Parallel Bud Mutation Sequencing Reveals that Fruit Sugar and Acid Metabolism Potentially Influence Stress in Malus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Distinct Components of Sugar and Acid Between Jonathan and Sweet Jonathan during Fruit Development
2.2. Whole Genome Sequencing of Jonathan and Sweet Jonathan Enables Identification of Extensive Genetic Mutations between Jonathan and Sweet Jonathan
2.3. The Transcriptome Profiles of Jonatha’ and Sweet Jonathan during Fruit Development
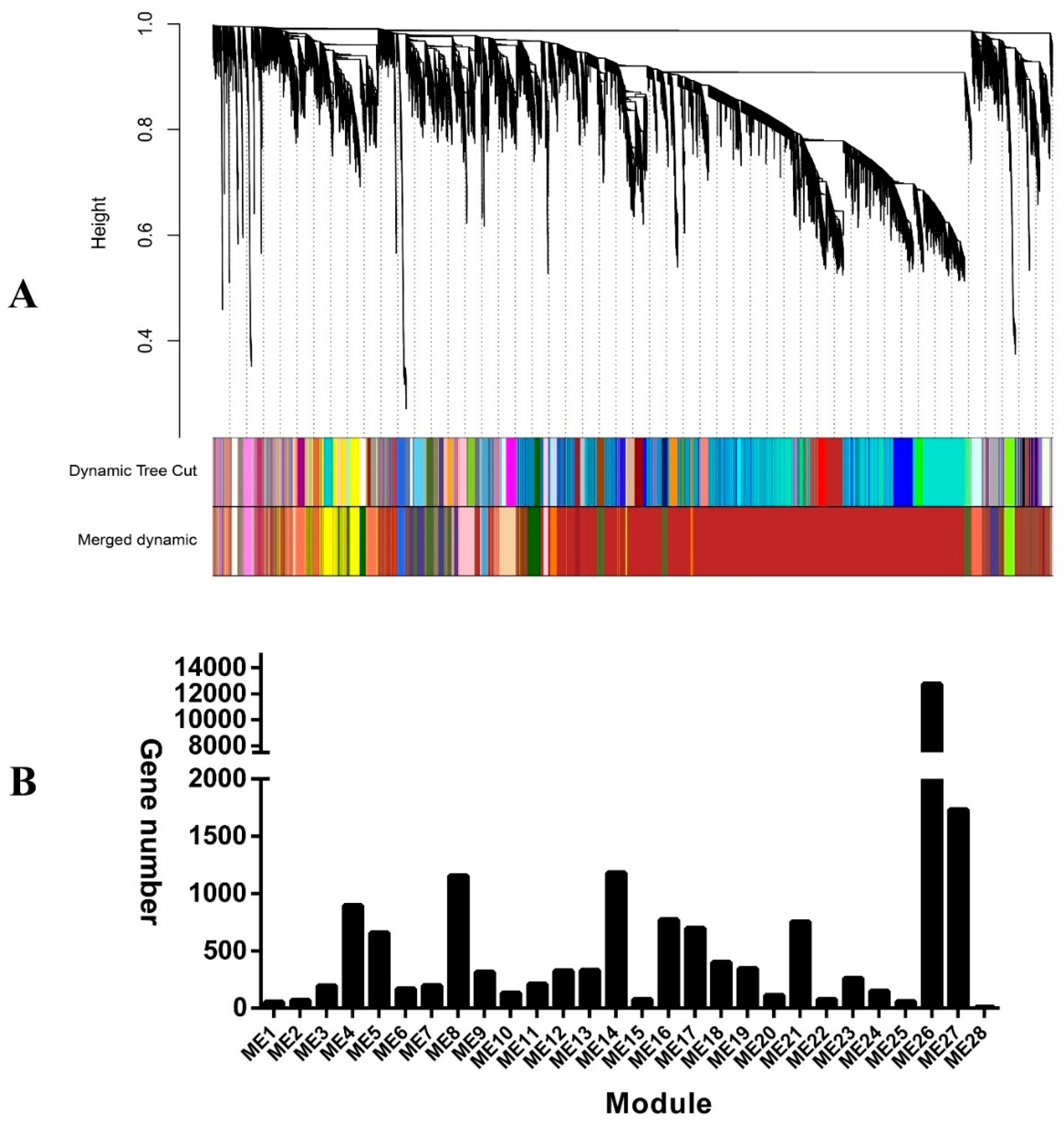
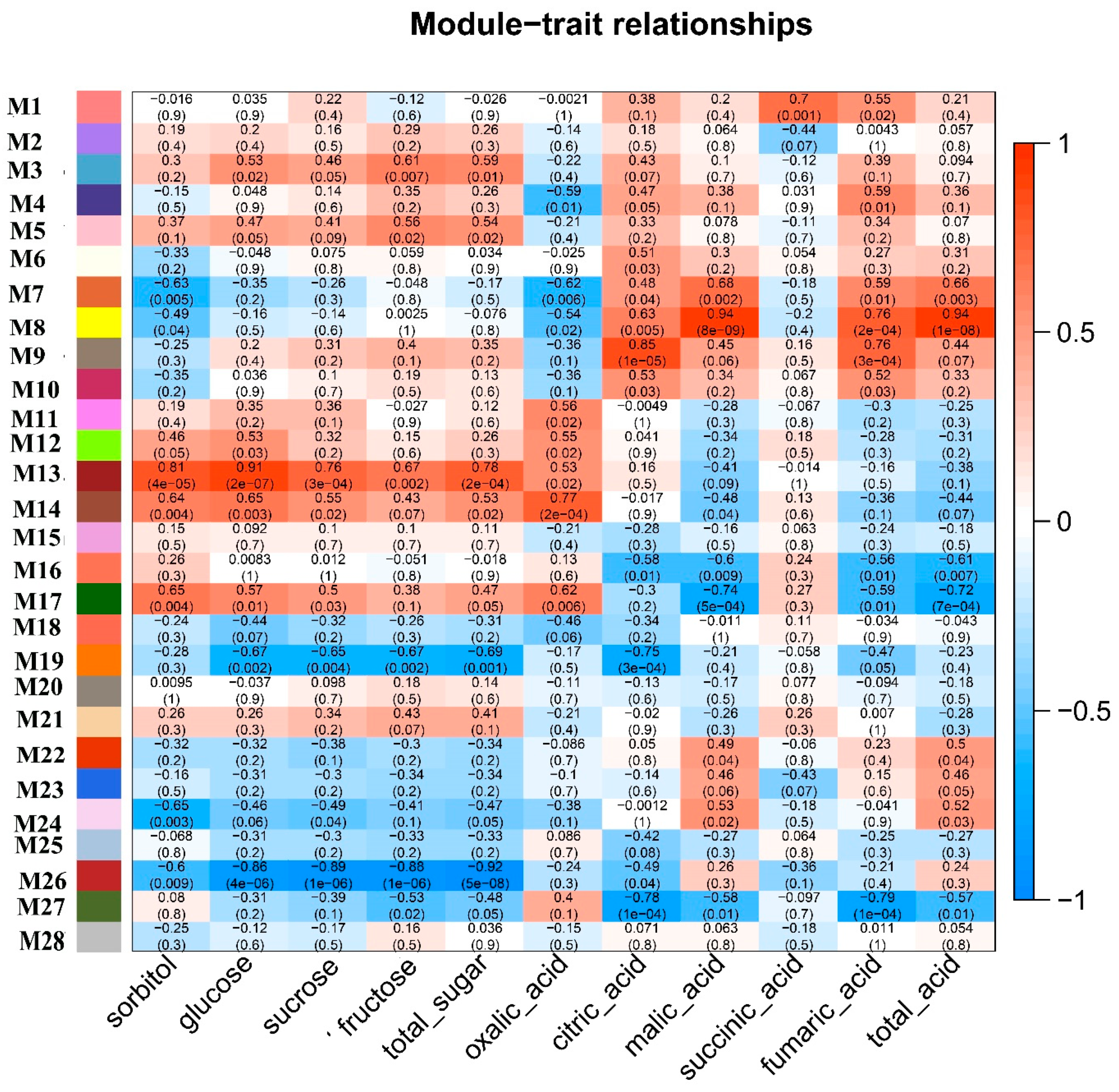
2.4. Identification of Co-Expression Modules
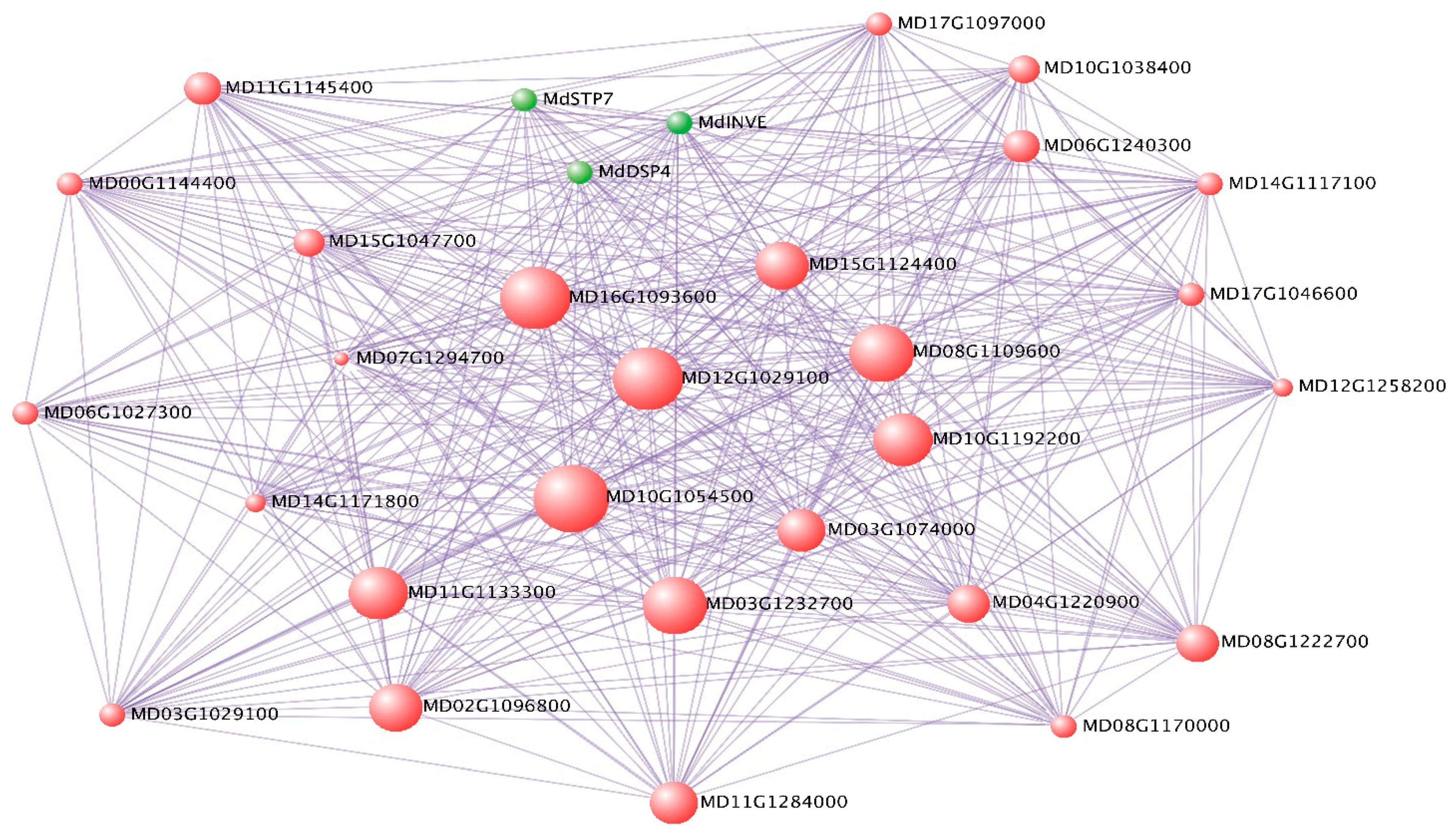
2.5. Deciphering Key Co-Expression Modules
2.6. Sugar Metabolism and Accumulation in Apple Fruit Development
2.7. The Stop Gain Mutation in MdMa1 may Partly Explain the Low Acid Content in Sweet Jonathan
2.8. Dramatic Upregulation of ABC Transporter Genes in Sweet Jonathan is Potentially Regulated by MdBPC6
2.9. PlantHhormone Signal Transduction
2.10. Disease Resistance Protein Genes
3. Discussion
3.1. Huge Numbers of Mutations Identified by Whole Genome Resequencing Suggested Variable Levels of Genetic Diversity among Apple Bud Mutant Cultivars
3.2. Identifying Hub Genes Associated with Sugar and Acid Accumulation by Deciphering Co-Expression Modules
3.3. Sugar and Acid in Apple Fruit may Influence Stress during Fruit Development
3.4. TranscriptFfactor MdBCP6 Could Regulate ABC Transporter Genes and Potentially Participate in Fruit Development or Stress Response
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. Measurement of Sugars and Acid
4.3. Resequencing of Jonathan and Sweet Jonathan
4.4. RNA-Seq Library Construction, Sequencing, and Data Processing
4.5. Identification of Co-Expression Modules
4.6. Visualization of Hub Genes
4.7. Gene Expression Analysis
4.8. Validation of SNPs and InDels
4.9. Identification of Cis-Motifs
4.10. Yeast One-Hybrid Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Granhall, I. Spontaneous and induced bud mutations in fruit trees. Acta Agric. Scand. 1954, 4, 594–600. [Google Scholar] [CrossRef]
- Hoying, S.A.; Rosenberger, D.A.; Lamont, G. The apple industry in China. N. Y. Fruit Q. 2006, 14, 13–20. [Google Scholar]
- El-Sharkawy, I.; Liang, D.; Xu, K. Transcriptome analysis of an apple (Malus × domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 2015, 66, 7359–7376. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.L.; Xi, F.F.; Yu, Y.H.; Zhang, X.Y.; Zhang, G.H.; Zhong, G.Y. Comparative RNA-Seq profiling of berry development between table grape ‘Kyoho’ and its early-ripening mutant ‘Fengzao’. BMC Genom. 2016, 17, 795. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.M.; Platzer, A.; Kazda, A.; Akimcheva, S.; Valuchova, S.; Nizhynska, V.; Nordborg, M.; Riha, K. Germline replications and somatic mutation accumulation are independent of vegetative life span in Arabidopsis. Proc. Natl. Acad. Sci. USA 2016, 113, 12226–12231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Z.; Wang, N.; Gao, Y.; Liu, Y.; Wu, Y.; Bai, Y.; Zhang, Z.; Lin, X.; Dong, Y.; et al. Tissue culture-induced heritable genomic variation in rice, and their phenotypic implications. PLoS ONE 2014, 9, e96879. [Google Scholar] [CrossRef]
- Li, M.; Feng, F.; Cheng, L. Expression patterns of genes involved in sugar metabolism and accumulation during apple fruit development. PLoS ONE 2012, 7, e33055. [Google Scholar] [CrossRef]
- Jia, D.; Shen, F.; Wang, Y.; Wu, T.; Xu, X.; Zhang, X.; Han, Z. Apple fruit acidity is genetically diversified by natural variations in three hierarchical epistatic genes MdSAUR37, MdPP2CH and MdALMTII. Plant J. 2018, 95, 427–443. [Google Scholar] [CrossRef]
- Ma, B.; Liao, L.; Fang, T.; Peng, Q.; Ogutu, C.; Zhou, H.; Ma, F.; Han, Y. A Ma10 gene encoding P-type ATPase is involved in fruit organic acid accumulation in apple. Plant Biotechnol. J. 2018. [Google Scholar] [CrossRef]
- Bai, Y.; Dougherty, L.; Cheng, L.L.; Zhong, G.Y.; Xu, K.N. Uncovering co-expression gene network modules regulating fruit acidity in diverse apples. BMC Genomics 2015, 16, 6–12. [Google Scholar] [CrossRef]
- Hu, D.G.; Sun, C.H.; Ma, Q.J.; You, C.X.; Cheng, L.; Hao, Y.J. MdMYB1 regulates anthocyanin and malate accumulation by directly facilitating their transport into vacuoles in apples. Plant Physiol. 2016, 170, 1315–1330. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, Y.; Zheng, Y.; Fei, Z.; Dandekar, A.M.; Xu, K.; Han, Z.; Cheng, L. Suppressing sorbitol synthesis substantially alters the lobal expression profile of stress response genes in apple (Malus domestica) Leaves. Plant Cell Physiol. 2015, 56, 1748–1761. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liu, F.; Chen, C.; Ma, F.; Li, M. The Malus domestica sugar transporter gene family: Identifications based on genome and expression profiling related to the accumulation of fruit sugars. Front. Plant Sci. 2014, 5, 569. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Jayanty, S.; Beaudry, R.; Loescher, W. Sorbitol transporter expression in apple sink tissues: Implications for fruit sugar accumulation and watercore development. J. Am. Soc. Hortic. Sci. 2005, 130, 261–268. [Google Scholar] [CrossRef]
- Fan, R.C.; Peng, C.C.; Xu, Y.H.; Wang, X.F.; Li, Y.; Shang, Y.; Du, S.Y.; Zhao, R.; Zhang, X.Y.; Zhang, L.Y.; et al. Apple sucrose transporter SUT1 and sorbitol transporter SOT6 interact with cytochrome b5 to regulate their affinity for substrate sugars. Plant Physiol. 2009, 150, 1880–1901. [Google Scholar] [CrossRef] [PubMed]
- Rosa, M.; Prado, C.; Podazza, G.; Interdonato, R.; Gonzalez, J.A.; Hilal, M.; Prado, F.E. Soluble sugars--metabolism, sensing and abiotic stress: A complex network in the life of plants. Plant Signal. Behav. 2009, 4, 388–393. [Google Scholar] [CrossRef]
- Meng, D.; Li, C.; Park, H.J.; Gonzalez, J.; Wang, J.; Dandekar, A.M.; Turgeon, B.G.; Cheng, L. Sorbitol modulates resistance to Alternaria alternata by regulating the expression of an NLR resistance gene in apple. Plant Cell 2018, 30, 1562–1581. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Etienne, A.; Génard, M.; Lobit, P.; Mbeguié-A-Mbéguié, D.; Bugaud, C. What controls fleshy fruit acidity? A review of malate and citrate accumulation in fruit cells. J. Exp. Bot. 2013, 64, 1451–1469. [Google Scholar] [CrossRef]
- Chern, M.; Canlas, P.E.; Fitzgerald, H.A.; Ronald, P.C. Rice NRR, a negative regulator of disease resistance, interacts with Arabidopsis NPR1 and rice NH1. Plant J. 2005, 43, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Glazebrook, J.; Clarke, J.D.; Volko, S.; Dong, X. The Arabidopsis NPR1 gene that controls systemic acquired resistance encodes a novel protein containing ankyrin repeats. Cell 1997, 88, 57–63. [Google Scholar] [CrossRef]
- Krichevsky, A.; Zaltsman, A.; Kozlovsky, S.V.; Tian, G.W.; Citovsky, V. Regulation of root elongation by histone acetylation in Arabidopsis. J. Mol. Biol. 2009, 385, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Kuusk, S.; Sohlberg, J.J.; Magnus Eklund, D.; Sundberg, E. Functionally redundant SHI family genes regulate Arabidopsis gynoecium development in a dose-dependent manner. Plant J. 2006, 47, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P.C. The basic helix-loop-helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, N.; Jiao, Y.; Li, R.; Xiao, D.; Wang, Z. The grapevine basic helix-loop-helix (bHLH) transcription factor positively modulates CBF-pathway and confers tolerance to cold-stress in Arabidopsis. Mol. Biol. Rep. 2014, 41, 5329–5342. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.K.; Dagenais, N.; Chory, J.; Weigel, D. Regulation of auxin response by the protein kinase PINOID. Cell 2000, 100, 469–478. [Google Scholar] [CrossRef]
- Bao, F.; Azhakanandam, S.; Franks, R.G. SEUSS and SEUSS-LIKE transcriptional adaptors regulate floral and embryonic development in Arabidopsis. Plant Physiol. 2010, 152, 821–836. [Google Scholar] [CrossRef]
- Shrestha, B.; Guragain, B.; Sridhar, V.V. Involvement of co-repressor LUH and the adapter proteins SLK1 and SLK2 in the regulation of abiotic stress response genes in Arabidopsis. BMC Plant Biol. 2014, 14, 54. [Google Scholar] [CrossRef]
- Yu, H.; Luo, N.; Sun, L.; Liu, D. HPS4/SABRE regulates plant responses to phosphate starvation through antagonistic interaction with ethylene signalling. J. Exp. Bot. 2012, 63, 4527–4538. [Google Scholar] [CrossRef]
- Bai, Y.; Dougherty, L.; Li, M.J.; Fazio, G.; Cheng, L.L.; Xu, K.N. A natural mutation-led truncation in one of the two aluminum-activated malate transporter-like genes at the Ma locus is associated with low fruit acidity in apple. Mol. Genet. Genom. 2011, 287, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Elkan, C. The value of prior knowledge in discovering motifs with MEME. Proceedings International Conference on Intelligent Syst. Mol. Biol. 1995, 3, 21–29. [Google Scholar]
- Shen, F.; Huang, Z.; Zhang, B.; Wang, Y.; Zhang, X.; Wu, T.; Xu, X.; Zhang, X.; Han, Z. Mapping gene markers for apple fruit ring rot disease resistance using a multi-omics approach. G3 Genes Genomes Genet. 2019, 9, 1663–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrier, G.; Le Cunff, L.; Dereeper, A.; Legrand, D.; Sabot, F.; Bouchez, O.; Audeguin, L.; Boursiquot, J.M.; This, P. Transposable elements are a major cause of somatic polymorphism in Vitis vinifera L. PLoS ONE 2012, 7, e32973. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Kim, G.H.; Kwon, S.I.; Kim, J.H.; Kwon, Y.S.; Choi, C. Analysis of ‘Fuji’ apple somatic variants from next-generation sequencing. Genet. Mol. Res. 2016, 15, 17–36. [Google Scholar] [CrossRef]
- Yang, T.; Li, K.T.; Hao, S.X.; Zhang, J.; Song, T.T.; Tian, J.; Yao, Y.C. The use of RNA sequencing and correlation network analysis to study potential regulators of crabapple leaf color transformation. Plant Cell Physiol. 2018, 59, 1027–1042. [Google Scholar] [CrossRef]
- Dietrich, K.; Weltmeier, F.; Ehlert, A.; Weiste, C.; Stahl, M.; Harter, K.; Droege-Laser, W. Heterodimers of the Arabidopsis transcription factors bZIP1 and bZIP53 reprogram amino acid metabolism during low energy stress. Plant Cell 2011, 23, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Ma, J.; Tan, M.; Mao, J.; An, N.; Sha, G.; Zhang, D.; Zhao, C.; Han, M. Transcriptome analysis reveals the effects of sugar metabolism and auxin and cytokinin signaling pathways on root growth and development of grafted apple. BMC Genom. 2016, 17, 150. [Google Scholar] [CrossRef] [Green Version]
- Soulaiman, S.; Ming, W.; Fabienne, D.; Maria, P.; Laurent, O.; Latifa, H.; Rossitza, A. The sugar-signaling hub: Overview of regulators and interaction with the hormonal and metabolic network. Int. J. Mol. Sci. 2018, 19, 2506. [Google Scholar]
- Lobit, P.; Genard, M.; Soing, P.; Habib, R. Modelling malic acid accumulation in fruits: Relationships with organic acids, potassium, and temperature. J. Exp. Bot. 2006, 57, 1471–1483. [Google Scholar] [CrossRef] [Green Version]
- Ceusters, J.; Borland, A.M.; De Proft, M.P. Drought adaptation in plants with crassulacean acid metabolism involves the flexible use of different storage carbohydrate pools. Plant Signal. Behav. 2009, 4, 212–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.L.; Wang, C.R.; Li, X.Y.; Yao, Y.X.; Hao, Y.J. Modifications of Kyoho grape berry quality under long-term NaCl treatment. Food Chem. 2013, 139, 31–937. [Google Scholar] [CrossRef] [PubMed]
- Monfared, M.M.; Simon, M.K.; Meister, R.J.; Roig-Villanova, I.; Kooiker, M.; Colombo, L.; Fletcher, J.C.; Gasser, C.S. Overlapping and antagonistic activities of BASIC PENTACYSTEINE genes affect a range of developmental processes in Arabidopsis. Plant J. 2011, 66, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, F.L.; Kerr, I.D. ABC transporter research: Going strong 40 years on. Biochem. Soc. Trans. 2015, 43, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Chang, Y.S.; Yang, F.Q.; Wang, Y.; Li, H.; Zhao, Y.; Chen, D.; Wu, T.; Zhang, X.; Han, Z. A dense SNP genetic map constructed using restriction site-associated DNA sequencing enables detection of QTLs controlling apple fruit quality. BMC Genom. 2015, 16, 747. [Google Scholar] [CrossRef] [Green Version]
- Janssen, B.J.; Thodey, K.; Schaffer, R.J.; Alba, R.; Balakrishnan, L.; Bishop, R.; Bowen, J.H.; Crowhurst, R.N.; Gleave, A.P.; Ledger, S.; et al. Global gene expression analysis of apple fruit development from the floral bud to ripe fruit. BMC Plant Biol. 2008, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing S. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Improving SNP discovery by base alignment quality. Bioinformatics 2011, 27, 1157–1158. [Google Scholar] [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stutz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Rombauts, S.; Dehais, P.; Van Montagu, M.; Rouze, P. PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999, 27, 295–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Gatto, P.; Moser, M.; Pindo, M.; Velasco, R. Isolation of functional RNA from small amounts of different grape and apple tissues. Mol. Biotechnol. 2004, 26, 95–100. [Google Scholar] [CrossRef]
- Dai, M.; Thompson, R.C.; Maher, C.; Contreras-Galindo, R.; Kaplan, M.H.; Markovitz, D.M.; Omenn, G.; Meng, F. NGSQC: Cross-platform quality analysis pipeline for deep sequencing data. BMC Genomics 2010, 11, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. International J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yu, H.; Liu, B.H.; Zhao, Z.; Liu, L.; Ma, L.X.; Li, Y.X.; Li, Y.Y. DCGL v2.0: An R package for unveiling differential regulation from differential co-expression. PLoS ONE 2013, 8, e79729. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T) (-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, 260–266. [Google Scholar] [CrossRef] [Green Version]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Shen, F.; Gao, Y.; Wang, D.; Wang, K. Parallel Bud Mutation Sequencing Reveals that Fruit Sugar and Acid Metabolism Potentially Influence Stress in Malus. Int. J. Mol. Sci. 2019, 20, 5988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235988
Zhao J, Shen F, Gao Y, Wang D, Wang K. Parallel Bud Mutation Sequencing Reveals that Fruit Sugar and Acid Metabolism Potentially Influence Stress in Malus. International Journal of Molecular Sciences. 2019; 20(23):5988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235988
Chicago/Turabian StyleZhao, Jirong, Fei Shen, Yuan Gao, Dajiang Wang, and Kun Wang. 2019. "Parallel Bud Mutation Sequencing Reveals that Fruit Sugar and Acid Metabolism Potentially Influence Stress in Malus" International Journal of Molecular Sciences 20, no. 23: 5988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235988