Optimization of Data-Independent Acquisition Mass Spectrometry for Deep and Highly Sensitive Proteomic Analysis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
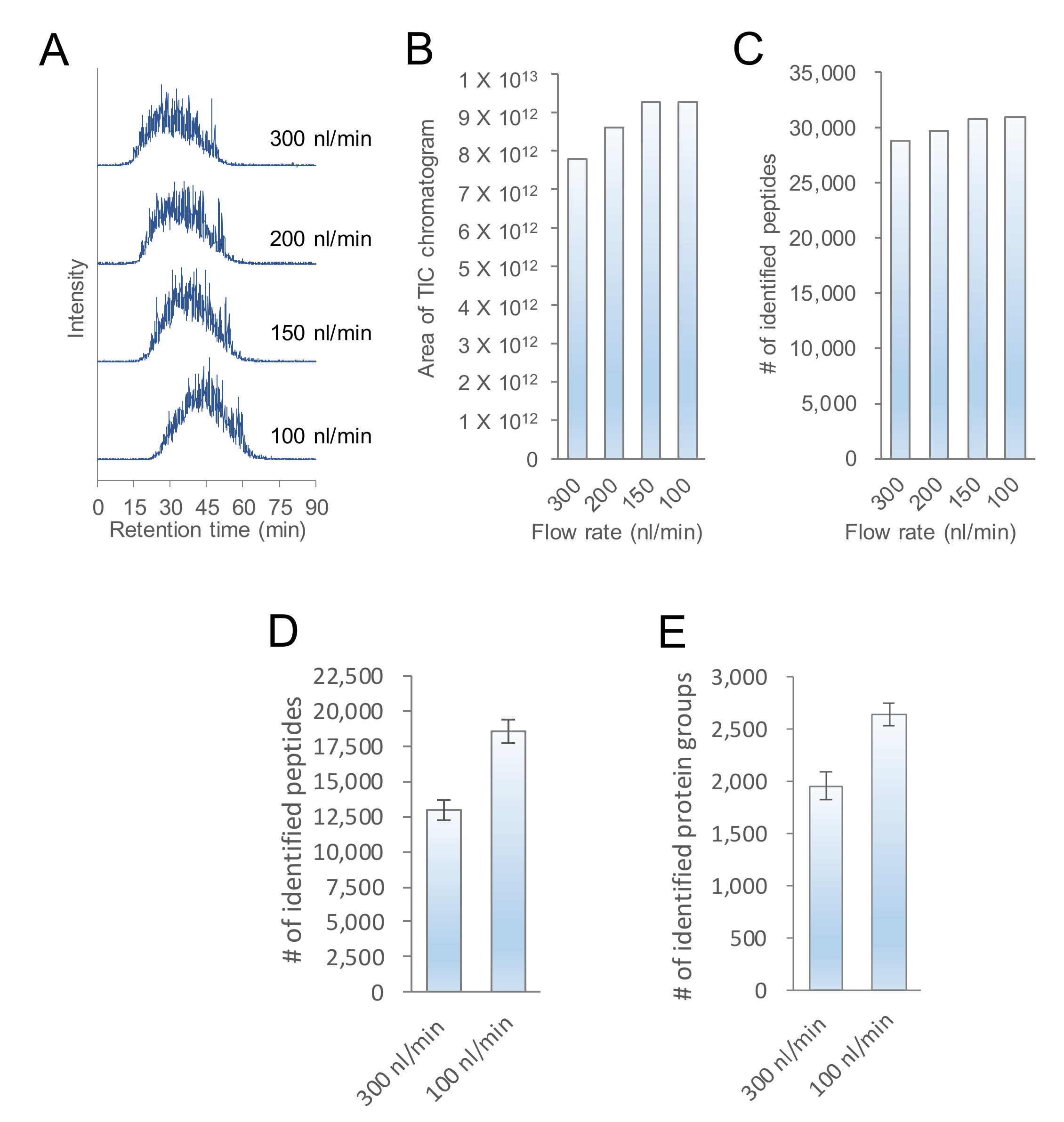
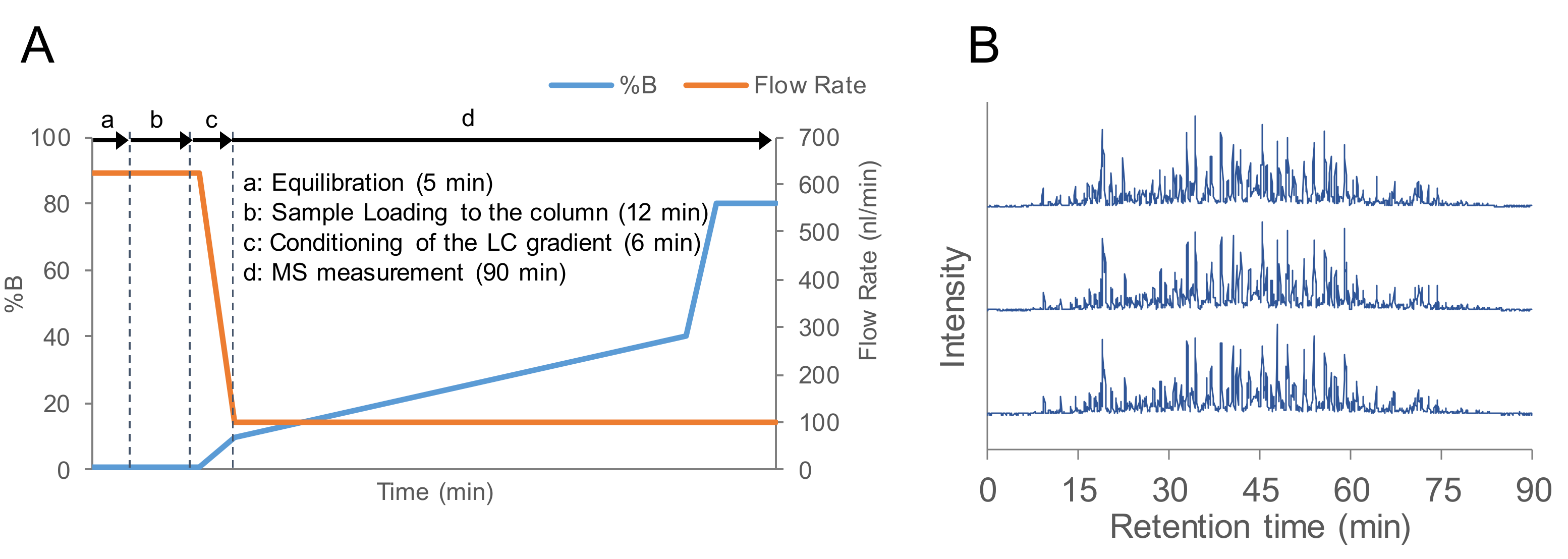
2.1. Evaluation of the Flow Rate of nanoLC-MS/MS
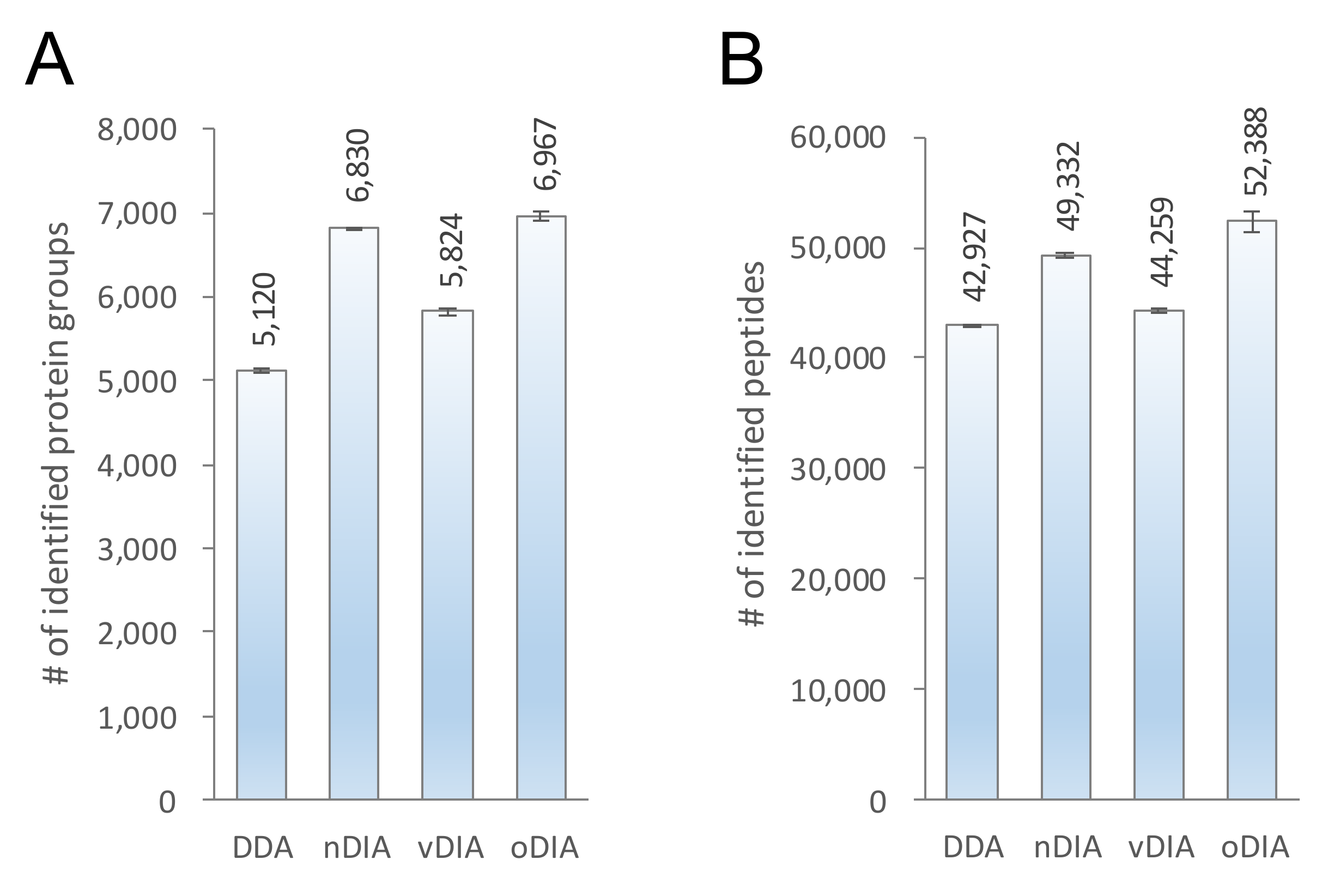
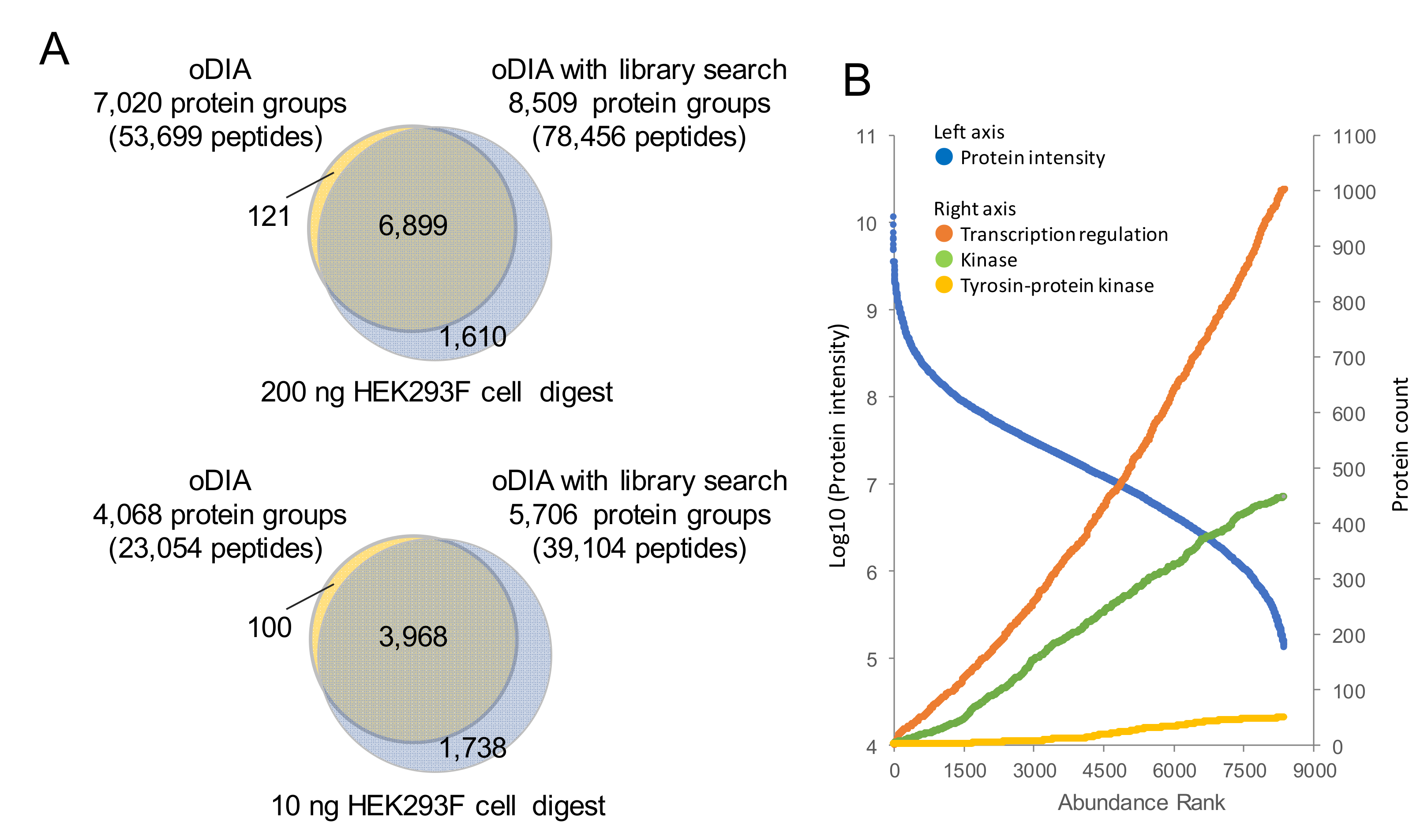
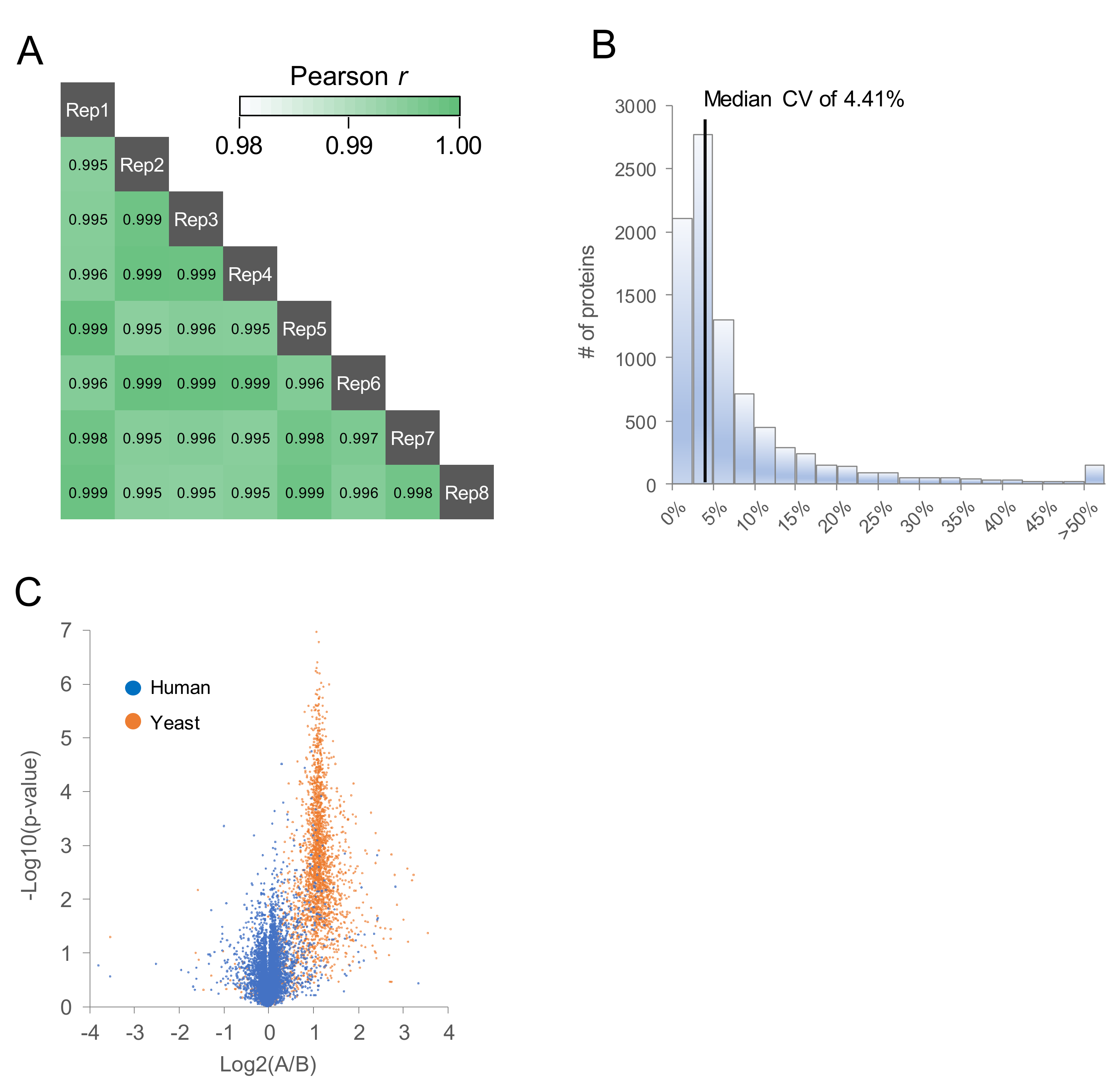
2.2. Comparison of MS/MS Acquisition Methods by Single-Shot Proteomics
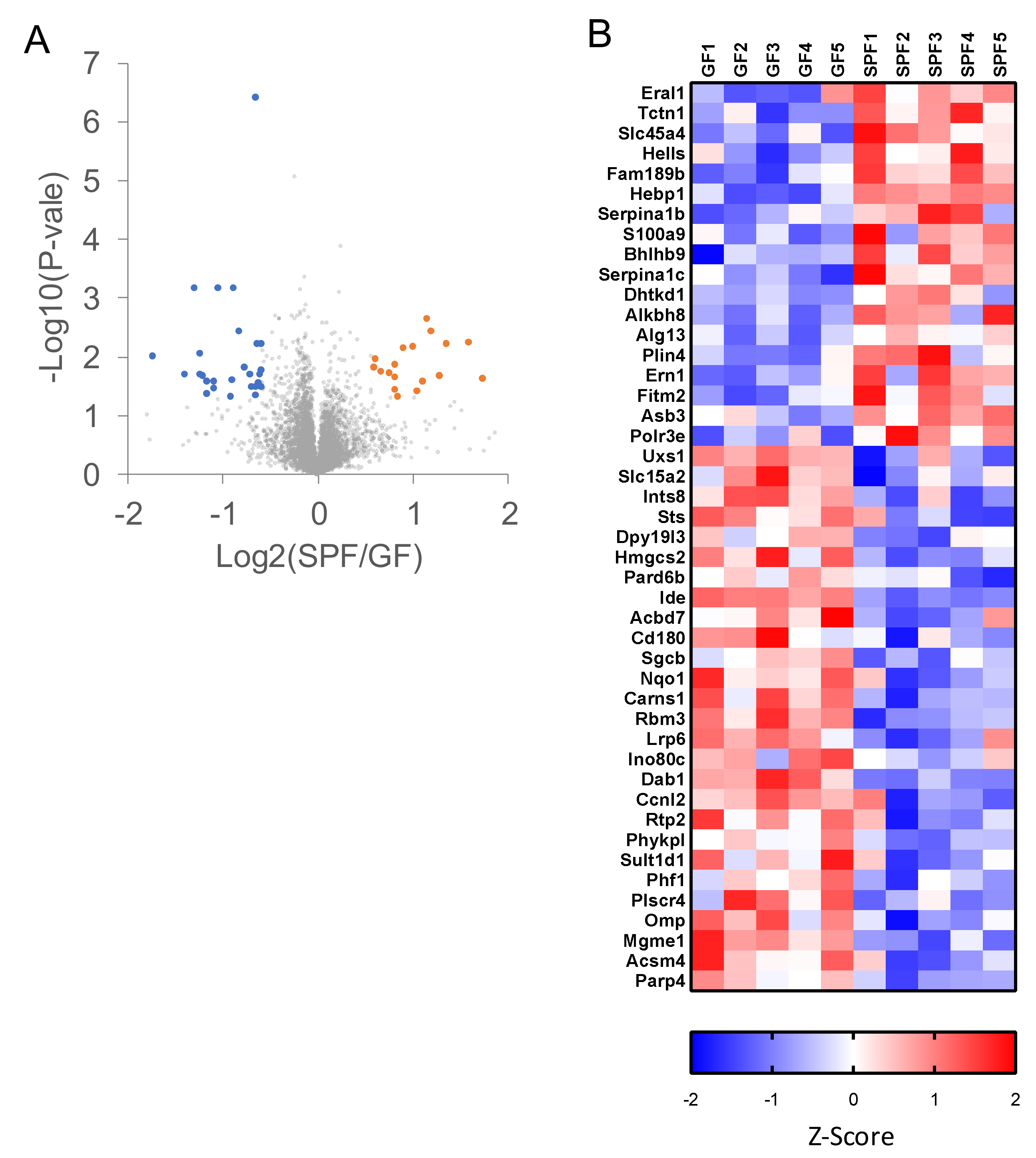
2.3. Proteomic Analyses of GF and SPF Mouse Cerebrums
3. Materials and Methods
3.1. Cell Culture
3.2. Animal Study
3.3. Sample Preparation for Proteomic Analysis
3.4. LC-MS/MS
3.5. Protein Identification by Searching a Protein Sequence Database
3.6. Protein Identification by Searching a Chromatogram Library
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MS | Mass spectrometry |
| DIA | Liquid chromatography |
| HEK293F | Floating human embryonic kidney cells 293 |
| DIA | Data-independent acquisition |
| DDA | Data-dependent acquisition |
| CV | Coefficients of variation |
| MudPIT | Multi-dimensional protein identification technology |
| TIC | Total ion current |
| nDIA | Normal-window data-independent acquisition |
| vDIA | Variable-window data-independent acquisition |
| oDIA | Overlapping-window data-independent acquisition |
| GF | Germ-free |
| SPF | Specific-pathogen-free |
References
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Paabo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [Google Scholar] [CrossRef]
- Adachi, J.; Hashiguchi, K.; Nagano, M.; Sato, M.; Sato, A.; Fukamizu, K.; Ishihama, Y.; Tomonaga, T. Improved Proteome and Phosphoproteome Analysis on a Cation Exchanger by a Combined Acid and Salt Gradient. Anal. Chem. 2016, 88, 7899–7903. [Google Scholar] [CrossRef]
- Masuda, T.; Tomita, M.; Ishihama, Y. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J. Proteome Res. 2008, 7, 731–740. [Google Scholar] [CrossRef]
- Chen, Q.; Yan, G.; Gao, M.; Zhang, X. Ultrasensitive Proteome Profiling for 100 Living Cells by Direct Cell Injection, Online Digestion and Nano-LC-MS/MS Analysis. Anal. Chem. 2015, 87, 6674–6680. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Ohara, O. Development of a NanoLC-MS/MS System Using a Nonporous Reverse Phase Column for Ultrasensitive Proteome Analysis. Anal. Chem. 2018, 90, 12334–12338. [Google Scholar] [CrossRef]
- Muraoka, S.; Kume, H.; Adachi, J.; Shiromizu, T.; Watanabe, S.; Masuda, T.; Ishihama, Y.; Tomonaga, T. In-depth membrane proteomic study of breast cancer tissues for the generation of a chromosome-based protein list. J. Proteome Res. 2013, 12, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Miyata, J.; Watanabe, T.; Shioya, J.; Arita, M.; Ohara, O. Proteogenomic Analyses of Cellular Lysates Using a Phenol-Guanidinium Thiocyanate Reagent. J. Proteome Res. 2019, 18, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shi, J.; Wang, Y.; Chen, Y.; Li, Y.; Kong, D.; Chang, L.; Liu, F.; Lv, Z.; Zhou, Y.; et al. Multiproteases Combined with High-pH Reverse-Phase Separation Strategy Verified Fourteen Missing Proteins in Human Testis Tissue. J. Proteome Res. 2018, 17, 4171–4177. [Google Scholar] [CrossRef] [PubMed]
- Dou, M.; Tsai, C.F.; Piehowski, P.D.; Wang, Y.; Fillmore, T.L.; Zhao, R.; Moore, R.J.; Zhang, P.; Qian, W.J.; Smith, R.D.; et al. Automated Nanoflow Two-Dimensional Reversed-Phase Liquid Chromatography System Enables In-Depth Proteome and Phosphoproteome Profiling of Nanoscale Samples. Anal. Chem. 2019, 91, 9707–9715. [Google Scholar] [CrossRef]
- Krieger, J.R.; Wybenga-Groot, L.E.; Tong, J.; Bache, N.; Tsao, M.S.; Moran, M.F. Evosep One Enables Robust Deep Proteome Coverage Using Tandem Mass Tags while Significantly Reducing Instrument Time. J. Proteome Res. 2019, 18, 2346–2353. [Google Scholar] [CrossRef]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Guo, T.; Aebersold, R. Mass spectrometry-based proteomic quest for diabetes biomarkers. Biochim. Biophys. Acta 2015, 1854, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Noble, W.S.; Wolf-Yadlin, A. Technical advances in proteomics: New developments in data-independent acquisition. F1000Res 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, J.; Thoming, J.G.; Pohl, S.; Pich, A.; Lenz, C.; Haussler, S. The Core Proteome of Biofilm-Grown Clinical Pseudomonas aeruginosa Isolates. Cells 2019, 8, 1129. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Sweet, S.M.M.; Egertson, J.D.; Sedgewick, A.J.; Woo, S.; Liao, W.L.; Merrihew, G.E.; Searle, B.C.; Vaske, C.; Heaton, R.; et al. Data-Independent Acquisition Mass Spectrometry To Quantify Protein Levels in FFPE Tumor Biopsies for Molecular Diagnostics. J. Proteome Res. 2019, 18, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Miyata, J.; Fukunaga, K.; Kawashima, Y.; Watanabe, T.; Saitoh, A.; Hirosaki, T.; Araki, Y.; Kikawada, T.; Betsuyaku, T.; Ohara, O.; et al. Dysregulated fatty acid metabolism in nasal polyp-derived eosinophils from patients with chronic rhinosinusitis. Allergy 2019, 74, 1113–1124. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell Proteom. 2012, 11, O111 016717. [Google Scholar] [CrossRef]
- Zhang, Y.; Bilbao, A.; Bruderer, T.; Luban, J.; Strambio-De-Castillia, C.; Lisacek, F.; Hopfgartner, G.; Varesio, E. The Use of Variable Q1 Isolation Windows Improves Selectivity in LC-SWATH-MS Acquisition. J. Proteome Res. 2015, 14, 4359–4371. [Google Scholar] [CrossRef]
- Amodei, D.; Egertson, J.; MacLean, B.X.; Johnson, R.; Merrihew, G.E.; Keller, A.; Marsh, D.; Vitek, O.; Mallick, P.; MacCoss, M.J. Improving Precursor Selectivity in Data-Independent Acquisition Using Overlapping Windows. J. Am. Soc. Mass Spectrom. 2019, 30, 669–684. [Google Scholar] [CrossRef]
- Tsou, C.C.; Avtonomov, D.; Larsen, B.; Tucholska, M.; Choi, H.; Gingras, A.C.; Nesvizhskii, A.I. DIA-Umpire: Comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 2015, 12, 258–264. [Google Scholar] [CrossRef]
- Tsou, C.C.; Tsai, C.F.; Teo, G.C.; Chen, Y.J.; Nesvizhskii, A.I. Untargeted, spectral library-free analysis of data-independent acquisition proteomics data generated using Orbitrap mass spectrometers. Proteomics 2016, 16, 2257–2271. [Google Scholar] [CrossRef] [PubMed]
- Ting, Y.S.; Egertson, J.D.; Bollinger, J.G.; Searle, B.C.; Payne, S.H.; Noble, W.S.; MacCoss, M.J. PECAN: Library-free peptide detection for data-independent acquisition tandem mass spectrometry data. Nat. Methods 2017, 14, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Searle, B.C.; Pino, L.K.; Egertson, J.D.; Ting, Y.S.; Lawrence, R.T.; MacLean, B.X.; Villen, J.; MacCoss, M.J. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 2018, 9, 5128. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sugiyama, N.; Tomita, M.; Ishihama, Y. Microscale phosphoproteome analysis of 10,000 cells from human cancer cell lines. Anal. Chem. 2011, 83, 7698–7703. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, R.; Piehowski, P.D.; Moore, R.J.; Lim, S.; Orphan, V.J.; Pasa-Tolic, L.; Qian, W.J.; Smith, R.D.; Kelly, R.T. Subnanogram proteomics: Impact of LC column selection, MS instrumentation and data analysis strategy on proteome coverage for trace samples. Int. J. Mass Spectrom. 2018, 427, 4–10. [Google Scholar] [CrossRef]
- Ficarro, S.B.; Zhang, Y.; Lu, Y.; Moghimi, A.R.; Askenazi, M.; Hyatt, E.; Smith, E.D.; Boyer, L.; Schlaeger, T.M.; Luckey, C.J.; et al. Improved electrospray ionization efficiency compensates for diminished chromatographic resolution and enables proteomics analysis of tyrosine signaling in embryonic stem cells. Anal. Chem. 2009, 81, 3440–3447. [Google Scholar] [CrossRef]
- Wakabayashi, M.; Yoshihara, H.; Masuda, T.; Tsukahara, M.; Sugiyama, N.; Ishihama, Y. Phosphoproteome analysis of formalin-fixed and paraffin-embedded tissue sections mounted on microscope slides. J. Proteome Res. 2014, 13, 915–924. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef]
- Hebert, A.S.; Prasad, S.; Belford, M.W.; Bailey, D.J.; McAlister, G.C.; Abbatiello, S.E.; Huguet, R.; Wouters, E.R.; Dunyach, J.J.; Brademan, D.R.; et al. Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal. Chem. 2018, 90, 9529–9537. [Google Scholar] [CrossRef]
- Muntel, J.; Gandhi, T.; Verbeke, L.; Bernhardt, O.M.; Treiber, T.; Bruderer, R.; Reiter, L. Surpassing 10,000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol. Omics. 2019, 15, 348–360. [Google Scholar] [CrossRef]
- Gerhardt, S.; Mohajeri, M.H. Changes of Colonic Bacterial Composition in Parkinson’s Disease and Other Neurodegenerative Diseases. Nutrients 2018, 10, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wekerle, H. Brain Autoimmunity and Intestinal Microbiota: 100 Trillion Game Changers. Trends Immunol. 2017, 38, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Xiayu, X.; Shi, C.; Chen, W.; Song, N.; Fu, X.; Zhou, R.; Xu, Y.F.; Huang, L.; et al. Altered Gut Microbiota in a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 1241–1257. [Google Scholar] [CrossRef] [PubMed]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Iashchishyn, I.A.; Kara, J.; Fodera, V.; Vetri, V.; Sancataldo, G.; Marklund, N.; Morozova-Roche, L.A. Proinflammatory and amyloidogenic S100A9 induced by traumatic brain injury in mouse model. Neurosci. Lett. 2019, 699, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Gruden, M.A.; Davydova, T.V.; Kudrin, V.S.; Wang, C.; Narkevich, V.B.; Morozova-Roche, L.A.; Sewell, R.D.E. S100A9 Protein Aggregates Boost Hippocampal Glutamate Modifying Monoaminergic Neurochemistry: A Glutamate Antibody Sensitive Outcome on Alzheimer-like Memory Decline. ACS Chem. Neurosci. 2018, 9, 568–577. [Google Scholar] [CrossRef]
- Manavalan, A.; Mishra, M.; Sze, S.K.; Heese, K. Brain-site-specific proteome changes induced by neuronal P60TRP expression. Neurosignals 2013, 21, 129–149. [Google Scholar] [CrossRef]
- Mishra, M.; Heese, K. P60TRP interferes with the GPCR/secretase pathway to mediate neuronal survival and synaptogenesis. J. Cell Mol. Med. 2011, 15, 2462–2477. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.T.; Zhu, B.L.; Lai, Y.J.; Long, Y.; Zha, J.S.; Hu, X.T.; Zhang, J.H.; Chen, G.J. HMGCS2 promotes autophagic degradation of the amyloid-beta precursor protein through ketone body-mediated mechanisms. Biochem Biophys Res. Commun. 2017, 486, 492–498. [Google Scholar] [CrossRef]
- Shi, L.; Zhao, D.; Hou, C.; Peng, Y.; Liu, J.; Zhang, S.; Liu, J.; Long, J. Early interleukin-6 enhances hepatic ketogenesis in APPSWE/PSEN1dE9 mice via 3-hydroxy-3-methylglutary-CoA synthase 2 signaling activation by p38/nuclear factor kappaB p65. Neurobiol. Aging 2017, 56, 115–126. [Google Scholar] [CrossRef]
- Inoue, Y.; Ueda, M.; Masuda, T.; Misumi, Y.; Yamashita, T.; Ando, Y. Memantine, a Noncompetitive N-Methyl-D-Aspartate Receptor Antagonist, Attenuates Cerebral Amyloid Angiopathy by Increasing Insulin-Degrading Enzyme Expression. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Delikkaya, B.; Moriel, N.; Tong, M.; Gallucci, G.; de la Monte, S.M. Altered expression of insulin-degrading enzyme and regulator of calcineurin in the rat intracerebral streptozotocin model and human apolipoprotein E-epsilon4-associated Alzheimer’s disease. Alzheimers Dement. 2019, 11, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Beaver, S.K.; Mesa-Torres, N.; Pey, A.L.; Timson, D.J. NQO1: A target for the treatment of cancer and neurological diseases, and a model to understand loss of function disease mechanisms. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Kang, S.S.; Wang, Z.H.; Liu, X.; Day, J.X.; Wu, Z.; Peng, J.; Xiang, D.; Springer, W.; Ye, K. Akt Phosphorylates NQO1 and Triggers its Degradation, Abolishing Its Antioxidative Activities in Parkinson’s Disease. J. Neurosci. 2019, 39, 7291–7305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barca, A.; Ippati, S.; Urso, E.; Vetrugno, C.; Storelli, C.; Maffia, M.; Romano, A.; Verri, T. Carnosine modulates the Sp1-Slc31a1/Ctr1 copper-sensing system and influences copper homeostasis in murine CNS-derived cells. Am. J. Physiol. Cell Physiol. 2019, 316, C235–C245. [Google Scholar] [CrossRef]
- Peretti, D.; Bastide, A.; Radford, H.; Verity, N.; Molloy, C.; Martin, M.G.; Moreno, J.A.; Steinert, J.R.; Smith, T.; Dinsdale, D.; et al. RBM3 mediates structural plasticity and protective effects of cooling in neurodegeneration. Nature 2015, 518, 236–239. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.J.; Zhuang, R.J.; Li, Y.B.; Li, T.; Yuan, X.; Lei, B.B.; Xie, Y.F.; Wang, M. Cold-inducible protein RBM3 mediates hypothermic neuroprotection against neurotoxin rotenone via inhibition on MAPK signalling. J. Cell Mol. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Wang, P.; Zhang, S.; Ren, L.; Lv, Y.; Yin, R.; Bi, J. Neuroprotective effect of melatonin on soluble Abeta1-42-induced cortical neurodegeneration via Reelin-Dab1 signaling pathway. Neurol. Res. 2017, 39, 621–631. [Google Scholar] [CrossRef]
- Trotter, J.; Lee, G.H.; Kazdoba, T.M.; Crowell, B.; Domogauer, J.; Mahoney, H.M.; Franco, S.J.; Muller, U.; Weeber, E.J.; D’Arcangelo, G. Dab1 is required for synaptic plasticity and associative learning. J. Neurosci. 2013, 33, 15652–15668. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Ogasawara, A.; Heese, K. [Possible mechanisms of A beta(1-40)- or A beta(1-42)-induced cell death and their rescue factors]. Nihon Yakurigaku Zasshi 2004, 124, 135–143. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawashima, Y.; Watanabe, E.; Umeyama, T.; Nakajima, D.; Hattori, M.; Honda, K.; Ohara, O. Optimization of Data-Independent Acquisition Mass Spectrometry for Deep and Highly Sensitive Proteomic Analysis. Int. J. Mol. Sci. 2019, 20, 5932. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235932
Kawashima Y, Watanabe E, Umeyama T, Nakajima D, Hattori M, Honda K, Ohara O. Optimization of Data-Independent Acquisition Mass Spectrometry for Deep and Highly Sensitive Proteomic Analysis. International Journal of Molecular Sciences. 2019; 20(23):5932. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235932
Chicago/Turabian StyleKawashima, Yusuke, Eiichiro Watanabe, Taichi Umeyama, Daisuke Nakajima, Masahira Hattori, Kenya Honda, and Osamu Ohara. 2019. "Optimization of Data-Independent Acquisition Mass Spectrometry for Deep and Highly Sensitive Proteomic Analysis" International Journal of Molecular Sciences 20, no. 23: 5932. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235932