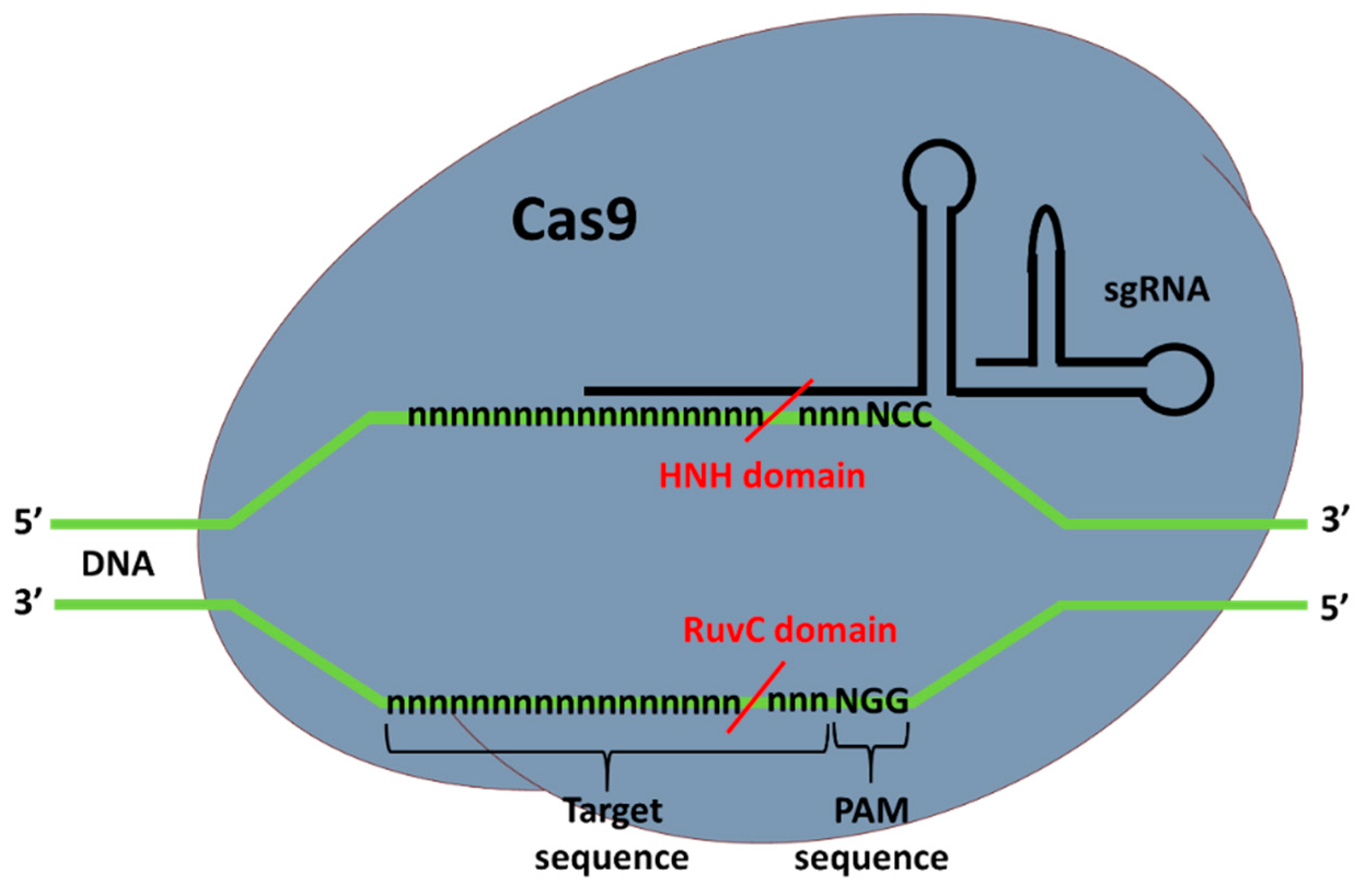
Type II CRISPR-Cas9 complex, as the name suggest, is composed of the Cas9 nuclease and a guide RNA (gRNA). When bound to DNA correctly, the Cas9 catalyzes a double-strand break (DSB) at the target site. Depending of the organism of origin, the Cas9 endonuclease can be structurally and functionally different. The most widely used and tested Cas9 is the Streptococcus pyogenes Cas9 (SpCas9), which requires a PAM (protospacer-adjacent motif) sequence downstream and adjacent of the target site to properly bind to the DNA sequence, which will then cut. For SpCas9 the PAM sequence is NGG, where N is any DNA nucleotide and G is guanine. This sequence occurs at a high frequency in the genome, allowing CRISPR-Cas9 to target virtually any gene. HNH and RuvC are the nuclease domains of Cas9 that allow for the DSB of the DNA [40]. When one of the domains is mutated, preventing the Cas9 from making DSB, a nickaseCas9 (nCas9) is produced. nCas9 is used to nick DNA, or to decrease off-targeting using two gRNAs complexed with nCas9 for DSB [41].
If both domains are inactive, the Cas9 complex acts as an inhibitor, preventing transcription of a certain gene by binding to the DNA area that constitutes that gene, but without cutting. This type of Cas9 tool is commonly known as dead Cas9 (dCas9) [42].
In addition to Cas9, a guide RNA is also necessary for targeting the DNA sequence. Together, CRISPR RNA (crRNA) and transactivating crRNA (tracrRNA) form the gRNA (two RNA chains bounded). This can also be fully synthesized in one piece, known as a synthetic single-guide RNA (sgRNA), composed of one RNA chain. gRNA binds to the Cas9 in order to form the ribonucleoproteic complex known as CRISPR-Cas9. The gRNA is complementary to the complementary chain of the target sequence and the crRNA part is composed of 20 nucleotides that bind to the DNA (Figure 2). The conserved 3′-end scaffold of gRNA (tracrRNA) binds to Cas9 due to the presence of a stem-loop structure in is end [40].
Even though there is the need for a PAM adjacent to the target site, the sequence (5′-NGG-3′) highly occurs in the genomes, thus, CRISPR-Cas9 can target virtually any gene. Even though this tool has many advantages, it also has its cons, a major one is the possibility of off-targeting due to homology with other sites [40].
The cleavage of the DNA creates a double-strand break (DSB) that is naturally repaired by the cell using one of two mechanisms. Usually it is repaired by the nonhomologous end joining (NHEJ) pathway, which introduces a small insertion or deletion (InDel) at the DSB site. However, the InDel normally disrupts the gene, which could subsequently impact structure and function of the resulting protein. The other pathway of repair, called homology-directed repair (HDR), uses, if existent (sister chromatid [43]) or added, a donor DNA fragment with homology to the flanking sequence that is integrated into the genome at the DSB site, thus repairing the broken DNA [44].
One of the limitations of using CRISPR-Cas9 technology for permanent gene modification is the efficiency of integration of the complex and donor template in living cells and the possible loss of cell viability due to the transfection process.
CRISPR-Cas9 integration can be carried out with plasmids or transcripts capable of expressing Cas9 and gRNA genes, or by directly transfecting the ribonucleoprotein (RNP) complex previously assembled in vitro [45].
The transformation can be achieved with electroporation, lipofection, polymeric nanoparticles, cell-penetrating peptides, or virus-like retroviruses and lentiviruses. However, disruption of the cell membrane is an aggressive procedure that results in many cell casualties. Furthermore, liposomes and viruses are limited by efficiency and size, and in vitro complexes can be toxic to the cells due to their exogenous nature [45].
Depending on the model and objective, the genome editing approach can change, but the highest nuclease activity is achieved with RNP. The RNP approach is reliable not only in cells, but also in embryos and in vitro assays. This approach is the most convenient and functional, since the complex synthesis is not dependent on DNA or mRNA expression by the cells to be edited. Moreover, it is the best method to avoid off-targeting due to the shorter exposure to the genome [46].
When a mutation in a specific gene leads to a nonfunctional protein, gene editing has the potential to repair this issue. In cases where a few mutations represent a large proportion of the causal alleles, it is possible to correct each mutation independently. Examples of such an approach exist in other pathologies, such as Duchenne muscular dystrophy [47], cystic fibrosis [48], chronic myeloid leukaemia [49], dominant progressive hearing loss [50], and autosomal dominant retinitis pigmentosa [51]. In brief, with CRISPR-Cas9, DSB can be performed in the mutated gene to correct the nonfunctional gene and potentially re-establish enzyme synthesis, and with an added donor DNA template (with a correct gene sequence), it is also possible to use DSB to insert a functional gene for synthesis of a functional enzyme.
Recently, a new revolutionary CRISPR-Cas9-based editing tool has been developed. This technology is known as Prime editing and can mediate targeted insertions, deletions, all 12 possible base-to-base conversions, and combinations in human cells without requiring DSBs or donor DNA templates.
This tool is composed of a nickase Cas9 fused with a reverse transcriptase (RT) that targets DNA using a guide RNA containing a spacer sequence that hybridizes with the target DNA. However, the sgRNA is engineered to have specificity to the DNA target and the new genetic information that replaces target DNA nucleotides. The DNA is nicked at the target site to expose a 3′-hydroxyl group that primes the reverse transcription of an edit-encoding extension on the engineered guide RNA (prime editing guide RNA—pegRNA) directly into the target site. This creates a heteroduplex with an edited and a nonedited strand. Subsequently, the cell’s natural repair mechanisms kicks in. To increase the chances of the DNA reparation mechanisms using the edited strand as template for reparation, a nick is created in the nonedited DNA. The nick is made with a normal sgRNA and nickase Cas9. This circumvents the heteroduplex issue by preferentially replacing the nonedited strand in the reparation process with a copy of the edited strand.
Prime editors1 (PE) use a RT fused to a RNA-programmable nickase and a pegRNA. PE2 uses an engineered RT to increase editing efficiencies, and PE3 nicks the nonedited strand to induce its replacement and further increase editing efficiency, typically to 20%–50% with 1%–10% indel formation (percentages of human embryonic kidney (HEK) 293T cell testing). PE3b is a PE3 system that additionally uses sgRNAs with spacers that match the edited strand, but not the original allele. This allows for a nick in the nonedited strand only after editing, minimizing concurrent nicks, DSB, and indel formation. Mismatches between the spacer and the unedited allele disfavor sgRNA nicking until editing of the PAM strand is completed. This system results in 13-fold lower average indels (0.74%) compared to PE3, without a decrease in edition efficiency [52].
3. CRISPR-Cas9 Edition for Disease Models and Therapeutic Approaches of Sphingolipidoses
Gene editing with CRISPR-Cas9 has been carried out to generate models of sphingolipidoses, and in a few such diseases, as an attempt at therapy (Table 1). In diseases where a partial reestablishment of function is enough to prevent the deleterious effect (such as many LSDs), CRISPR-Cas9 could be more widely used.
With CRISPR-Cas9, DSB can be performed in the mutated gene to correct the nonfunctional gene and potentially re-establish enzyme synthesis. With an added donor DNA template with a correct gene sequence, it is also possible to use DSB in order to insert a functional gene for synthesis of a functional enzyme. The use of Prime editing is also an option for the correction of specific nucleotide mutations, without the need of a DNA donor template or DSB.
3.1. Gaucher Disease
CRISPR-Cas9 has not been used yet for gene therapy in GD, but models have been generated (Table 1). Specifically, HEK 293T cells and A549 (adenocarcinomic human alveolar basal epithelial) cells have been used for disease modelling by knocking out the GBA1 gene, using CRISPR-Cas9. The study aimed at observing the importance of the GCase enzyme in endocytic trafficking of viruses and exocytic transport. The results suggest diminished infection in both knockout cell lines when infected with influenza virus [53].
In addition to cellular models, in vivo models have been created. Recently, two zebrafish models that exhibit glucocerebrosidase deficiency have been generated using CRISPR-Cas9. Knockout of the genes GBA1 and GBA2 has been carried out in wild-type zebrafish AB/TL strain embryos. Once reaching adulthood, the knockout models were outcrossed to ABTL WT zebrafish. GBA1 zebrafish mutants were heterozygous and GBA2 mutants were homozygous. Double mutant larvae were then generated by crossing both GBA1 and GBA2 mutants. These models were used to study glucosylsphingosine and glucosylated cholesterol level variations in the presence of glucocerebrosidase deficiency [54].
3.2. Fabry Disease
Several researchers have applied CRISPR-Cas9 to FD. In 2016, researchers used CRISPR-Cas9 for gene knockout of GLA (in exon 1) in HEK 293T cells to create in vitro drug screening models for FD. More specifically, they aimed to design a FD model to study enzyme replacement therapy (ERT) with recombinant human α-Gal-A (rhα-GLA) [55].
A similar study also employed CRISPR-Cas9 to create a FD cell model to evaluate the amenability to chaperone therapy. In this case, the target was exon 3 of the GLA gene in HEK 293T cells. Although the target site was specific, single-cell analysis revealed different GLA mutations triggered by CRISPR-Cas9, partially leading to a complete loss of enzymatic function. Therefore, this model was successful in acquiring a disrupted GLA gene, but the method was not very encouraging since multiple mutations were found for a supposedly specific cutting tool [56].
In addition to HEK 293T cells, human embryonic stem cells (hESCs) have also been used to create FD models. A GLA knockout was generated for further differentiation into cardiomyocytes and proteome study. The model was made in order to study autophagic dysfunction and exosome secretion in FD-associated hypertrophic cardiomyopathy [57].
Other cases of FD models have been generated with other cell lines, namely human immortalized podocyte model of FD [58] and a human endothelial cell line, both with GLA disruption using CRISPR-Cas9 tools [59].
CRISPR-Cas9 technology has also been used to generate in vivo models including the GLA-knockout rat. The rat model is completely deficient in α-Gal-A activity; accumulates the established GSL biomarkers, Gb3 and lyso-Gb3; and develops mechanical pain behavior. This model was used to study the cation channel transient receptor potential ankyrin 1 (TRPA1) as a potential target to treat the pain of FD patients. Using the TRPA1 antagonist HC-030031, the mechanical pain behavior in Fabry rats was reversed with success [60].
Although CRISPR-Cas9 was, in these cases, used for disease model generation and not for gene/cell therapy, this work in models provides important insight for precise gene targeting of GLA. This background information can be used in future gene therapy and integration of a nonmutated GLA gene. Additionally, a good example of off-targeting occurrence, even in the presence of a highly promising off-targeting score, has been reported. Off-targeting is more than a prevision, it is always a real possibility.
In addition to disease modelling, gene editing for restoration of enzyme activity has also been done using CRISPR-Cas9. In 2017, Sheng-Kai Chang and collaborators restored GLA enzyme activity in FD patients’ fibroblasts after deleting the GLA IVS4 + 919 G > A mutation using CRISPR-Cas9. This specific mutation seems to be related to the presentation of cardiac FD issues, with greater rate of incidence in the Taiwanese population. The mutation interferes with the normal RNA splicing, producing a truncated GLA protein with no enzyme activity. The therapy was performed with CRISPR-Cas9, using two sgRNAs for the mutation deletion in FD patient fibroblasts, and demonstrated the proof of principle as an increase in GLA activity and clearance of intracellular Gb3 was observed. This work showed clear evidence that CRISPR-Cas9 has the potential for gene therapy directed to genetic diseases [61].
3.3. Tay–Sachs and Sandhoff Diseases
Both diseases are caused by mutations of HEXA or/and HEXB genes. Michael Tropak et al. achieved the disruption of HEXA and HEXB genes with CRISPR-Cas9 in HEK 293 cell line. Ultimately, they aimed to modulate the disease with the intent of testing a lab-made chimeric enzyme with a single hybrid µ-subunit. This subunit had a α-subunit active site, a stable β-subunit interface, and unique areas in each subunit for GM2AP (GM2 activator protein) interaction [62].
Allende and collaborators successfully performed gene correction using CRISPR-Cas9 to treat SD in patients’ cells. Fibroblasts of infantile SD patients were reprogramed to generate iPS cells that would be used for modelling the disease. The reprograming of diseased fibroblasts was first carried out via transfection of episomal vectors encoding the four reprogramming factors, OCT-3/4, SOX2, KLF4, and L-MYC. The resulting iPSCs had normal karyotype, pluripotent markers, and were able to form embryoid bodies that differentiated into the three embryonic germ layers. SD iPSCs showed reduced β-hexosaminidase activity compared with control iPSCs, which were purchased. Once the SD iPSCs model was prepared and tested, gene correction was performed. In order to correct the intron 10 acceptor splice-site mutation of HEXA gene, a plasmid was designed to express the sgRNA and the Cas9 nuclease for DSB catalysis and selection with puromycin. For correction, a single-stranded oligodeoxynucleotide of 181 bp was used as donor template. The template had a G to A correction, a silent point mutation to disrupt the PAM sequence for no Cas9 recutting, and two silent point mutations to create a KpnI restriction enzyme site for screening. Edited and not edited SD IPSCs were used for cerebral organoid formation that would mimic the first trimester of neurodevelopment. The result was accumulation of GM2 ganglioside, higher cellular size, and proliferation only in the unedited iPSCs [63].
Recent work using Prime editing, a new technique based on CRISPR-Cas9, has allowed for the correction of the most common frequent causal mutation in the Ashkenazi Jewish population. The mutation is a 4 bp insertion in HEXA (HEXA 1278 + TATC). PE3 prime editing was used to recreate the mutation by installation of the 4 bp insertion into HEXA gene of lipoinfected HEK 293T cell lines, resulting in 31% efficiency and 0.8% indels. Two isolated edited cells homozygous for HEXA 1278 + TATC mutation were then used to test 43 pegRNAs and three nicking sgRNAs with PE3 or PE3b systems for correction of the pathogenic insertion. Nineteen of the pegRNAs resulted in ≥20% successful editing reaching the best pegRNA, 33% efficiency, and 0.32% indels using PE3b [52].
3.4. Niemann–Pick Disease
In any disease, a cellular model is necessary to carry out gene and drug therapy tests. Although NPD type C1 is not exactly a sphingolipidosis, but rather a lipid transport disease, CRISPR-Cas9 applications in this pathology are worth mentioning. Many developments have been made in disease modelling. Descriptions of these developments are present in multiple types of sources, including articles and protocol books. One example is “Cholesterol Homeostasis: Methods and Protocols, Methods in Molecular Biology”, which describes with precision the generation of cellular NPC cholesterol storage phenotypes in HeLa cells using CRISPR-Cas9 to disrupt the NPC1 gene [64].
Additionally, in vivo models, such as zebrafish NPC1-null mutants, have been generated using CRISPR-Cas9 gene targeting. Two models were made, one for early liver NPC1 disease and one for later neurological phenotype. sgRNAs, targeting exon 2 and 7, respectively, were injected with Cas9 mRNA into wild-type zebrafish embryos at the first cell stage. For mutation screening, the fish were raised to adulthood and outcrossed with wild-type adults for later PCR and fragment analysis [65]. A similar work has been also performed by Yusheng Lin et al. [66].
3.5. Krabbe Disease
In 2019, a work was published showing that GALC-expressing human neural stem cells (hNSCs) can be engineered to secrete lysosomal enzymes that are able to cross-correct Krabbe fibroblasts in vitro. To increase precision, multiple loci were targeted using Cas9 mRNA with modified synthetic gRNAs, along with DNA donor templates. The transplantation of the altered hNSCs into oligodendrocyte mutant shiverer-immunodeficient mice demonstrated migration and differentiation into astrocytes, neurons, and myelin-producing oligodendrocytes. They also generated altered hNSCs with GALC overexpression, capable of cross-correcting Galc enzyme activity via the mannose-6-phosphate receptor pathway. Since the GALC gene is overexpressed, it would be of great interest to use such cells in cell therapy for KD [67].
KD animal models have also been generated. For example, a gene knockdown of galactocerebrosidase in zebrafish was obtained in two GALC co-orthologs (Galca and Galcb) [68].
3.6. GM1-Gangliosidosis and Morquio Syndrome
Yvonne Latour et al. generated in 2019, a GM1-gangliosidosis model with purchased iPSCs, and consequently tested AAV9 gene therapy. Using CRISPR-Cas9, the researchers targeted exons 2 and 6 with different sgRNAs. The all-in-one plasmid method was used for expression of the complex in the cells after electroporation. The GLB1 knockout was confirmed by β-gal enzyme activity assays and DNA sequencing. The knockout iPSCs were then used to generate cerebral organoids that revealed progressive accumulation of GM1 ganglioside. Finally, AAVrh9 (AAV9) vectors carrying the human β-gal gene (AAV9-GLB1) or GFP (AAV9-GFP) were injected in GLB1 knockout organoids. The result was an increase in β-gal activity and concomitant reduction in GM1 ganglioside content in the AAV9-GLB1-injected organoids when compared with AAV9-GFP-injected organoids. This work contributed to the completion of the preclinical studies and progression to clinical trials using the AAV9-GLB1 vector as capable of reversing the phenotype [69].
Another recent work was able to generate GM1-gangliosidose and Morquio syndrome type B in mice models. Using CRISPR-Cas9, mutation W273L (position 274 in mice) was introduced into the GLB1 gene to generate a Morquio B model, while for GM1-gangliosidosis, a 20bp mutation was generated to remove the catalytic nucleophile of β-gal (β-gal-/-). The mice were generated by embryo microinjection of Cas9 protein, sgRNA targeting exon 8, and in the case of Morquio syndrome type B mice, a donor oligonucleotide to introduce a 2 bp mutation.
The generated Morquio syndrome type B mice showed reduction in β-gal enzyme activity, but no marked phenotype after one year. β-gal-/- mice lost β-gal enzyme activity, resulting in ganglioside accumulation and severe cellular vacuolation in the central nervous system (CNS), leading to neuromotor and neurocognitive dysfunction with disease progression. This was the first model of β-galactosidase deficiency with residual enzyme activity [70].
4. Conclusions
Since CRISPR-Cas9 was discovered, many advances have been made in gene editing for disease treatment that were previously impossible. The main targets of research are the most common and severe pathologies, such as cancer [71], which partly explains why there are still few gene therapies based in CRISPR-Cas9 for the rare sphingolipidoses [72,73].
CRISPR-Cas9 has been widely applied to generate knockouts for the study of the mechanisms underlying several diseases. The models created using CRISPR-Cas9 have already provided important insights. Additionally, this tool is being used to test the involvement of infection agents in the pathophysiology of diseases and the causality of novel genetic variants discovered through next-generation sequencing (Table 1 summarizes several applications of CRISPR-Cas9).
Commonly used therapies in sphingolipidoses have limitations, such as not being able to cross the blood–brain barrier or missing the target molecules. Some of these limitations can be circumvented by CRISPR-Cas9 genome editing. This tool has the potential to alter, with a high degree of precision, very specific sites of the genome. However, the off-target effects that arise from CRISPR-Cas9 editing cannot be disregarded as they can have deleterious effects. As with other techniques, multiple checkpoints are required at the gene and protein levels for the careful assessment of the results.
Undoubtedly, progress in the field of gene editing is rapid and further studies, not subjects of the present work, demonstrated that other approaches to gene therapy, such as RNA [74] or viral-based therapeutic approaches [75], provide useful insights for future applications. Prime editing already provides an improved tool for genome editing, based on Cas9, with the advantage of being less prone to off-targeting [52].
CRISPR-Cas9 has great potential for successful disease modelling, as well as for therapeutic approaches, avoiding problems inherent to other treatments. In light of the versatility of this technique, it is possible to envisage the development of other applications that have yet to be explored.
Funding
Fundação para a Ciência e a Tecnologia (Portugal, FCT) project PTDC/BIM-MEC/4762/2014.
Acknowledgments
We acknowledge the collaboration of Meg Quint in the proofreading of the manuscript and the team at the Unit of R&D of DGH at INSA-Porto for the agreeable environment provided.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cox, T.M.; Cachon-Gonzalez, M.B. The cellular pathology of lysosomal diseases. J. Pathol. 2012, 226, 241–254. [Google Scholar] [CrossRef]
- Platt, F.M.; D’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Sandhoff, K. Sphingolipidoses. J. Clin. Pathol. Suppl. 1974, 8, 94–105. [Google Scholar] [CrossRef]
- Platt, F.M. Sphingolipid lysosomal storage disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Arenz, C. Recent advances and novel treatments for sphingolipidoses. Future Med. Chem. 2017, 9, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Ysselstein, D.; Shulman, J.M.; Krainc, D. Emerging links between pediatric lysosomal storage diseases and adult parkinsonism. Mov Disord. 2019, 34, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Melum, E.; Jiang, X.; Baker, K.D.; Macedo, M.F.; Fritsch, J.; Dowds, C.M.; Wang, J.; Pharo, A.; Kaser, A.; Tan, C.; et al. Control of CD1d-restricted antigen presentation and inflammation by sphingomyelin. Nat. Immunol. 2019, 20, 1644–1655. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Palmieri Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Do, J.; Tayebi, N.; Sidransky, E. Can GBA1-Associated Parkinson Disease Be Modeled in the Mouse? Trends Neurosci. 2019, 42, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Elstein, D.; Zimran, A.; Goker-Alpan, O. New Directions in Gaucher Disease. Hum. Mutat. 2016, 37, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.L.; Mohan, D. Gaucher Disease and Its Treatment Options. Ann. Pharmacother. 2013, 47, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Belmatoug, N.; Bembi, B.; Deegan, P.; Elstein, D.; Fernandez-Sasso, D.; Giraldo, P.; Goker-Alpan, O.; Lau, H.; Lukina, E.; et al. GOS Study group.Demographics and patient characteristics of 1209 patients with Gaucher disease: Descriptive analysis from the Gaucher Outcome Survey (GOS). Am. J. Hematol. 2018, 93, 205–212. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Horowitz, M.; Bendikov-Bar, I. Gaucher disease paradigm: From ERAD to comorbidity. Hum. Mutat. 2012, 33, 1398–1407. [Google Scholar] [CrossRef]
- Maor, G.; Rapaport, D.; Horowitz, M. The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet. 2019, 28, 1768–1781. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Wegrzynowicz, M.; Rusconi, R.; Deangeli, G.; Di Monte, D.A.; Spillantini, M.G.; Schapira, A.H.V. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain 2017, 140, 2706–2721. [Google Scholar] [CrossRef]
- Duro, G.; Zizzo, C.; Cammarata, G.; Burlina, A.; Burlina, A.; Polo, G.; Scalia, S.; Oliveri, R.; Sciarrino, S.; Francofonte, D.; et al. Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease? Int. J. Mol. Sci. 2018, 19, 3726. [Google Scholar] [CrossRef]
- Germain, D.P.; Elliott, P.M.; Falissard, B.; Fomin, V.V.; Hilz, M.J.; Jovanovic, A.; Kantola, I.; Linhart, A.; Mignani, R.; Namdar, M.; et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol. Genet. Metab. Rep. 2019, 19, 100454. [Google Scholar] [CrossRef]
- Mehta, A.; Widmer, U. Chapter 19 Natural history of Fabry disease. In Fabry Dis Ease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Waldek, S.; Feriozzi, S. Fabry nephropathy: A review—How can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014, 15, 72. [Google Scholar] [CrossRef]
- Vu, M.; Li, R.; Baskfield, A.; Lu, B.; Farkhondeh, A.; Gorshkov, K.; Motabar, O.; Beers, J.; Chen, G.; Zou, J.; et al. Neural stem cells for disease modeling and evaluation of therapeutics for Tay-Sachs disease. Orphanet J. Rare Dis. 2018, 13, 152. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.; Minnich, S.; Teigen, C.; Raymond, K. Diagnosing Lysosomal Storage Disorders: The GM2 Gangliosidoses. Curr. Protoc. Hum. Genet. 2014, 83, 16.1–16.8. [Google Scholar] [PubMed]
- Karimzadeh, P.; Jafari, N.; Biglari, H.N.; Dari, S.J.; Abadi, F.A.; Alaee, M.-R.; Nemati, H.; Saket, S.; Tonekaboni, S.H.; Taghdiri, M.-M.; et al. GM2-Gangliosidosis (Sandhoff and Tay Sachs disease): Diagnosis and Neuroimaging Findings (An Iranian Pediatric Case Series). Iran. J. Child Neurol. 2014, 8, 55–60. [Google Scholar] [PubMed]
- Solovyeva, V.V.; Shaimardanova, A.A.; Chulpanova, D.S.; Kitaeva, K.V.; Chakrabarti, L.; Rizvanov, A.A. New Approaches to Tay-Sachs Disease Therapy. Front. Physiol. 2018, 9, 1663. [Google Scholar] [CrossRef]
- Duarte, A.J.; Ribeiro, D.; Oliveira, P.; Amaral, O. Mutation Frequency of Three Neurodegenerative Lysosomal Storage Diseases: From Screening to Treatment? Arch. Med. Res. 2017, 48, 263–269. [Google Scholar] [CrossRef]
- Kaback, M.M.; Desnick, R.J. Hexosaminidase A Deficiency. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Vanier, M.T. Niemann-Pick diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar]
- Pineda, M.; Juríčková, K.; Karimzadeh, P.; Kolnikova, M.; Malinova, V.; Insua, J.L.; Velten, C.; Kolb, S.A. Disease characteristics, prognosis and miglustat treatment effects on disease progression in patients with Niemann-Pick disease Type C: An international, multicenter, retrospective chart review. Orphanet J. Rare Dis. 2019, 14, 32. [Google Scholar] [CrossRef]
- Simonaro, C.M.; Park, J.-H.; Eliyahu, E.; Shtraizent, N.; McGovern, M.M.; Schuchman, E.H. Imprinting at the SMPD1 Locus: Implications for Acid Sphingomyelinase—Deficient Niemann-Pick Disease. Am. J. Hum. Genet. 2006, 78, 865–870. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef]
- Graziano, A.C.E.; Cardile, V. History, genetic, and recent advances on Krabbe disease. Gene 2015, 555, 2–13. [Google Scholar] [CrossRef]
- Spratley, S.J.; Hill, C.H.; Viuff, A.H.; Edgar, J.R.; Skjødt, K.; Deane, J.E. Molecular Mechanisms of Disease Pathogenesis Differ in Krabbe Disease Variants. Traffic 2016, 17, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Bascou, N.; Derenzo, A.; Poe, M.D.; Escolar, M.L. A prospective natural history study of Krabbe disease in a patient cohort with onset between 6 months and 3 years of life. Orphanet J. Rare Dis. 2018, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Oshima, A.; Namba, E. β-Galactosidase deficiency (β-galactosidosis) GM1 gangliosidosis and Morquio B disease. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3775–3809. [Google Scholar]
- Morita, M.; Saito, S.; Ikeda, K.; Ohno, K.; Sugawara, K.; Suzuki, T.; Togawa, T.; Sakuraba, H. Structural bases of GM1 gangliosidosis and Morquio B disease. J. Hum. Genet. 2009, 54, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caciotti, A.; Garman, S.C.; Rivera-Colón, Y.; Procopio, E.; Catarzi, S.; Ferri, L.; Guido, C.; Martelli, P.; Parini, R.; Antuzzi, D.; et al. GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim. Biophys. Acta 2011, 1812, 782–790. [Google Scholar] [CrossRef]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement therapy for inherited enzyme deficiency—Macrophage-targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR—Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Trevino, A.E.; Zhang, F. Genome editing using Cas9 nickases. Methods Enzymol. 2014, 546, 161–174. [Google Scholar]
- Jensen, M.K. Design principles for nuclease-deficient CRISPR-based transcriptional regulators. FEMS Yeast Res. 2018, 18, foy039. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Oberdoerffer, P. Chromatin dynamics in DNA double-strand break repair. Biochim. Biophys. Acta 2012, 1819, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2019, 9, 691. [Google Scholar] [CrossRef]
- Han, X.; Liu, Z.; Jo, M.C.; Zhang, K.; Li, Y.; Zeng, Z.; Li, N.; Zu, Y.; Qin, L. CRISPR-Cas9 delivery to hard-to-transfect cells via membrane deformation. Sci. Adv. 2015, 1, e1500454. [Google Scholar] [CrossRef] [Green Version]
- Kouranova, E.; Forbes, K.; Zhao, G.; Warren, J.; Bartels, A.; Wu, Y.; Cui, X. CRISPRs for Optimal Targeting: Delivery of CRISPR Components as DNA, RNA, and Protein into Cultured Cells and Single-Cell Embryos. Hum. Gene Ther. 2016, 27, 464–475. [Google Scholar] [CrossRef]
- Zhang, Y.; Long, C.; Li, H.; McAnally, J.R.; Baskin, K.K.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3, e1602814. [Google Scholar] [CrossRef] [Green Version]
- Sanz, D.J.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS ONE 2017, 12, e0184009. [Google Scholar] [CrossRef] [Green Version]
- García-Tuñón, I.; Hernández-Sánchez, M.; Ordoñez, J.L.; Alonso-Pérez, V.; Álamo-Quijada, M.; Benito, R.; Guerrero, C.; Hernández-Rivas, J.M.; Sánchez-Martín, M. The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitro and in a xenograft model of chronic myeloid leukemia. Oncotarget 2017, 8, 26027–26040. [Google Scholar] [CrossRef] [Green Version]
- György, B.; Nist-Lund, C.; Pan, B.; Asai, Y.; Karavitaki, K.D.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Solanes, P.; Spataro, S.; et al. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat. Med. 2019, 25, 1123–1130. [Google Scholar] [CrossRef]
- Diakatou, M.; Manes, G.; Bocquet, B.; Meunier, I.; Kalatzis, V. Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 20, 2542. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019. [Google Scholar] [CrossRef]
- Drews, K.; Calgi, M.P.; Harrison, W.C.; Drews, C.M.; Costa-Pinheiro, P.; Shaw, J.J.P.; Jobe, K.A.; Nelson, E.A.; Han, J.D.; Fox, T.; et al. Glucosylceramidase Maintains Influenza Virus Infection by Regulating Endocytosis. J. Virol. 2019, 93, e00017–e00019. [Google Scholar] [CrossRef] [Green Version]
- Lelieveld, L.T.; Mirzaian, M.; Kuo, C.-L.; Artola, M.; Ferraz, M.J.; Peter, R.E.A.; Akiyama, H.; Greimel, P.; Berg, R.J.B.H.N.V.D.; Overkleeft, H.S.; et al. Role of β-glucosidase 2 in aberrant glycosphingolipid metabolism: Model of glucocerebrosidase deficiency in zebrafish. J. Lipid Res. 2019, 60, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Song, H.-Y.; Chiang, H.-C.; Tseng, W.-L.; Wu, P.; Chien, C.-S.; Leu, H.-B.; Yang, Y.-P.; Wang, M.-L.; Jong, Y.-J.; Chen, C.-H.; et al. Using CRISPR/Cas9-Mediated GLA Gene Knockout as an In Vitro Drug Screening Model for Fabry Disease. Int. J. Mol. Sci. 2016, 17, 2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenders, M.; Stappers, F.; Niemietz, C.; Schmitz, B.; Boutin, M.; Ballmaier, P.J.; Zibert, A.; Schmidt, H.; Brand, S.-M.; Auray-Blais, C.; et al. Mutation-specific Fabry disease patient-derived cell model to evaluate the amenability to chaperone therapy. J. Med. Genet. 2019, 56, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Song, H.Y.; Chien, C.S.; Yarmishyn, AA.; Chou, S.J.; Yang, Y.P.; Wang, M.L.; Wang, C.Y.; Leu, H.B.; Yu, W.C.; Chang, Y.L.; et al. Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy. Cells 2019, 8, 327. [Google Scholar] [CrossRef] [Green Version]
- Pereira, E.M.; Labilloy, A.; Eshbach, M.L.; Roy, A.; Subramanya, A.R.; Monte, S.; Labilloy, G.; Weisz, O.A. Characterization and phosphoproteomic analysis of a human immortalized podocyte model of Fabry disease generated using CRISPR/Cas9 technology. Am. J. Physiol. Physiol. 2016, 311, F1015–F1024. [Google Scholar] [CrossRef]
- Kang, J.J.; Kaissarian, N.M.; Desch, K.C.; Kelly, R.J.; Shu, L.; Bodary, P.F.; Shayman, J.A. α-galactosidase A deficiency promotes von Willebrand factor secretion in models of Fabry disease. Kidney Int. 2019, 95, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.J.; Aoki, K.; Moehring, F.; Murphy, C.A.; O’Hara, C.L.; Tiemeyer, M.; Stucky, C.L.; Dahms, N.M. Neuropathic pain in a Fabry disease rat model. JCI Insight 2018, 3, e99171. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.K.; Lu, Y.H.; Chen, Y.R.; Hsieh, Y.P.; Lin, W.J.; Hsu, T.; Niu, D.M. AB043. Correction of the GLA IVS4 + 919 G > A mutation with CRISPR/Cas9 deletion strategy in fibroblasts of Fabry disease. Ann. Transl. Med 2017, 5 (Suppl. 2), AB043. [Google Scholar] [CrossRef] [Green Version]
- Tropak, M.B.; Yonekawa, S.; Karumuthil-Melethil, S.; Thompson, P.; Wakarchuk, W.; Gray, S.J.; Walia, J.S.; Mark, B.L.; Mahuran, D. Construction of a hybrid β-hexosaminidase subunit capable of forming stable homodimers that hydrolyze GM2 ganglioside in vivo. Mol. Ther. Methods Clin. Dev. 2016, 3, 15057. [Google Scholar] [CrossRef]
- Allende, M.L.; Cook, E.K.; Larman, B.C.; Nugent, A.; Brady, J.M.; Golebiowski, D.; Sena-Esteves, M.; Tifft, C.J.; Proia, R.L. Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J. Lipid Res. 2018, 59, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Lukmantara, I.; Yang, H. CRISPR/Cas9-Mediated Generation of Niemann–Pick C1 Knockout Cell Line. Methods Mol. Biol. 2017, 1583, 73–83. [Google Scholar] [PubMed]
- Tseng, W.-C.; Loeb, H.E.; Pei, W.; Tsai-Morris, C.-H.; Xu, L.; Cluzeau, C.V.; Wassif, C.A.; Feldman, B.; Burgess, S.M.; Pavan, W.J.; et al. Modeling Niemann-Pick disease type C1 in zebrafish: A robust platform for in vivo screening of candidate therapeutic compounds. Dis. Model. Mech. 2018, 11, dmm034165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Cai, X.; Wang, G.; Ouyang, G.; Cao, H.; Caia, X. Model construction of Niemann-Pick type C disease in zebrafish. Biol. Chem. 2018, 399, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Dever, D.P.; Scharenberg, S.G.; Camarena, J.; Kildebeck, E.J.; Clark, J.T.; Martin, R.M.; Bak, R.O.; Tang, Y.; Dohse, M.; Birgmeier, J.A.; et al. CRISPR/Cas9 Genome Engineering in Engraftable Human Brain-Derived Neural Stem Cells. iScience 2019, 15, 524–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zizioli, D.; Guarienti, M.; Tobia, C.; Gariano, G.; Borsani, G.; Bresciani, R.; Ronca, R.; Giacopuzzi, E.; Preti, A.; Gaudenzi, G.; et al. Molecular cloning and knockdown of galactocerebrosidase in zebrafish: New insights into the pathogenesis of Krabbe’s disease. Biochim. Biophys. Acta 2014, 1842, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latour, Y.L.; Yoon, R.; Thomas, S.E.; Grant, C.; Li, C.; Sena-Esteves, M.; Allende, M.L.; Proia, R.L.; Tifft, C.J.; et al. Human GLB1 knockout cerebral organoids: A model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis. Mol. Genet. Metab. Rep. 2019, 21, 100513. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, M.J.; Ou, L.; Tăbăran, A.-F.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; O’Sullivan, M.G.; Whitley, C.B. Comprehensive behavioral and biochemical outcomes of novel murine models of GM1-gangliosidosis and Morquio syndrome type B. Mol. Genet. Metab. 2019, 126, 139–150. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.-J.; Zhu, L.-Y.; Yan, Z.-Y.; Xu, Y.; Wang, Q.-L.; Lu, X.-J. CRISPR-Cas9 for in vivo Gene Therapy: Promise and Hurdles. Mol. Ther. Nucleic Acids 2016, 5, e349. [Google Scholar] [CrossRef] [Green Version]
- Mollanoori, H.; Teimourian, S. Therapeutic applications of CRISPR/Cas9 system in gene therapy. Biotechnol. Lett. 2018, 40, 907–914. [Google Scholar] [CrossRef]
- Lundstrom, K. Special Issue: Gene Therapy with Emphasis on RNA Interference. Viruses 2015, 7, 4482–4487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.S.; Searle, P.F.; Onion, D.; Mautner, V. Viral gene therapy strategies: From basic science to clinical application. J. Pathol. 2006, 208, 299–318. [Google Scholar] [CrossRef] [PubMed]
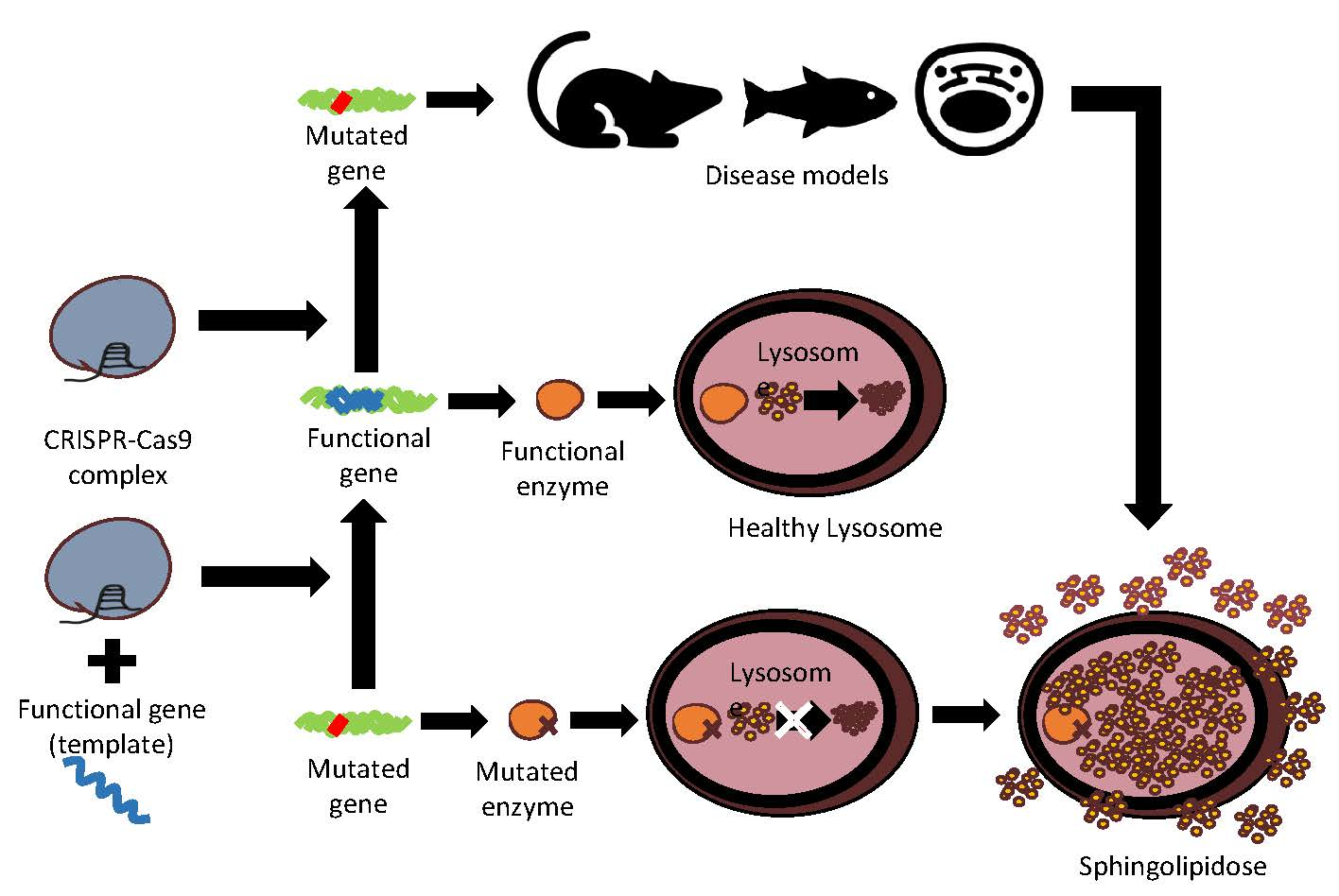
Figure 1.
GM1-gangliosidosis degradation pathway, sphingolipid synthesis pathway, and enzyme activity recovery with CRISPR-Cas9 and DNA template. The red lines describe the interrupted pathway of degradation due to a specific dysfunctional enzyme. In bold are the names of the sphingolipidoses caused by the mutated enzymes and the consequent interrupted pathways. Scheme drawn on the basis of the bibliography used in this review.
Figure 1.
GM1-gangliosidosis degradation pathway, sphingolipid synthesis pathway, and enzyme activity recovery with CRISPR-Cas9 and DNA template. The red lines describe the interrupted pathway of degradation due to a specific dysfunctional enzyme. In bold are the names of the sphingolipidoses caused by the mutated enzymes and the consequent interrupted pathways. Scheme drawn on the basis of the bibliography used in this review.

Figure 2.
CRISPR-Cas9 structure and DNA binding for target sequence cleavage. “n” and “N” represent any nucleotide. The red lines indicate the cleavage locations made by Cas9 nuclease after CRISPR-Cas9 complex that binds to the target.
Figure 2.
CRISPR-Cas9 structure and DNA binding for target sequence cleavage. “n” and “N” represent any nucleotide. The red lines indicate the cleavage locations made by Cas9 nuclease after CRISPR-Cas9 complex that binds to the target.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Sphingolipidoses and CRISPR-Cas9 use in disease models and gene/cell therapy.
| Genes | Enzymes | Substrates | Disease | CRISPR-Cas9 for Disease Models: | CRISPR-Cas9 Gene/Cell Therapy | References | |
|---|---|---|---|---|---|---|---|
| Cell Models | In vivo Models | ||||||
| GBA1 | Glucocerebrosidase | Glucosylceramide | Gaucher disease | - HEK cells - A549 cells | - Zebrafish | X | [53,54] |
| GLA | α-galactosidase A | Globotriaosylceramide | Fabry disease | - HEK cells - hESCs - Podocytes - Human endothelialcells | - Rat - Mouse | - Restoration of GLA enzyme activity in FD patient′s Fibroblasts with mutation knockout | [55,56,57,58,59,60,61] |
| HEXA | α-hexosaminidase s-hexosaminidase | GM2-ganglioside | Tay-Sachs disease | - HEK cells | X | - Gene correction of HEXA 1278+TATC mutation in HEK293T cell using Prime editing | [62] |
| HEXB | α-hexosaminidase β-hexosaminidase | GM2-ganglioside | Sandhoff disease | - iPSCs | X | - No accumulation of GM2 ganglioside in iPSCs generated from fibroblasts of infantile SD patients due to template knockin | [52,63] |
| SMPD | Acid sphingomyelinase | Sphingomyelin | Niemann-pick diseases | - HeLa cells | - Zebrafish | X | [64,65,66] |
| GALC | β-galactocerebrosidase | Galactolipids | Krabbe disease | X | - Zebrafish | - Cell therapy with CRISPR-Cas9 edited hNSCs with correct enzyme activity | [67,68] |
| GLB1 | β-galactosidase | GM1-ganglioside | GM1-gangliosidosis | - iPSCs | - Mouse | X | [69,70] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Santos, R.; Amaral, O. Advances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy. Int. J. Mol. Sci. 2019, 20, 5897. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235897
AMA Style
Santos R, Amaral O. Advances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy. International Journal of Molecular Sciences. 2019; 20(23):5897. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235897
Chicago/Turabian StyleSantos, Renato, and Olga Amaral. 2019. "Advances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy" International Journal of Molecular Sciences 20, no. 23: 5897. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235897
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.