Overcoming Hurdles in Nanoparticle Clinical Translation: The Influence of Experimental Design and Surface Modification

 , ,
, ,
Abstract
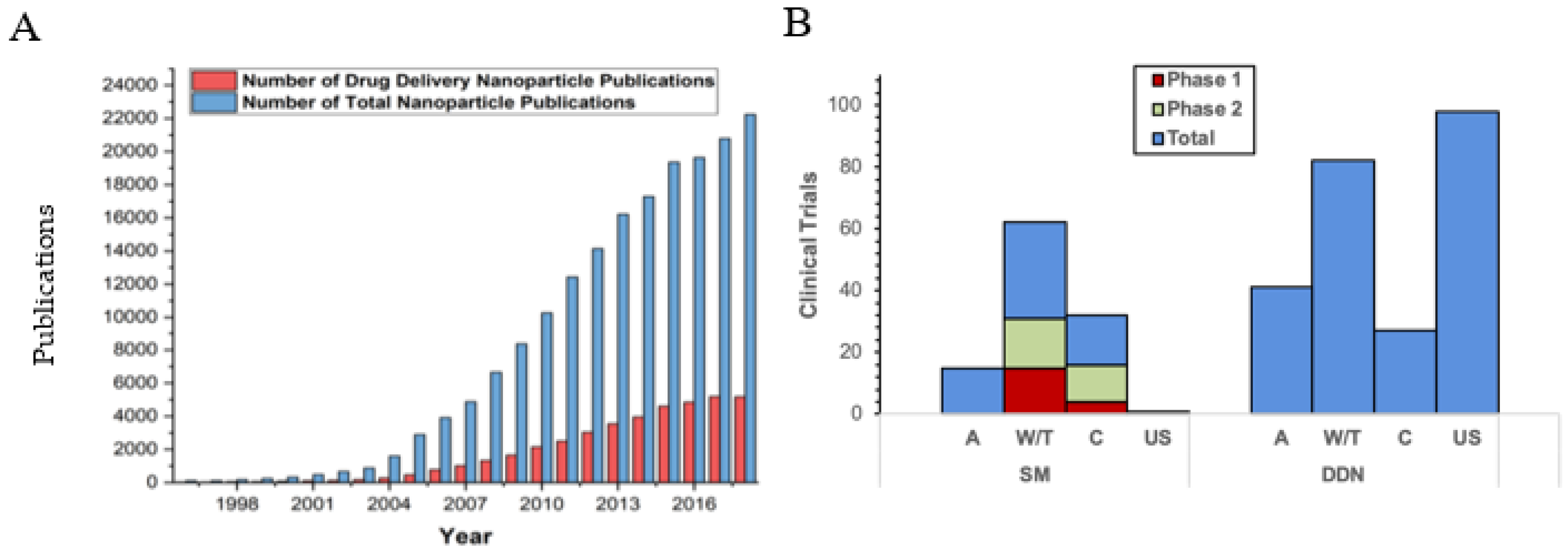
:1. Introduction
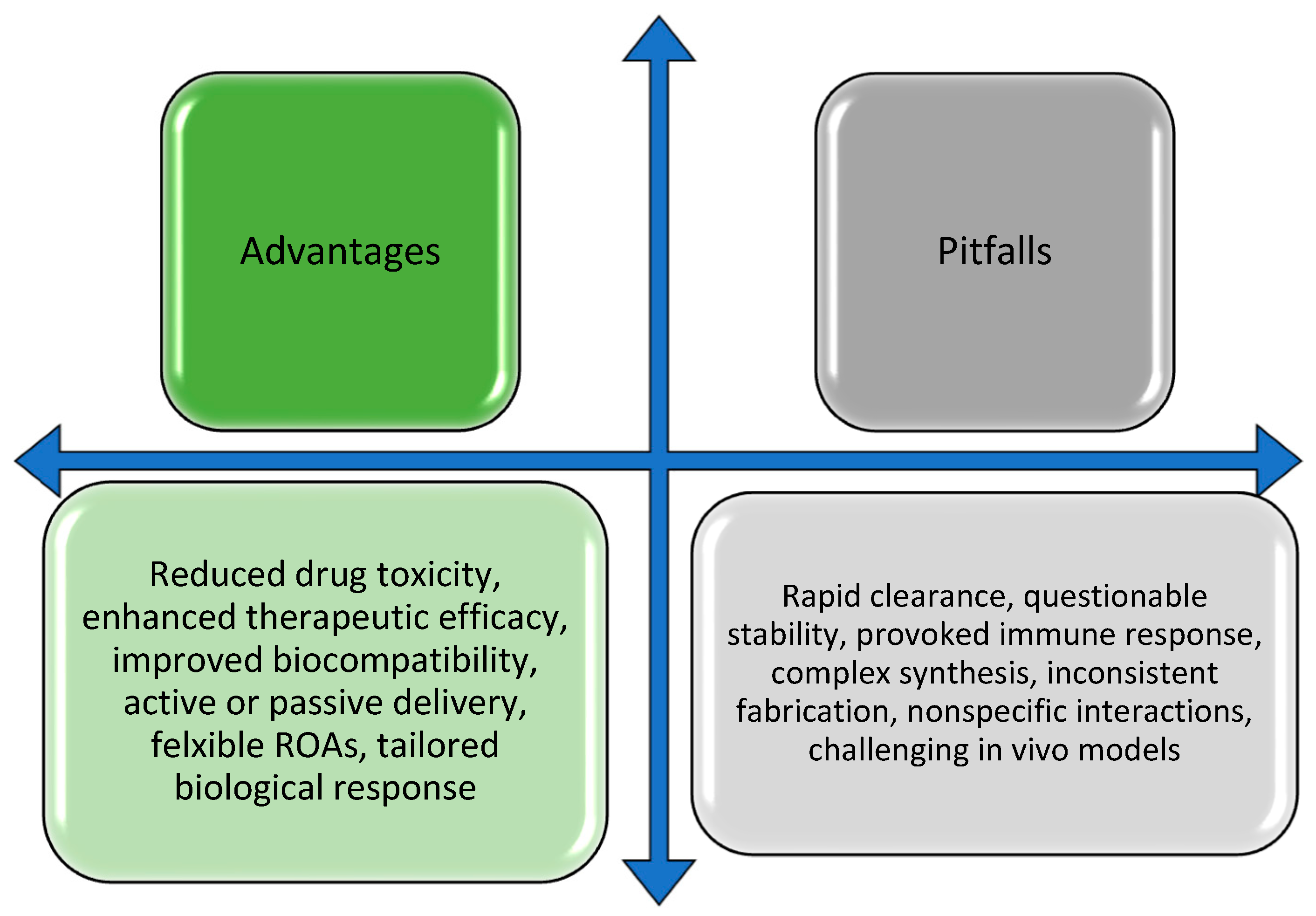
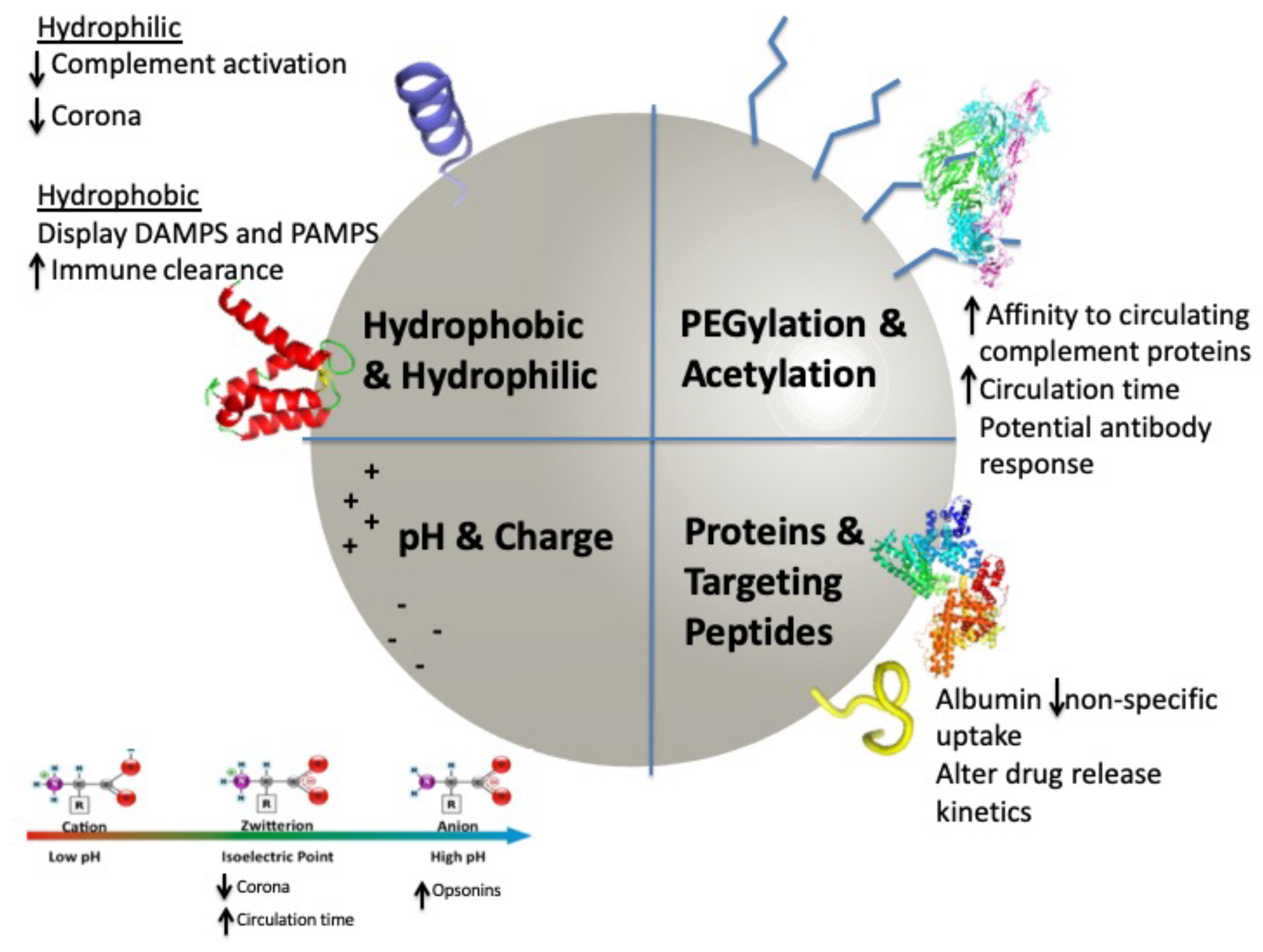
2. Surface Modifications
2.1. Modifications to Enhance Circulation
2.2. Modifications for Cellular Targeting and Retention
2.3. Modifications Targeting Payload Release
3. Corona Development
Mechanisms of Clearance
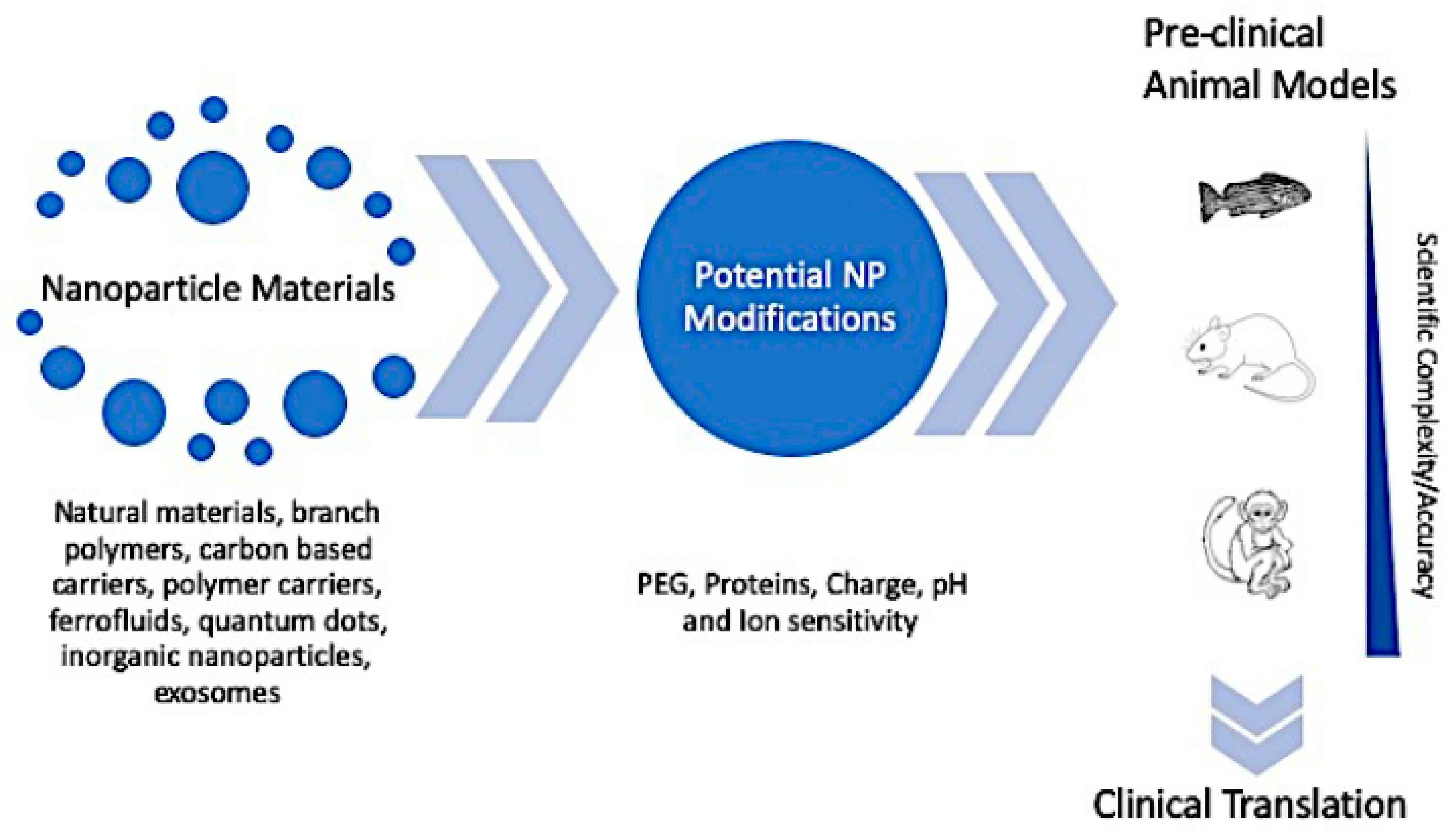
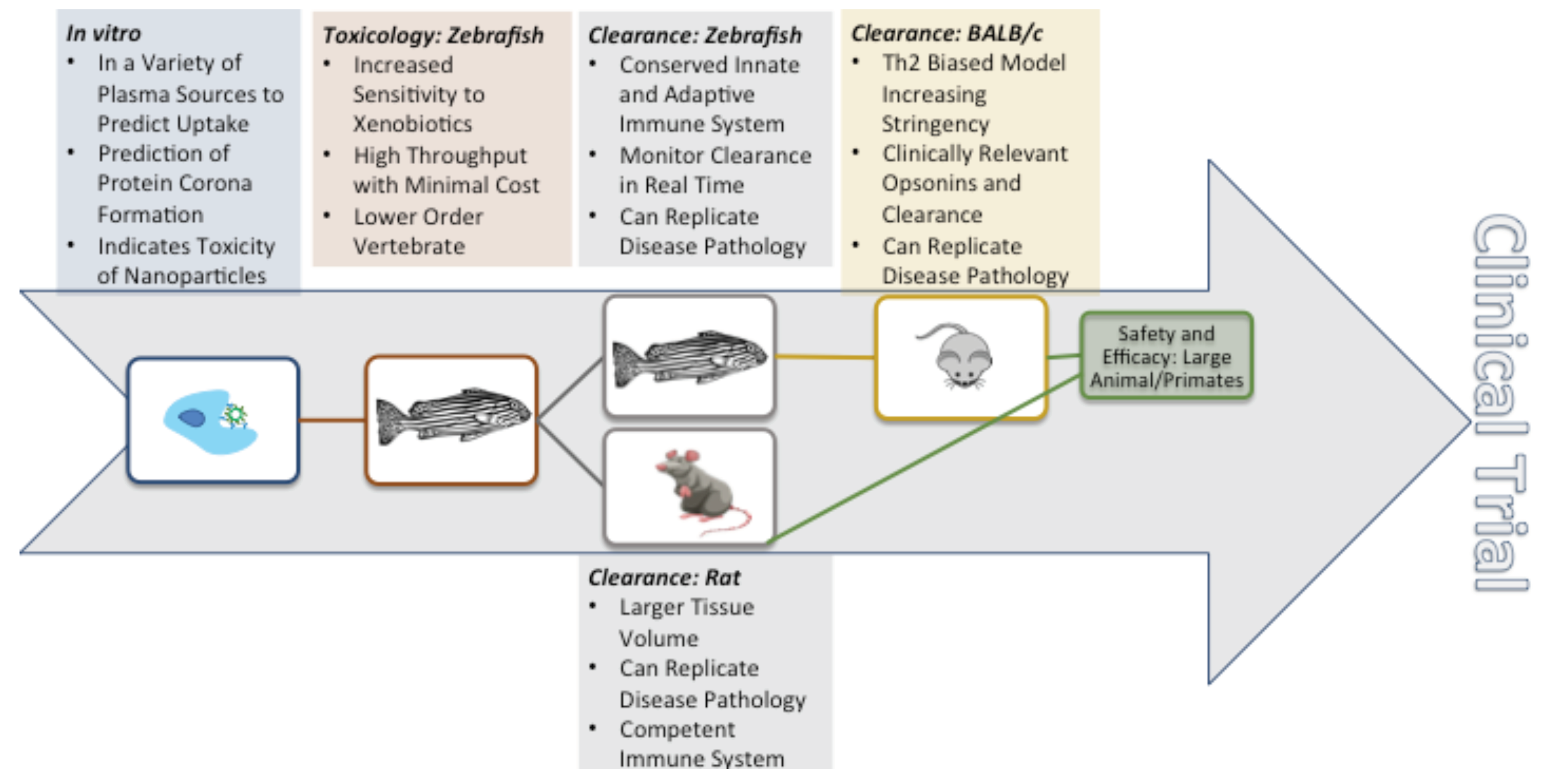
4. In Vivo Nanoparticle Assessment
4.1. A Comparison of Preclinical In Vivo Models
4.1.1. Zebrafish
4.1.2. Rodents
4.2. Route of Administration
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PEG | Polyethylene Glycol |
| mPEG | Methoxy Polyethylene Glycol |
| FDA | Food and Drug Administration |
| ROA | Rout of Administration |
| TPP | Triphenylphosphine |
| PVA | Polyvinylalcohol |
| Ig | Immunoglobulins |
| MPS | Mononuclear Phagocyte System |
| PC: Chol | Phosphatidyl-choline: Cholesterol |
| ADME | Absorption, Distribution, Metabolism, and Excretion |
| NSG | NOD Skid Gamma mice |
| PDX | Patient Derived Xenograft |
| IV | Intravenous Injection |
| LAM | Lung-Associated Macrophages |
| OA | Osteoarthritis |
References
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E.K. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Bamberger, D.; Schupp, J.; Weilbächer, M.; Kaps, L.; Strobl, S.; Radi, L.; Diken, M.; Strand, D.; Tuettenberg, A.; et al. Dextran-based therapeutic nanoparticles for hepatic drug delivery. Nanomedicine 2016, 11, 2663–2677. [Google Scholar] [CrossRef] [PubMed]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef] [PubMed]
- Karmali, P.P.; Kotamraju, V.R.; Kastantin, M.; Black, M.; Missirlis, D.; Tirrell, M.; Ruoslahti, E. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomedicine 2009, 5, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Exosomes as Drug Carriers for Cancer Therapy|Molecular Pharmaceutics. Available online: https://0-pubs-acs-org.brum.beds.ac.uk/doi/abs/10.1021/acs.molpharmaceut.9b00104 (accessed on 11 November 2019).
- Pardo, J.; Peng, Z.; Leblanc, R.M. Cancer Targeting and Drug Delivery Using Carbon-Based Quantum Dots and Nanotubes. Molecules 2018, 23, 378. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lin, Y.; Han, N.; Li, X.; Geng, H.; Wang, X.; Cui, Y.; Wang, S. Mesoporous carbon nanomaterials in drug delivery and biomedical application. Drug Deliv. 2017, 24, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.; Lim, V. Colon targeted drug delivery of branch-chained disulphide cross-linked polymers: Design, synthesis, and characterisation studies. Chem. Cent. J. 2016, 10. [Google Scholar] [CrossRef]
- Karandish, F.; Haldar, M.K.; You, S.; Brooks, A.E.; Brooks, B.D.; Guo, B.; Choi, Y.; Mallik, S. Prostate-Specific Membrane Antigen Targeted Polymersomes for Delivering Mocetinostat and Docetaxel to Prostate Cancer Cell Spheroids. Acs Omega 2016, 1, 952–962. [Google Scholar] [CrossRef]
- Yoshida, T.; Lai, T.C.; Kwon, G.S.; Sako, K. pH- and ion-sensitive polymers for drug delivery. Expert Opin. Drug Deliv. 2013, 10, 1497–1513. [Google Scholar] [CrossRef]
- Janagam, D.R.; Wu, L.; Lowe, T.L. Nanoparticles for drug delivery to the anterior segment of the eye. Adv. Drug Deliv. Rev. 2017, 122, 31–64. [Google Scholar] [CrossRef]
- Kim, H.Y.; Ryu, J.H.; Chu, C.W.; Son, G.M.; Jeong, Y.-I.; Kwak, T.-W.; Kim, D.H.; Chung, C.-W.; Rhee, Y.H.; Kang, D.H.; et al. Paclitaxel-incorporated nanoparticles using block copolymers composed of poly(ethylene glycol)/poly(3-hydroxyoctanoate). Nanosc. Res. Lett. 2014, 9, 525. [Google Scholar] [CrossRef] [PubMed]
- Uhlirova, D.; Stankova, M.; Docekalova, M.; Hosnedlova, B.; Kepinska, M.; Ruttkay-Nedecky, B.; Ruzicka, J.; Fernandez, C.; Milnerowicz, H.; Kizek, R. A Rapid Method for the Detection of Sarcosine Using SPIONs/Au/CS/SOX/NPs for Prostate Cancer Sensing. Int. J. Mol. Sci. 2018, 19, 722. [Google Scholar] [CrossRef] [PubMed]
- Javanbakht, T.; Laurent, S.; Stanicki, D.; Wilkinson, K.J. Relating the Surface Properties of Superparamagnetic Iron Oxide Nanoparticles (SPIONs) to Their Bactericidal Effect towards a Biofilm of Streptococcus mutans. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar]
- Van Haute, D.; Berlin, J.M. Challenges in realizing selectivity for nanoparticle biodistribution and clearance: Lessons from gold nanoparticles. Ther. Deliv. 2017, 8, 763–774. [Google Scholar] [CrossRef]
- Yang, G.; Phua, S.Z.F.; Bindra, A.K.; Zhao, Y. Degradability and Clearance of Inorganic Nanoparticles for Biomedical Applications. Adv. Mater. 2019, 31, 1805730. [Google Scholar] [CrossRef]
- Yu, K.; Sun, C.; Zhang, B.; Hassan, M.; He, Y. Size-dependent adsorption of antibiotics onto nanoparticles in a field-scale wastewater treatment plant. Environ. Pollut. 2019, 248, 1079–1087. [Google Scholar] [CrossRef]
- De Jong, W.H.; Borm, P.J. Drug delivery and nanoparticles: Applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef]
- Fang, C.; Shi, B.; Pei, Y.-Y.; Hong, M.-H.; Wu, J.; Chen, H.-Z. In vivo tumor targeting of tumor necrosis factor-α-loaded stealth nanoparticles: Effect of MePEG molecular weight and particle size. Eur. J. Pharm. Sci. 2006, 27, 27–36. [Google Scholar] [CrossRef]
- Papi, M.; Caputo, D.; Palmieri, V.; Coppola, R.; Palchetti, S.; Bugli, F.; Martini, C.; Digiacomo, L.; Pozzi, D.; Caracciolo, G. Clinically approved PEGylated nanoparticles are covered by a protein corona that boosts the uptake by cancer cells. Nanoscale 2017, 9, 10327–10334. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Rattan, R.; Bhattacharjee, S.; Zong, H.; Swain, C.; Siddiqui, M.A.; Visovatti, S.H.; Kanthi, Y.; Desai, S.; Pinsky, D.J.; Goonewardena, S.N. Nanoparticle-macrophage interactions: A balance between clearance and cell-specific targeting. Bioorg. Med. Chem. 2017, 25, 4487–4496. [Google Scholar] [CrossRef] [PubMed]
- PEG-Intron (Peginterferon alfa-2b) Package Insert. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2001/pegsche080701LB.htm (accessed on 4 August 2019).
- MACUGEN—(Pegaptanib Sodium Injection). Available online: http://www.bauschretinarx.com/macugen/ecp/about? (accessed on 4 August 2019).
- Genentech: Pegasys® (peginterferon alfa-2a)—Information for Patients. Available online: https://www.gene.com/patients/medicines/pegasys (accessed on 4 August 2019).
- Learn About Neulasta® (pegfilgrastim). Find Important Product Information at Neulasta.com. Available online: https://www.neulasta.com/ (accessed on 4 August 2019).
- About SOMAVERT. Available online: https://www.somavert.com/about-somavert (accessed on 4 August 2019).
- Dinndorf, P.A.; Gootenberg, J.; Cohen, M.H.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Pegaspargase (Oncaspar®) for the First-Line Treatment of Children with Acute Lymphoblastic Leukemia (ALL). Oncologist 2007, 12, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Guttmann, A.; Krasnokutsky, S.; Pillinger, M.H.; Berhanu, A. Pegloticase in gout treatment—Safety issues, latest evidence and clinical considerations. Ther. Adv. Drug Saf. 2017, 8, 379–388. [Google Scholar] [CrossRef] [PubMed]
- English, C.; Aloi, J.J. New FDA-Approved Disease-Modifying Therapies for Multiple Sclerosis. Clin. Ther. 2015, 37, 691–715. [Google Scholar] [CrossRef] [PubMed]
- ADYNOVATE® [Antihemophilic Factor (Recombinant), PEGylated]. Available online: http://ssshare.it/kYIT (accessed on 4 August 2019).
- Kivitz, A.J.; Conaghan, P.G.; Cinar, A.; Lufkin, J.; Kelley, S.D. Rescue Analgesic Medication Use by Patients Treated with Triamcinolone Acetonide Extended-Release for Knee Osteoarthritis Pain: Pooled Analysis of Three Phase 2/3 Randomized Clinical Trials. Pain Ther. 2019, 8, 1–10. [Google Scholar] [CrossRef]
- Bradbury, M.S.; Phillips, E.; Montero, P.H.; Cheal, S.M.; Stambuk, H.; Durack, J.C.; Sofocleous, C.T.; Meester, R.J.C.; Wiesner, U.; Patel, S. Clinically-translated silica nanoparticles as dual-modality cancer-targeted probes for image-guided surgery and interventions. Integr. Biol. (Camb.) 2013, 5, 74–86. [Google Scholar] [CrossRef]
- Andreopoulou, E.; Gaiotti, D.; Kim, E.; Downey, A.; Mirchandani, D.; Hamilton, A.; Jacobs, A.; Curtin, J.; Muggia, F. Pegylated liposomal doxorubicin HCL (PLD; Caelyx/Doxil®): Experience with long-term maintenance in responding patients with recurrent epithelial ovarian cancer. Ann. Oncol. 2007, 18, 716–721. [Google Scholar] [CrossRef]
- Sieber, S.; Grossen, P.; Uhl, P.; Detampel, P.; Mier, W.; Witzigmann, D.; Huwyler, J. Zebrafish as a predictive screening model to assess macrophage clearance of liposomes in vivo. Nanomed.-Nanotechnol. 2019, 17, 82–93. [Google Scholar] [CrossRef]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R.H. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Verhoef, J.J.F.; Anchordoquy, T.J. Questioning the Use of PEGylation for Drug Delivery. Drug Deliv. Transl. Res. 2013, 3, 499–503. [Google Scholar] [CrossRef] [PubMed]
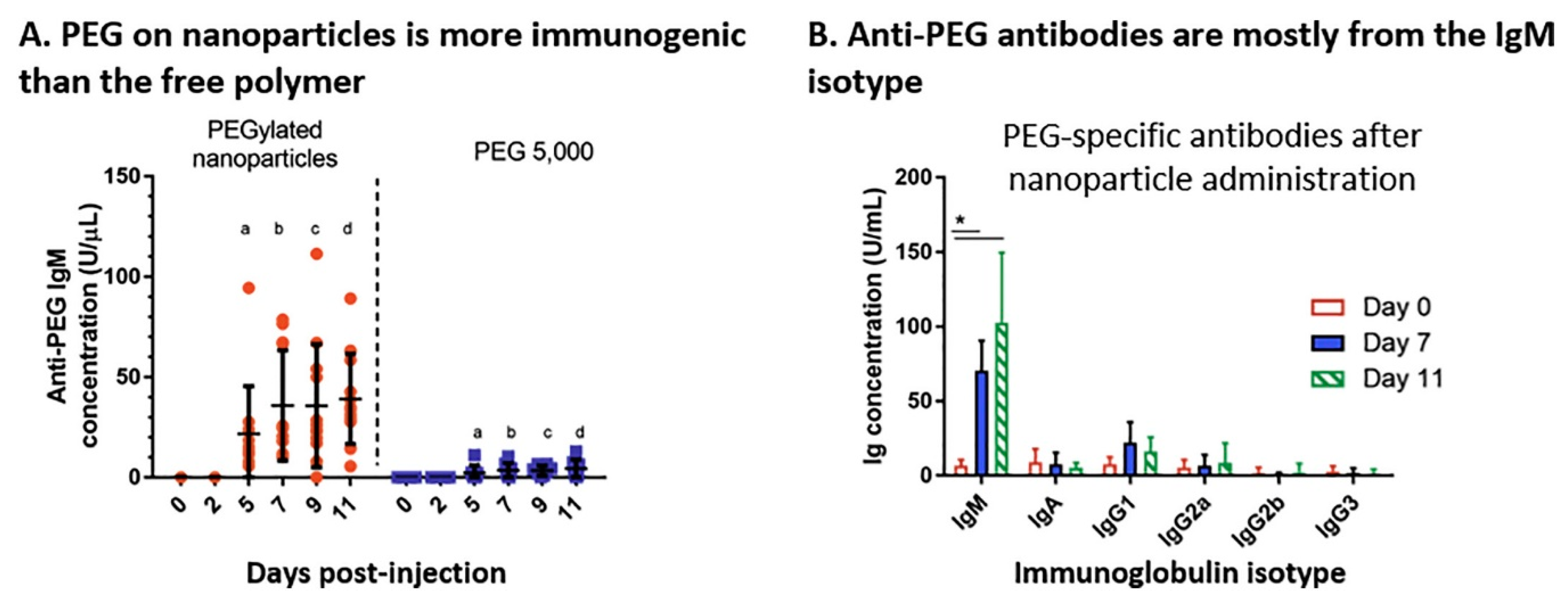
- Shimizu, T.; Ichihara, M.; Yoshioka, Y.; Ishida, T.; Nakagawa, S.; Kiwada, H. Intravenous Administration of Polyethylene Glycol-Coated (PEGylated) Proteins and PEGylated Adenovirus Elicits an Anti-PEG Immunoglobulin M Response. Biol. Pharm. Bull. 2012, 35, 1336–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poppenborg, S.M.; Wittmann, J.; Walther, W.; Brandenburg, G.; Krähmer, R.; Baumgart, J.; Leenders, F. Impact of anti-PEG IgM antibodies on the pharmacokinetics of pegylated asparaginase preparations in mice. Eur. J. Pharm. Sci. 2016, 91, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Toyofuku, W.M.; Scott, M.D. Immunogenicity of murine mPEG-red blood cells and the risk of anti-PEG antibodies in human blood donors. Exp. Hematol. 2017, 47, 36–47. [Google Scholar] [CrossRef]
- Yang, Q.; Lai, S.K. Anti-PEG immunity: Emergence, characteristics, and unaddressed questions. Wires. Nanomed. Nanobi. 2015, 7, 655–677. [Google Scholar] [CrossRef] [Green Version]
- Garay, R.P.; El-Gewely, R.; Armstrong, J.K.; Garratty, G.; Richette, P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin. Drug Deliv. 2012, 9, 1319–1323. [Google Scholar] [CrossRef]
- Cheng, T.-L.; Wu, P.-Y.; Wu, M.-F.; Chern, J.-W.; Roffler, S.R. Accelerated Clearance of Polyethylene Glycol-Modified Proteins by Anti-Polyethylene Glycol IgM. Bioconjugate Chem. 1999, 10, 520–528. [Google Scholar] [CrossRef]
- Wang, C.; Cheng, X.; Su, Y.; Pei, Y.; Song, Y.; Jiao, J.; Huang, Z.; Ma, Y.; Dong, Y.; Yao, Y.; et al. Accelerated blood clearance phenomenon upon cross-administration of PEGylated nanocarriers in beagle dogs. Int. J. Nanomed. 2015, 10, 3533–3545. [Google Scholar]
- Ishida, T.; Ichihara, M.; Wang, X.; Yamamoto, K.; Kimura, J.; Majima, E.; Kiwada, H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control Release 2006, 112, 15–25. [Google Scholar] [CrossRef]
- Lubich, C.; Allacher, P.; de la Rosa, M.; Bauer, A.; Prenninger, T.; Horling, F.M.; Siekmann, J.; Oldenburg, J.; Scheiflinger, F.; Reipert, B.M. The Mystery of Antibodies Against Polyethylene Glycol (PEG)—What do we Know? Pharm. Res. 2016, 33, 2239–2249. [Google Scholar] [CrossRef]
- Grenier, P.; de Oliveira Viana, I.M.; Lima, E.M.; Bertrand, N. Anti-polyethylene glycol antibodies alter the protein corona deposited on nanoparticles and the physiological pathways regulating their fate in vivo. J. Control. Release 2018, 287, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kiwada, H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm. 2008, 354, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, K.; Hamano, M.; Ma, H.; Kawano, K.; Maitani, Y.; Aoshi, T.; Ishii, K.J.; Yokoyama, M. Hydrophobic blocks of PEG-conjugates play a significant role in the accelerated blood clearance (ABC) phenomenon. J. Control Release 2013, 165, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.; Piperigkou, Z.; Karamanou, K.; Engin, A.B.; Docea, A.O.; Constantin, C.; Negrei, C.; Nikitovic, D.; Tsatsakis, A. Protein bio-corona: Critical issue in immune nanotoxicology. Arch. Toxicol. 2017, 91, 1031–1048. [Google Scholar] [CrossRef] [Green Version]
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine (Lond.) 2016, 11, 81–100. [Google Scholar] [CrossRef] [Green Version]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.-C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Bhushan, B.; Khanadeev, V.; Khlebtsov, B.; Khlebtsov, N.; Gopinath, P. Impact of albumin based approaches in nanomedicine: Imaging, targeting and drug delivery. Adv. Colloid Interface Sci. 2017, 246, 13–39. [Google Scholar] [CrossRef]
- Guan, X.; Chang, Y.; Sun, J.; Song, J.; Xie, Y. Engineered Hsp Protein Nanocages for siRNA Delivery. Macromol. Biosci. 2018, 18, 1800013. [Google Scholar] [CrossRef]
- Florczak, A.; Mackiewicz, A.; Dams-Kozlowska, H. Functionalized Spider Silk Spheres As Drug Carriers for Targeted Cancer Therapy. Biomacromolecules 2014, 15, 2971–2981. [Google Scholar] [CrossRef]
- González, A.; Tártara, L.I.; Palma, S.D.; Alvarez Igarzabal, C.I. Crosslinked soy protein films and their application as ophthalmic drug delivery system. Mater. Sci. Eng. C 2015, 51, 73–79. [Google Scholar] [CrossRef]
- McMasters, J.; Panitch, A. Collagen-Binding Nanoparticles for Extracellular Anti-Inflammatory Peptide Delivery Decrease Platelet Activation, Promote Endothelial Migration, and Suppress Inflammation. Acta Biomater. 2017, 49, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhandhukia, J.P.; Shi, P.; Peddi, S.; Li, Z.; Aluri, S.; Ju, Y.; Brill, D.; Wang, W.; Janib, S.M.; Lin, Y.-A.; et al. Bifunctional Elastin-like Polypeptide Nanoparticles Bind Rapamycin and Integrins and Suppress Tumor Growth in Vivo. Bioconjug. Chem. 2017, 28, 2715–2728. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Xia, S.; Wu, K.; Huang, Z.; Chen, H.; Chen, J.; Zhang, J. A pH/Enzyme-responsive tumor-specific delivery system for doxorubicin. Biomaterials 2010, 31, 6309–6316. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Xiao, W.; Lam, K.S. Tumor-targeting peptides from combinatorial libraries. Adv. Drug Deliv. Rev. 2017, 110–111, 13–37. [Google Scholar] [CrossRef]
- Le Joncour, V.; Laakkonen, P. Seek & Destroy, use of targeting peptides for cancer detection and drug delivery. Bioorganic Med. Chem. 2018, 26, 2797–2806. [Google Scholar]
- Feron, O. Tumor-Penetrating Peptides: A Shift from Magic Bullets to Magic Guns. Sci. Transl. Med. 2010, 2, ps26–ps34. [Google Scholar] [CrossRef]
- Yin, H.; Yang, J.; Zhang, Q.; Yang, J.; Wang, H.; Xu, J.; Zheng, J. iRGD as a tumor-penetrating peptide for cancer therapy (Review). Mol. Med. Rep. 2017, 15, 2925–2930. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, P.; Haldar, M.K.; Karandish, F.; Confeld, M.; Hossain, R.; Borowicz, P.; Gange, K.; Xia, L.; Sarkar, K.; Mallik, S. Tissue-Penetrating, Hypoxia-Responsive Echogenic Polymersomes For Drug Delivery To Solid Tumors. Chem. A Eur. J. 2018, 24, 12490–12494. [Google Scholar] [CrossRef]
- Carter, T.; Mulholland, P.; Chester, K. Antibody-targeted nanoparticles for cancer treatment. Immunotherapy 2016, 8, 941–958. [Google Scholar] [CrossRef]
- Su, C.-Y.; Chen, M.; Chen, L.-C.; Ho, Y.-S.; Ho, H.-O.; Lin, S.-Y.; Chuang, K.-H.; Sheu, M.-T. Bispecific antibodies (anti-mPEG/anti-HER2) for active tumor targeting of docetaxel (DTX)-loaded mPEGylated nanocarriers to enhance the chemotherapeutic efficacy of HER2-overexpressing tumors. Drug Deliv. 2018, 25, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Al-Bayati, K.; Ho, E.A. Development of antibody-modified chitosan nanoparticles for the targeted delivery of siRNA across the blood-brain barrier as a strategy for inhibiting HIV replication in astrocytes. Drug Deliv. Transl. Res. 2017, 7, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Gothwal, A.; Kesharwani, P.; Alsaab, H.; Iyer, A.K.; Gupta, U. Dendrimer nanoarchitectures for cancer diagnosis and anticancer drug delivery. Drug Discov. Today 2017, 22, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, Q.; Chang, H.; Xiao, J.; Cheng, Y. Stimuli-responsive dendrimers in drug delivery. Biomater. Sci. 2016, 4, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, H.H.; Cerqueira, S.R.; Sousa, N.; Oliveira, J.M.; Reis, R.L.; Zorec, R. The uptake, retention and clearance of drug-loaded dendrimer nanoparticles in astrocytes – electrophysiological quantification. Biomater. Sci. 2018, 6, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Haque, S.; McLeod, V.M.; Jones, S.; Fung, S.; Whittaker, M.; McIntosh, M.; Pouton, C.; Owen, D.J.; Porter, C.J.H.; Kaminskas, L.M. Effect of increased surface hydrophobicity via drug conjugation on the clearance of inhaled PEGylated polylysine dendrimers. Eur. J. Pharm. Biopharm. 2017, 119, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Loftus, A.L.; Mulley, G.; Jenkins, A.T.A. A Thin Film Detection/Response System for Pathogenic Bacteria. J. Am. Chem. Soc. 2010, 132, 6566–6570. [Google Scholar] [CrossRef]
- Xiong, M.-H.; Li, Y.-J.; Bao, Y.; Yang, X.-Z.; Hu, B.; Wang, J. Bacteria-Responsive Multifunctional Nanogel for Targeted Antibiotic Delivery. Adv. Mater. 2012, 24, 6175–6180. [Google Scholar] [CrossRef]
- De la Rica, R.; Aili, D.; Stevens, M.M. Enzyme-responsive nanoparticles for drug release and diagnostics. Adv. Drug Deliv. Rev. 2012, 64, 967–978. [Google Scholar] [CrossRef]
- Enzyme-responsive materials for wound infection diagnosis. Biotechnol. Bioeng. 2016, 113, 2534. [CrossRef] [Green Version]
- Cai, X.; Luo, Y.; Zhang, W.; Du, D.; Lin, Y. pH-Sensitive ZnO Quantum Dots–Doxorubicin Nanoparticles for Lung Cancer Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2016, 8, 22442–22450. [Google Scholar] [CrossRef] [PubMed]
- Landarani-Isfahani, A.; Moghadam, M.; Mohammadi, S.; Royvaran, M.; Moshtael-Arani, N.; Rezaei, S.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I. Elegant pH-Responsive Nanovehicle for Drug Delivery Based on Triazine Dendrimer Modified Magnetic Nanoparticles. Langmuir 2017, 33, 8503–8515. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Luo, Z.; Zhang, J.; Luo, T.; Zhou, J.; Zhao, X.; Cai, K. Hollow mesoporous silica nanoparticles facilitated drug delivery via cascade pH stimuli in tumor microenvironment for tumor therapy. Biomaterials 2016, 83, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Haldar, M.K.; Katti, P.; Dawes, C.; You, S.; Choi, Y.; Mallik, S. Hypoxia Responsive, Tumor Penetrating Lipid Nanoparticles for Delivery of Chemotherapeutics to Pancreatic Cancer Cell Spheroids. Bioconjug. Chem. 2016, 27, 1830–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, P.; Haldar, M.K.; You, S.; Choi, Y.; Mallik, S. Hypoxia-Responsive Polymersomes for Drug Delivery to Hypoxic Pancreatic Cancer Cells. Biomacromolecules 2016, 17, 2507–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, K.; Konar, A.; Kumar, B.S.H.; Koul, V. Lactoferrin-conjugated pH and redox-sensitive polymersomes based on PEG-S-S-PLA-PCL-OH boost delivery of bacosides to the brain. Nanoscale 2018, 10, 17781–17798. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Liu, G.; Hu, J.; Liu, S. Photo- and Reduction-Responsive Polymersomes for Programmed Release of Small and Macromolecular Payloads. Biomacromolecules 2018, 19, 2071–2081. [Google Scholar] [CrossRef]
- Law, B.; Tung, C.-H. Proteolysis: A biological process adapted in drug delivery, therapy, and imaging. Bioconjug. Chem. 2009, 20, 1683–1695. [Google Scholar] [CrossRef]
- Woo, G.L.Y.; Mittelman, M.W.; Santerre, J.P. Synthesis and characterization of a novel biodegradable antimicrobial polymer. Biomaterials 2000, 21, 1235–1246. [Google Scholar] [CrossRef]
- Minelli, C.; Lowe, S.B.; Stevens, M.M. Engineering nanocomposite materials for cancer therapy. Small 2010, 6, 2336–2357. [Google Scholar] [CrossRef]
- Said, S.S.; El-Halfawy, O.M.; El-Gowelli, H.M.; Aloufy, A.K.; Boraei, N.A.; El-Khordagui, L.K. Bioburden-responsive antimicrobial PLGA ultrafine fibers for wound healing. Eur. J. Pharm. Biopharm. 2012, 80, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, B.F. Proteases as Selective Activators of Triggered Drug Release: A Potential Answer to the Problem of Biomaterial-Associated Infections? J. Biotechnol. Biomater. 2012, 2, E111. [Google Scholar] [CrossRef]
- Tanihara, M.; Suzuki, Y.; Nishimura, Y.; Suzuki, K.; Kakimaru, Y.; Fukunishi, Y. A novel microbial infection-responsive drug release system. J. Pharm. Sci. 1999, 88, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Woo, G.L.Y.; Yang, M.L.; Yin, H.Q.; Jaffer, F.; Mittelman, M.W.; Santerre, J.P. Biological characterization of a novel biodegradable antimicrobial polymer synthesized with fluoroquinolones. J. Biomed. Mater. Res. 2002, 59, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Tanihara, M.; Nishimura, Y.; Suzuki, K.; Kakimaru, Y.; Shimizu, Y. A novel wound dressing with an antibiotic delivery system stimulated by microbial infection. ASAIO J. 1997, 43, M854–M857. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Wang, M.; Han, Y.; Hu, B.; Teng, L.; Zhou, J.; Zhang, H.; Chen, J. Enzyme-responsive mesoporous silica nanoparticles for tumor cells and mitochondria multistage-targeted drug delivery. Int. J. Nanomed. 2019, 14, 2533–2542. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Han, X.; Yang, Y.; Qiao, H.; Yu, Z.; Liu, Y.; Wang, J.; Tang, T. Bacteria-Targeting Nanoparticles with Microenvironment-Responsive Antibiotic Release To Eliminate Intracellular Staphylococcus aureus and Associated Infection. ACS Appl. Mater. Interfaces 2018, 10, 14299–14311. [Google Scholar] [CrossRef]
- Cai, R.; Chen, C. The Crown and the Scepter: Roles of the Protein Corona in Nanomedicine. Adv. Mater. 2018, 1805740. [Google Scholar] [CrossRef]
- Leroux, J.C.; De Jaeghere, F.; Anner, B.; Doelker, E.; Gurny, R. An investigation on the role of plasma and serum opsonins on the internalization of biodegradable poly(D,L-lactic acid) nanoparticles by human monocytes. Life Sci. 1995, 57, 695–703. [Google Scholar] [CrossRef]
- Nguyen, V.H.; Lee, B.-J. Protein corona: A new approach for nanomedicine design. Int. J. Nanomed. 2017, 12, 3137–3151. [Google Scholar] [CrossRef] [Green Version]
- Mahmoudi, M.; Lynch, I.; Ejtehadi, M.R.; Monopoli, M.P.; Bombelli, F.B.; Laurent, S. Protein-nanoparticle interactions: Opportunities and challenges. Chem. Rev. 2011, 111, 5610–5637. [Google Scholar] [CrossRef] [PubMed]
- Ehrenberg, M.S.; Friedman, A.E.; Finkelstein, J.N.; Oberdörster, G.; McGrath, J.L. The influence of protein adsorption on nanoparticle association with cultured endothelial cells. Biomaterials 2009, 30, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggård, T.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.C.; Giljohann, D.A.; Daniel, W.L.; Zheng, D.; Prigodich, A.E.; Mirkin, C.A. Scavenger Receptors Mediate Cellular Uptake of Polyvalent Oligonucleotide-Functionalized Gold Nanoparticles. Bioconjug. Chem. 2010, 21, 2250–2256. [Google Scholar] [CrossRef] [Green Version]
- Seong, S.-Y.; Matzinger, P. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 2004, 4, 469–478. [Google Scholar] [CrossRef]
- Moyano, D.F.; Liu, Y.; Peer, D.; Rotello, V.M. Modulation of Immune Response Using Engineered Nanoparticle Surfaces. Small 2016, 12, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Hulander, M.; Lundgren, A.; Berglin, M.; Ohrlander, M.; Lausmaa, J.; Elwing, H. Immune complement activation is attenuated by surface nanotopography. Int. J. Nanomed. 2011, 6, 2653–2666. [Google Scholar] [CrossRef] [Green Version]
- Caracciolo, G.; Palchetti, S.; Colapicchioni, V.; Digiacomo, L.; Pozzi, D.; Capriotti, A.L.; La Barbera, G.; Laganà, A. Stealth effect of biomolecular corona on nanoparticle uptake by immune cells. Langmuir 2015, 31, 10764–10773. [Google Scholar] [CrossRef]
- Pozzi, D.; Colapicchioni, V.; Caracciolo, G.; Piovesana, S.; Capriotti, A.L.; Palchetti, S.; De Grossi, S.; Riccioli, A.; Amenitsch, H.; Laganà, A. Effect of polyethyleneglycol (PEG) chain length on the bio-nano-interactions between PEGylated lipid nanoparticles and biological fluids: From nanostructure to uptake in cancer cells. Nanoscale 2014, 6, 2782–2792. [Google Scholar] [CrossRef]
- Aggarwal, P.; Hall, J.B.; McLeland, C.B.; Dobrovolskaia, M.A.; McNeil, S.E. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 2009, 61, 428–437. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; Neun, B.W.; Man, S.; Ye, X.; Hansen, M.; Patri, A.K.; Crist, R.M.; McNeil, S.E. Protein Corona Composition Does Not Accurately Predict Hematocompatibility of Colloidal Gold Nanoparticles. Nanomedicine 2014, 10, 1453–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, D.; Sundaram, S.K.; Teeguarden, J.G.; Riley, B.J.; Fifield, L.S.; Jacobs, J.M.; Addleman, S.R.; Kaysen, G.A.; Moudgil, B.M.; Weber, T.J. Adsorbed proteins influence the biological activity and molecular targeting of nanomaterials. Toxicol. Sci. 2007, 100, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyano, D.F.; Saha, K.; Prakash, G.; Yan, B.; Kong, H.; Yazdani, M.; Rotello, V.M. Fabrication of Corona-Free Nanoparticles with Tunable Hydrophobicity. ACS Nano 2014, 8, 6748–6755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Liu, S.; Bai, T.; Keefe, A.J.; Zhang, L.; Ella-Menye, J.-R.; Li, Y.; Jiang, S. Poly(carboxybetaine) nanomaterials enable long circulation and prevent polymer-specific antibody production. Nano Today 2014, 9, 10–16. [Google Scholar] [CrossRef]
- Clemments, A.M.; Botella, P.; Landry, C.C. Protein Adsorption From Biofluids on Silica Nanoparticles: Corona Analysis as a Function of Particle Diameter and Porosity. ACS Appl. Mater. Interfaces 2015, 7, 21682–21689. [Google Scholar] [CrossRef] [Green Version]
- Abdelkhaliq, A.; van der Zande, M.; Punt, A.; Helsdingen, R.; Boeren, S.; Vervoort, J.J.M.; Rietjens, I.M.C.M.; Bouwmeester, H. Impact of nanoparticle surface functionalization on the protein corona and cellular adhesion, uptake and transport. J. Nanobiotechnol. 2018, 16, 70. [Google Scholar] [CrossRef] [Green Version]
- Yatim, K.M.; Lakkis, F.G. A Brief Journey through the Immune System. Clin. J. Am. Soc. Nephrol. 2015, 10, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Hume, D.A. The mononuclear phagocyte system. Curr. Opin. Immunol. 2006, 18, 49–53. [Google Scholar] [CrossRef]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle uptake: The phagocyte problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [Green Version]
- Elci, S.G.; Jiang, Y.; Yan, B.; Kim, S.T.; Saha, K.; Moyano, D.F.; Yesilbag Tonga, G.; Jackson, L.C.; Rotello, V.M.; Vachet, R.W. Surface Charge Controls the Suborgan Biodistributions of Gold Nanoparticles. ACS Nano 2016, 10, 5536–5542. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.M.; Endres, R.G. The Mechanism of Phagocytosis: Two Stages of Engulfment. Biophys. J. 2014, 107, 1542–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Choi, H.; Zhou, R.; Chen, I.-W. RES blockade: A strategy for boosting efficiency of nanoparticle drug. Nano Today 2015, 10, 11–21. [Google Scholar] [CrossRef]
- Campbell, F.; Bos, F.L.; Sieber, S.; Arias-Alpizar, G.; Koch, B.E.; Huwyler, J.; Kros, A.; Bussmann, J. Directing Nanoparticle Biodistribution through Evasion and Exploitation of Stab2-Dependent Nanoparticle Uptake. ACS Nano 2018, 12, 2138–2150. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Mg, S.; Mayor, S. Endocytosis unplugged: Multiple ways to enter the cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-Independent Pathways of Endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance Properties of Nano-sized Particles and Molecules as Imaging Agents: Considerations and Caveats. Nanomedicine (Lond) 2008, 3, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Sadauskas, E.; Danscher, G.; Stoltenberg, M.; Vogel, U.; Larsen, A.; Wallin, H. Protracted elimination of gold nanoparticles from mouse liver. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 162–169. [Google Scholar] [CrossRef]
- Gad, S.C.; Sharp, K.L.; Montgomery, C.; Payne, J.D.; Goodrich, G.P. Evaluation of the Toxicity of Intravenous Delivery of Auroshell Particles (Gold–Silica Nanoshells). Int. J. Toxicol. 2012, 31, 584–594. [Google Scholar] [CrossRef]
- Huang, X.; Li, L.; Liu, T.; Hao, N.; Liu, H.; Chen, D.; Tang, F. The Shape Effect of Mesoporous Silica Nanoparticles on Biodistribution, Clearance, and Biocompatibility in Vivo. ACS Nano 2011, 5, 5390–5399. [Google Scholar] [CrossRef] [PubMed]
- Arvizo, R.R.; Miranda, O.R.; Moyano, D.F.; Walden, C.A.; Giri, K.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Reid, J.M.; Mukherjee, P. Modulating Pharmacokinetics, Tumor Uptake and Biodistribution by Engineered Nanoparticles. PLoS ONE 2011, 6, e24374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souris, J.S.; Lee, C.-H.; Cheng, S.-H.; Chen, C.-T.; Yang, C.-S.; Ho, J.A.; Mou, C.-Y.; Lo, L.-W. Surface Charge-Mediated Rapid Hepatobiliary Excretion of Mesoporous Silica Nanoparticles. Biomaterials 2010, 31, 5564–5574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA/CDER. Drug Products, Including Biological Products, that Contain Nanomaterials—Guidance for Industry; US Department of Health and Human Services: Silver Spring, MD, USA, 2017; Volume 29. [CrossRef]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine (Lond.) 2016, 11, 673–692. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug Delivery: Is the Enhanced Permeability and Retention Effect Sufficient for Curing Cancer? Bioconj. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef]
- Kagan, V.E.; Konduru, N.V.; Feng, W.; Allen, B.L.; Conroy, J.; Volkov, Y.; Vlasova, I.I.; Belikova, N.A.; Yanamala, N.; Kapralov, A.; et al. Carbon nanotubes degraded by neutrophil myeloperoxidase induce less pulmonary inflammation. Nat. Nanotechnol. 2010, 5, 354–359. [Google Scholar] [CrossRef]
- Sukhanova, A.; Bozrova, S.; Sokolov, P.; Berestovoy, M.; Karaulov, A.; Nabiev, I. Dependence of Nanoparticle Toxicity on Their Physical and Chemical Properties. Nanoscale Res. Lett. 2018, 13, 44. [Google Scholar] [CrossRef] [Green Version]
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Moghimi, S.M.; Simberg, D. Translational gaps in animal models of human infusion reactions to nanomedicines. Nanomedicine 2018, 13, 973–975. [Google Scholar] [CrossRef]
- Hua, S.; de Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front Pharm. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.K.; Simon, J.; Rosenauer, C.; Mailänder, V.; Morsbach, S.; Landfester, K. The Transferability from Animal Models to Humans: Challenges Regarding Aggregation and Protein Corona Formation of Nanoparticles. Biomacromolecules 2018, 19, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Haque, E.; Ward, A.C. Zebrafish as a Model to Evaluate Nanoparticle Toxicity. Nanomaterials (Basel) 2018, 8, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Pomeren, M.; Peijnenburg, W.J.G.M.; Vlieg, R.C.; van Noort, S.J.T.; Vijver, M.G. The biodistribution and immuno-responses of differently shaped non-modified gold particles in zebrafish embryos. Nanotoxicology 2019, 13, 558–571. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.-S. Zebrafish: A complete animal model to enumerate the nanoparticle toxicity. J. Nanobiotechnology 2016, 14, 65. [Google Scholar] [CrossRef] [Green Version]
- Sieber, S.; Grossen, P.; Detampel, P.; Siegfried, S.; Witzigmann, D.; Huwyler, J. Zebrafish as an early stage screening tool to study the systemic circulation of nanoparticulate drug delivery systems in vivo. J. Control. Release 2017, 264, 180–191. [Google Scholar] [CrossRef]
- Evensen, L.; Johansen, P.L.; Koster, G.; Zhu, K.; Herfindal, L.; Speth, M.; Fenaroli, F.; Hildahl, J.; Bagherifam, S.; Tulotta, C.; et al. Zebrafish as a model system for characterization of nanoparticles against cancer. Nanoscale 2015, 8, 862–877. [Google Scholar] [CrossRef] [Green Version]
- Novoa, B.; Figueras, A. Zebrafish: Model for the study of inflammation and the innate immune response to infectious diseases. Adv. Exp. Med. Biol. 2012, 946, 253–275. [Google Scholar]
- Meeker, N.D.; Trede, N.S. Immunology and zebrafish: Spawning new models of human disease. Dev. Comp. Immunol. 2008, 32, 745–757. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Farhangrazi, Z.S. Just so stories: The random acts of anti-cancer nanomedicine performance. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1661–1666. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Selvan, S.T.; Yang, Y.; Kim, M.J.; Yi, D.K.; Kwon, I.C.; Kim, K. Engineering nanoparticle strategies for effective cancer immunotherapy. Biomaterials 2018, 178, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Mink, J.G.; Radl, J.; van den Berg, P.; Haaijman, J.J.; van Zwieten, M.J.; Benner, R. Serum immunoglobulins in nude mice and their heterozygous littermates during ageing. Immunology 1980, 40, 539–545. [Google Scholar]
- Sobczynski, D.J.; Eniola-Adefeso, O. IgA and IgM protein primarily drive plasma corona-induced adhesion reduction of PLGA nanoparticles in human blood flow. Bioeng. Transl. Med. 2017, 2, 180–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, V.P.; Gifford, G.B.; Chen, F.; Benasutti, H.; Wang, G.; Groman, E.V.; Scheinman, R.; Saba, L.; Moghimi, S.M.; Simberg, D. Immunoglobulin deposition on biomolecule corona determines complement opsonisation efficiency of preclinical and clinical nanoparticles. Nat. Nanotechnol. 2019, 14, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Roberts, R.A.; Robbins, G.R.; Perry, J.L.; Kai, M.P.; Chen, K.; Bo, T.; Napier, M.E.; Ting, J.P.Y.; DeSimone, J.M.; et al. Nanoparticle clearance is governed by Th1/Th2 immunity and strain background. J. Clin. Investig. 2013, 123, 3061–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, S.; DeGiovanni, P.-J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Kai, M.P.; Brighton, H.E.; Fromen, C.A.; Shen, T.W.; Luft, J.C.; Luft, Y.E.; Keeler, A.W.; Robbins, G.R.; Ting, J.P.Y.; Zamboni, W.C.; et al. Tumor Presence Induces Global Immune Changes and Enhances Nanoparticle Clearance. ACS Nano 2016, 10, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Ipe, B.I.; Bawendi, M.G.; Frangioni, J.V. Renal Clearance of Nanoparticles. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Mangal, S.; Gao, W.; Li, T.; Zhou, Q.T. Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: Challenges and opportunities. Acta Pharmacol. Sin. 2017, 38, 782–797. [Google Scholar] [CrossRef]
- Palecanda, A.; Kobzik, L. Receptors for unopsonized particles: The role of alveolar macrophage scavenger receptors. Curr. Mol. Med. 2001, 1, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Brain, J.D. Mechanisms, measurement, and significance of lung macrophage function. Environ. Health Perspect. 1992, 97, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Izumi, H.; Yoshiura, Y.; Tomonaga, T.; Oyabu, T.; Myojo, T.; Kawai, K.; Yatera, K.; Shimada, M.; Kubo, M.; et al. Evaluation of Pulmonary Toxicity of Zinc Oxide Nanoparticles Following Inhalation and Intratracheal Instillation. Int. J. Mol. Sci. 2016, 17, 1241. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.; Hodgson, A.; Warren, J.; Guo, C.; Smith, R. Size-dependent deposition of inhaled nanoparticles in the rat respiratory tract using a new nose-only exposure system. Aerosol Sci. Technol. 2016, 50, 1–10. [Google Scholar] [CrossRef]
- Kreyling, W.G.; Möller, W.; Holzwarth, U.; Hirn, S.; Wenk, A.; Schleh, C.; Schäffler, M.; Haberl, N.; Gibson, N.; Schittny, J.C. Age-Dependent Rat Lung Deposition Patterns of Inhaled 20 Nanometer Gold Nanoparticles and their Quantitative Biokinetics in Adult Rats. ACS Nano 2018, 12, 7771–7790. [Google Scholar] [CrossRef] [Green Version]
- Oberdörster, G.; Kuhlbusch, T.A.J. In vivo effects: Methodologies and biokinetics of inhaled nanomaterials. NanoImpact 2018, 10, 38–60. [Google Scholar] [CrossRef]
- Takenaka, S.; Möller, W.; Semmler-Behnke, M.; Karg, E.; Wenk, A.; Schmid, O.; Stoeger, T.; Jennen, L.; Aichler, M.; Walch, A.; et al. Efficient Internalization and Intracellular Translocation of Inhaled Gold Nanoparticles in Rat Alveolar Macrophages. Available online: https://0-www-futuremedicine-com.brum.beds.ac.uk/doi/abs/10.2217/nnm.11.152 (accessed on 2 October 2019).
- Labens, R.; Daniel, C.; Hall, S.; Xia, X.-R.; Schwarz, T. Effect of intra-articular administration of superparamagnetic iron oxide nanoparticles (SPIONs) for MRI assessment of the cartilage barrier in a large animal model. PLoS ONE 2017, 12, e0190216. [Google Scholar] [CrossRef] [Green Version]
- Whitmire, R.E.; Wilson, D.S.; Singh, A.; Levenston, M.E.; Murthy, N.; García, A.J. Self-assembling nanoparticles for intra-articular delivery of anti-inflammatory proteins. Biomaterials 2012, 33, 7665–7675. [Google Scholar] [CrossRef] [Green Version]
- Geiger, B.C.; Grodzinsky, A.J.; Hammond, P.T. Designing Drug Delivery Systems for Articular Joints. Chem. Eng. Progress 2018, 114, 46–51. [Google Scholar]
- Holyoak, D.T.; Tian, Y.F.; van der Meulen, M.C.; Singh, A. Osteoarthritis: Pathology, mouse models, and nanoparticle injectable systems for targeted treatment. Ann. Biomed. Eng. 2016, 44, 2062–2075. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Agarwal, R.; Diaz-Ruiz, C.A.; Willett, N.J.; Wang, P.; Lee, L.A.; Wang, Q.; Guldberg, R.E.; García, A.J. Nano-engineered particles for enhanced intra-articular retention and delivery of proteins. Adv. Healthc Mater. 2014, 3, 1562–1567. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bendele, A.M.; Blanks, R.C.; Bodick, N. Sustained efficacy of a single intra-articular dose of FX006 in a rat model of repeated localized knee arthritis. Osteoarthr. Cartil. 2015, 23, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Jordan, J.M. Epidemiology of Osteoarthritis. Clin. Geriatr. Med. 2010, 26, 355–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.M. Animal Models of Osteoarthritis: Comparisons and Key Considerations. Vet. Pathol. 2015, 52, 803–818. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Domb, A.; Quellec, P.; Blunk, T.; Müller, R.H.; Verbavatz, J.M.; Langer, R. The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres. Adv. Drug Deliv. Rev. 1995, 16, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.; Gao, X.; Hu, Q.; Jiang, D.; Feng, X.; Zhang, X.; Song, Q.; Yao, L.; Huang, M.; Jiang, X.; et al. iNGR-modified PEG-PLGA nanoparticles that recognize tumor vasculature and penetrate gliomas. Biomaterials 2014, 35, 4319–4332. [Google Scholar] [CrossRef]
- Lohcharoenkal, W.; Wang, L.; Chen, Y.C.; Rojanasakul, Y. Protein Nanoparticles as Drug Delivery Carriers for Cancer Therapy. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Cucinotto, I.; Fiorillo, L.; Gualtieri, S.; Arbitrio, M.; Ciliberto, D.; Staropoli, N.; Grimaldi, A.; Luce, A.; Tassone, P.; Caraglia, M.; et al. Nanoparticle Albumin Bound Paclitaxel in the Treatment of Human Cancer: Nanodelivery Reaches Prime-Time? J. Drug Deliv. 2013, 2013. [Google Scholar] [CrossRef]
- Hoogenboezem, E.N.; Duvall, C.L. Harnessing albumin as a carrier for cancer therapies. Adv. Drug Deliv. Rev. 2018, 130, 73–89. [Google Scholar] [CrossRef]
- Jithan, A.; Madhavi, K.; Madhavi, M.; Prabhakar, K. Preparation and characterization of albumin nanoparticles encapsulating curcumin intended for the treatment of breast cancer. Int. J. Pharm. Investig. 2011, 1, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Chonn, A.; Cullis, P.R.; Devine, D.V. The role of surface charge in the activation of the classical and alternative pathways of complement by liposomes. J. Immunol. 1991, 146, 4234–4241. [Google Scholar] [PubMed]
- Nakase, I.; Futaki, S. Combined treatment with a pH-sensitive fusogenic peptide and cationic lipids achieves enhanced cytosolic delivery of exosomes. Sci. Rep. 2015, 5, 10112. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, Y.; Xie, R.; Gong, S. Tumor-targeted pH/redox dual-sensitive unimolecular nanoparticles for efficient siRNA delivery. J. Control Release 2017, 259, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalliola, S.; Repo, E.; Srivastava, V.; Heiskanen, J.P.; Sirviö, J.A.; Liimatainen, H.; Sillanpää, M. The pH sensitive properties of carboxymethyl chitosan nanoparticles cross-linked with calcium ions. Colloids Surf. B 2017, 153, 229–236. [Google Scholar] [CrossRef]
- Ma, J.; Hu, Z.; Wang, W.; Wang, X.; Wu, Q.; Yuan, Z. pH-Sensitive Reversible Programmed Targeting Strategy by the Self-Assembly/Disassembly of Gold Nanoparticles. Acs Appl. Mater. Interfaces 2017, 9, 16767–16777. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name | Formulation | Stage of Development | Targeted Disease State | Source |
|---|---|---|---|---|
| Mircera | Methoxypolyeletheneglycol-epoetin beta | FDA Approved | Anemia, chronic renal failure | [21] |
| PegIntron | Peginterferonalpha-2b (mPEG-interferon alpha-2b) | FDA Approved | HIV inflammation | [24] |
| Macugen®/Pegaptanib | PEGylated anti-VEGF aptamer (vascular endothelial growth factor) aptamer | FDA Approved | Anemia with chronic renal failure | [25] |
| Pegasys | PEGylated IFN alpha-2a protein | FDA Approved | Hepatitis B and C | [26] |
| Neulasta®/pegfilgrastim | PEGylated GCSF protein | FDA Approved | Leukopenia by chemotherapy | [27] |
| Somavert®/pegvisomant | PEGylated HGH receptor antagonist | FDA Approved | Acromegaly | [28] |
| Oncaspar®/pegaspargase | Polymer-protein conjugate PEGylated l-asparaginase | FDA Approved | Acute lymphocytic blood clot | [29] |
| Krystexxa®/pegloticase | Polymer-protein conjugate (PEGylated porcine-likeuricase) | FDA Approved | Chronic gout | [30] |
| Plegridy | Polymer-protein conjugate (PEGylated IFNbeta-1a) | FDA Approved | Multiple sclerosis | [31] |
| ADYNOVATE | Polymer-protein conjugate (PEGylated factor VIII) | FDA Approved | Hemophilia | [32] |
| ZILRETTA | triamcinolone acetonide encapsulated PLGA co-polymer matrix microspheres | FDA Approved | osteoarthritis-related knee pain | [33] |
| N/A | 124I-cRGDY-PEG-C dots | Clinical Trials | Cancer imaging | [34] |
| Caelyx/Doxil | Pegylated liposomal doxorubicin HCL | Clinical Trials | Ovarian Epithelial Cancer, Fallopian Tube Cancer, or Primary Peritoneal Cancer | [35] |
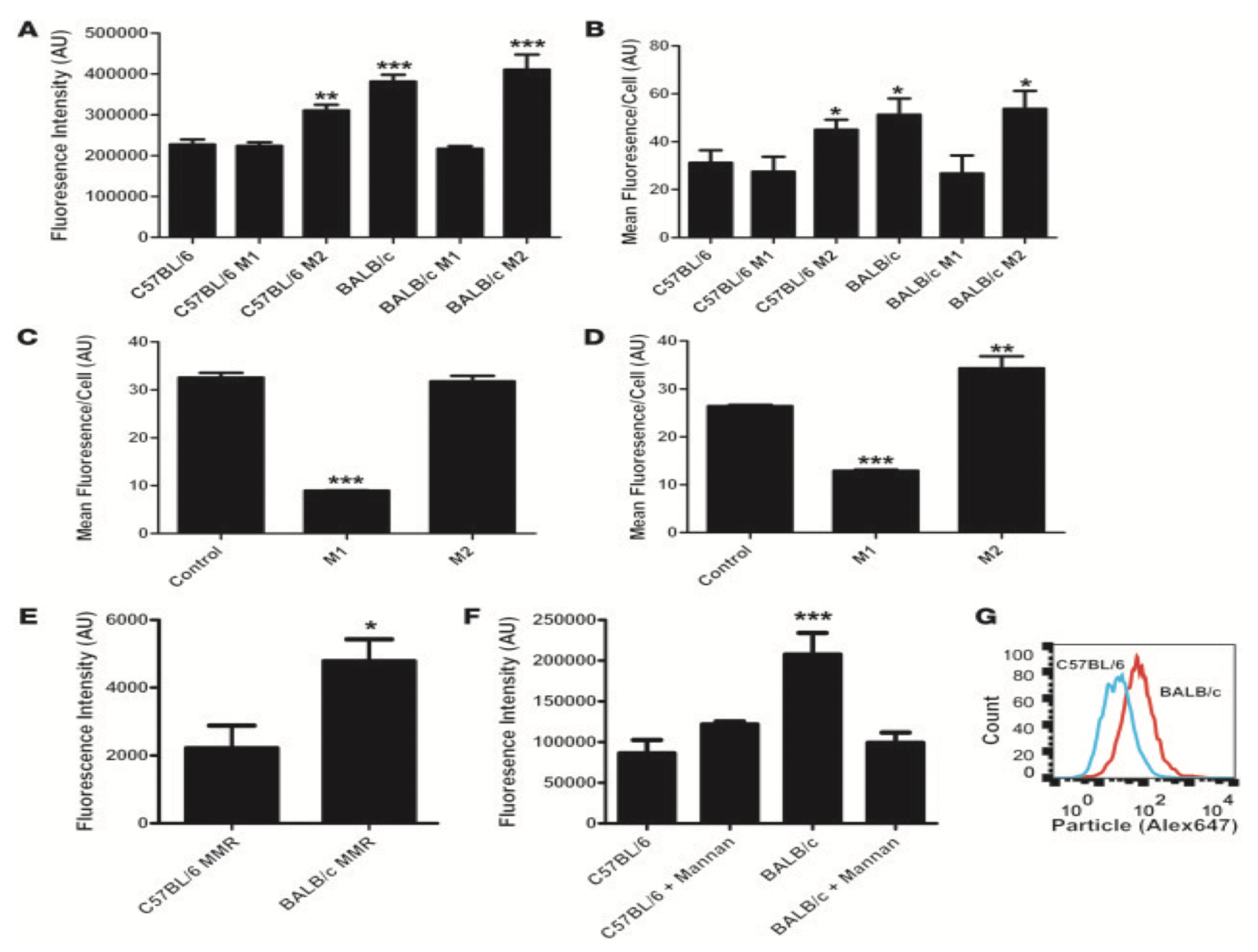
| Strain | Mature T Cells | Th1/Th2 | Mature B Cells | Immunoglobins | Macrophages | Rate of Clearance | |
|---|---|---|---|---|---|---|---|
| IgG | IgA | ||||||
| 0 | - | - | - | - | - | Defective | ↓↓ |
| Nude | - | - | Present/ Defective | - | - | + | ↓ |
| C57/Bl6 | + | Th1 Biased | + | + | + | + | ↑ |
| BALB/c | + | Th2 Biased | + | + | + | + | ↑↑ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shreffler, J.W.; Pullan, J.E.; Dailey, K.M.; Mallik, S.; Brooks, A.E. Overcoming Hurdles in Nanoparticle Clinical Translation: The Influence of Experimental Design and Surface Modification. Int. J. Mol. Sci. 2019, 20, 6056. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236056
Shreffler JW, Pullan JE, Dailey KM, Mallik S, Brooks AE. Overcoming Hurdles in Nanoparticle Clinical Translation: The Influence of Experimental Design and Surface Modification. International Journal of Molecular Sciences. 2019; 20(23):6056. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236056
Chicago/Turabian StyleShreffler, Jacob W., Jessica E. Pullan, Kaitlin M. Dailey, Sanku Mallik, and Amanda E. Brooks. 2019. "Overcoming Hurdles in Nanoparticle Clinical Translation: The Influence of Experimental Design and Surface Modification" International Journal of Molecular Sciences 20, no. 23: 6056. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236056