The Interplay between the Endocannabinoid System, Epilepsy and Cannabinoids


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Epilepsy and Neuroinflammation
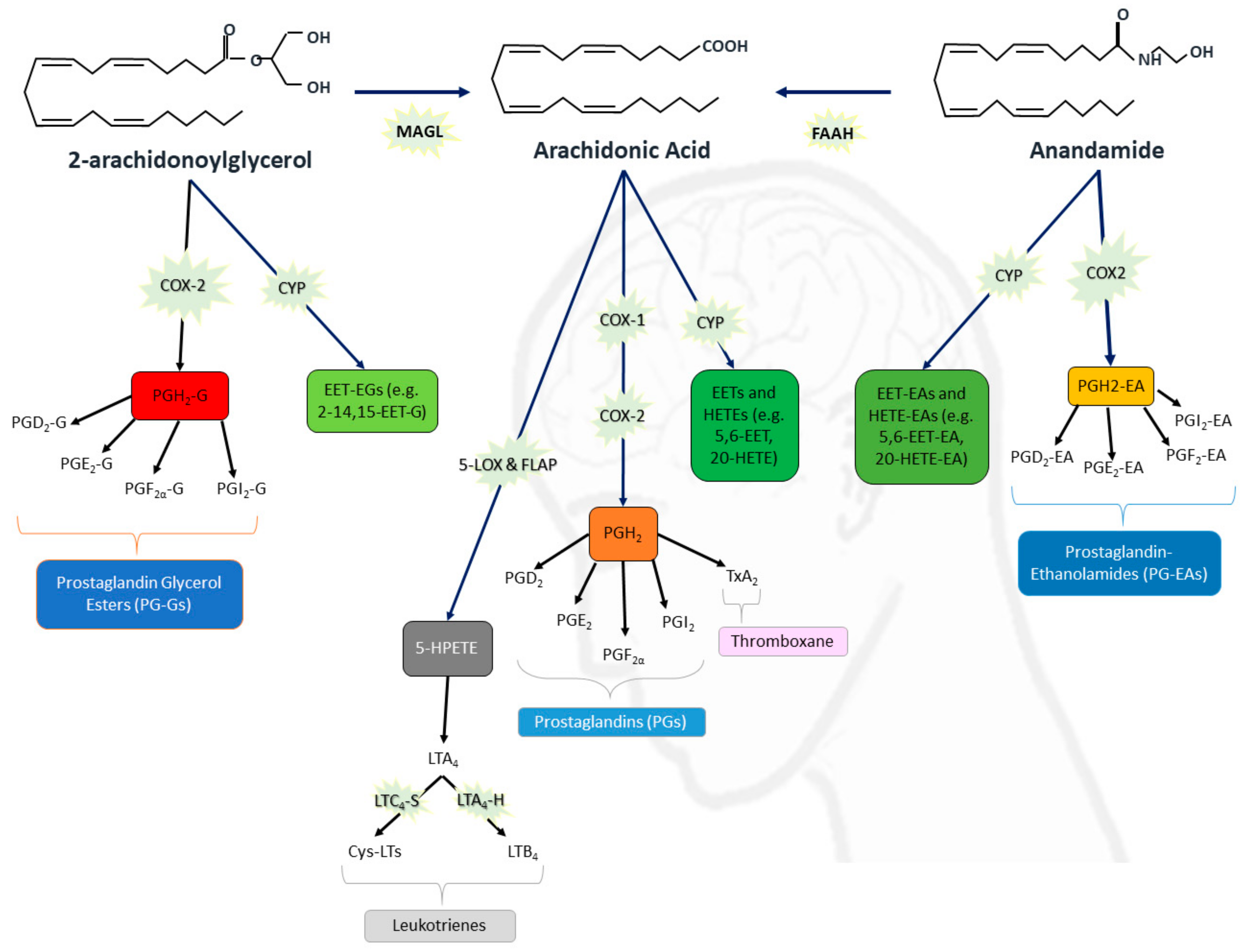
The Role of the Endocannabinoid System in Neuroinflammation and Epilepsy
3. Cannabinoids Are an Emerging Treatment for Drug-Resistant Epilepsy, Especially in Children
Cannabinoids Interact with the Endocannabinoid System
4. Biomarkers for Determining Neuroinflammation and Monitoring Cannabinoid Effects
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoylglycerol |
| 5-HTR | 5-hydroxytryptamine receptor |
| AA | Arachidonic acid |
| AEA | N-arachidonoyl-ethanolamine or anandamide |
| AEDs | Anti-epileptic drugs |
| CBD | Cannabidiol |
| CBDV | Cannabidivarin |
| Cys-LT | Cysteinyl leukotriene |
| CNS | Central nervous system |
| COX | Cyclooxygenase |
| CYP | Cytochrome P450 |
| EA | Ethanolamide |
| ECS | Endocannabinoid system |
| EET | Epoxyeicosatrienoic acid |
| ENT1 | Equilibrative nucleoside transporter |
| FAAH | Fatty acid amide hydrolase |
| FDA | Food and Drug Administration |
| GPR | G-protein coupled receptor |
| HETE | Hydroxyeicosatetraenoic acid |
| HPETE | 5-hydroperoxyeicosatetraenoic acid |
| IL-1β | Interleukin-1β |
| LOX | Lipoxygenase |
| LT | Leukotriene |
| MAGL | Monoacylglycerol lipase |
| mRNA | Messenger ribonucleic acid |
| miRNA | Micro ribonucleic acid |
| MRS | Magnetic Resonance Spectroscopy |
| PET | Positron Emission Tomography |
| PG | Prostaglandin |
| PGH2-G | Prostaglandin H2-glycerol ester |
| RNA | Ribonucleic acid |
| TGA | Therapeutic Goods Administration |
| TNF-α | Tumor necrosis factor-α |
| Δ9-THC or THC | Delta-9 tetrahydrocannabinol |
| TRPV | Transient Receptor Potential Vanilloid |
| TSPO | Translocator protein |
| VDAC | Voltage-dependent anion selective channel protein |
| WHO | World Health Organization |
References
- WHO. Neurological Disorders: Public Health Challenges; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- WHO. Epilepsy. Available online: https://www.who.int/en/news-room/fact-sheets/detail/epilepsy (accessed on 16 May 2019).
- Granata, T.; Marchi, N.; Carlton, E.; Ghosh, C.; Gonzalez-Martinez, J.; Alexopoulos, A.V.; Janigro, D. Management of the patient with medically refractory epilepsy. Expert Rev. Neurother. 2009, 9, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, M.J.; Corey, L.A.; Christensen, K.; Friis, M.L. Epileptic seizures and syndromes in twins: The importance of genetic factors. Epilepsy Res. 2003, 55, 137–146. [Google Scholar] [CrossRef]
- Helbig, I.; Scheffer, I.E.; Mulley, J.C.; Berkovic, S.F. Navigating the channels and beyond: Unravelling the genetics of the epilepsies. Lancet Neurol. 2008, 7, 231–245. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Rojas, A.; Chen, D.; Ganesh, T.; Varvel, N.H.; Dingledine, R. The COX-2/prostanoid signaling cascades in seizure disorders. Expert Opin. Ther. Targets 2019, 23, 1–13. [Google Scholar] [CrossRef]
- Hassan, S.; Eldeeb, K.; Millns, P.J.; Bennett, A.J.; Alexander, S.P.H.; Kendall, D.A. Cannabidiol enhances microglial phagocytosis via transient receptor potential (TRP) channel activation. Br. J. Pharmacol. 2014, 171, 2426–2439. [Google Scholar] [CrossRef]
- Ravizza, T.; Boer, K.; Redeker, S.; Spliet, W.G.M.; van Rijen, P.C.; Troost, D.; Vezzani, A.; Aronica, E. The IL-1β system in epilepsy-associated malformations of cortical development. Neurobiol. Dis. 2006, 24, 128–143. [Google Scholar] [CrossRef]
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef]
- Vezzani, A.; Lang, B.; Aronica, E. Immunity and Inflammation in Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 6, a022699. [Google Scholar] [CrossRef]
- Dow-Edwards, D.; Silva, L. Endocannabinoids in brain plasticity: Cortical maturation, HPA axis function and behavior. Brain Res. 2017, 1654, 157–164. [Google Scholar] [CrossRef]
- Soltesz, I.; Alger, B.E.; Kano, M.; Lee, S.-H.; Lovinger, D.M.; Ohno-Shosaku, T.; Watanabe, M. Weeding out bad waves: Towards selective cannabinoid circuit control in epilepsy. Nat. Rev. Neurosci. 2015, 16, 264. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef]
- Rodrigues, R.S.; Lourenço, D.M.; Paulo, S.L.; Mateus, J.M.; Ferreira, M.F.; Mouro, F.M.; Moreira, J.B.; Ribeiro, F.F.; Sebastião, A.M.; Xapelli, S. Cannabinoid Actions on Neural Stem Cells: Implications for Pathophysiology. Molecules 2019, 24, 1350. [Google Scholar] [CrossRef]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of Endogenous Cannabinoids in Synaptic Signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar] [CrossRef] [PubMed]
- Busquets-Garcia, A.; Bains, J.; Marsicano, G. CB1 Receptor Signaling in the Brain: Extracting Specificity from Ubiquity. Neuropsychopharmacology 2018, 43, 4–20. [Google Scholar] [CrossRef]
- Atwood, B.K.; Mackie, K. CB2: A cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 2010, 160, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.Q.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, ΨGPR53 and GPR55: GPR55 is extensively expressed in human brain. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef]
- Ludányi, A.; Erőss, L.; Czirják, S.; Vajda, J.; Halász, P.; Watanabe, M.; Palkovits, M.; Maglóczky, Z.; Freund, T.F.; Katona, I. Downregulation of the CB1 Cannabinoid Receptor and Related Molecular Elements of the Endocannabinoid System in Epileptic Human Hippocampus. J. Neurosci. 2008, 28, 2976–2990. [Google Scholar] [CrossRef] [PubMed]
- Romigi, A.; Bari, M.; Placidi, F.; Marciani, M.G.; Malaponti, M.; Torelli, F.; Izzi, F.; Prosperetti, C.; Zannino, S.; Corte, F.; et al. Cerebrospinal fluid levels of the endocannabinoid anandamide are reduced in patients with untreated newly diagnosed temporal lobe epilepsy. Epilepsia 2010, 51, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Stella, N.; Schweitzer, P.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Ives, D.; Ramesha, C.S. Synthesis of Prostaglandin E2 Ethanolamide from Anandamide by Cyclooxygenase-2. J. Biol. Chem. 1997, 272, 21181–21186. [Google Scholar] [CrossRef] [PubMed]
- Drazen, J.M.; Israel, E.; O’Byrne, P.M. Treatment of Asthma with Drugs Modifying the Leukotriene Pathway. N. Engl. J. Med. 1999, 340, 197–206. [Google Scholar] [CrossRef]
- Zelasko, S.; Arnold, W.R.; Das, A. Endocannabinoid metabolism by cytochrome P450 monooxygenases. Prostaglandins Other Lipid Mediat. 2015, 116, 112–123. [Google Scholar] [CrossRef]
- Urquhart, P.; Nicolaou, A.; Woodward, D.F. Endocannabinoids and their oxygenation by cyclo-oxygenases, lipoxygenases and other oxygenases. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 366–376. [Google Scholar] [CrossRef]
- Alhouayek, M.; Muccioli, G.G. COX-2-derived endocannabinoid metabolites as novel inflammatory mediators. Trends Pharmacol. Sci. 2014, 35, 284–292. [Google Scholar] [CrossRef]
- Kozak, K.R.; Rowlinson, S.W.; Marnett, L.J. Oxygenation of the Endocannabinoid, 2-Arachidonylglycerol, to Glyceryl Prostaglandins by Cyclooxygenase-2. J. Biol. Chem. 2000, 275, 33744–33749. [Google Scholar] [CrossRef]
- Carrasco, E.; Casper, D.; Werner, P. PGE2 receptor EP1 renders dopaminergic neurons selectively vulnerable to low-level oxidative stress and direct PGE2 neurotoxicity. J. Neurosci. Res. 2007, 85, 3109–3117. [Google Scholar] [CrossRef]
- Saleem, S.; Ahmad, A.S.; Maruyama, T.; Narumiya, S.; Doré, S. PGF2α FP Receptor Contributes to Brain Damage Following Transient Focal Brain Ischemia. Neurotox. Res. 2009, 15, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science (N. Y.) 2011, 334, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, M.J.; Blair, R.E.; Falenski, K.W.; Martin, B.R.; DeLorenzo, R.J. The Endogenous Cannabinoid System Regulates Seizure Frequency and Duration in a Model of Temporal Lobe Epilepsy. J. Pharmacol. Exp. Ther. 2003, 307, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugaya, Y.; Yamazaki, M.; Uchigashima, M.; Kobayashi, K.; Watanabe, M.; Sakimura, K.; Kano, M. Crucial Roles of the Endocannabinoid 2-Arachidonoylglycerol in the Suppression of Epileptic Seizures. Cell Rep. 2016, 16, 1405–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desjardins, P.; Sauvageau, A.; Bouthillier, A.; Navarro, D.; Hazell, A.S.; Rose, C.; Butterworth, R.F. Induction of astrocytic cyclooxygenase-2 in epileptic patients with hippocampal sclerosis. Neurochem. Int. 2003, 42, 299–303. [Google Scholar] [CrossRef] [Green Version]
- Serrano, G.E.; Lelutiu, N.; Rojas, A.; Cochi, S.; Shaw, R.; Makinson, C.D.; Wang, D.; FitzGerald, G.A.; Dingledine, R. Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J. Neurosci. 2011, 31, 14850–14860. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, V.; Strobel, H.W. Cytochrome P450-mediated metabolism in brain: Functional roles and their implications. Expert Opin. Drug Metab. Toxicol. 2013, 9, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Kommaddi, R.P.; Valli, K.; Ryder, D.; Hyde, T.M.; Kleinman, J.E.; Strobel, H.W.; Ravindranath, V. Drug metabolism in human brain: High levels of cytochrome P4503A43 in brain and metabolism of anti-anxiety drug alprazolam to its active metabolite. PLoS ONE 2008, 3, e2337. [Google Scholar] [CrossRef] [Green Version]
- Pai, H.V.; Kommaddi, R.P.; Chinta, S.J.; Mori, T.; Boyd, M.R.; Ravindranath, V. A Frameshift Mutation and Alternate Splicing in Human Brain Generate a Functional Form of the Pseudogene Cytochrome P4502D7 That Demethylates Codeine to Morphine. J. Biol. Chem. 2004, 279, 27383–27389. [Google Scholar] [CrossRef] [Green Version]
- Snider, N.T.; Kornilov, A.M.; Kent, U.M.; Hollenberg, P.F. Anandamide Metabolism by Human Liver and Kidney Microsomal Cytochrome P450 Enzymes to Form Hydroxyeicosatetraenoic and Epoxyeicosatrienoic Acid Ethanolamides. J. Pharmacol. Exp. Ther. 2007, 321, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Liu, J.-Y.; Timofeyev, V.; Qiu, H.; Hwang, S.H.; Tuteja, D.; Lu, L.; Yang, J.; Mochida, H.; Low, R.; et al. Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: Insight gained using metabolomic approaches. J. Mol. Cell. Cardiol. 2009, 47, 835–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science (N. Y.) 1999, 285, 1276–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westphal, C.; Konkel, A.; Schunck, W.-H. CYP-eicosanoids—A new link between omega-3 fatty acids and cardiac disease? Prostaglandins Other Lipid Mediat. 2011, 96, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Snider, N.T.; Nast, J.A.; Tesmer, L.A.; Hollenberg, P.F. A Cytochrome P450-Derived Epoxygenated Metabolite of Anandamide Is a Potent Cannabinoid Receptor 2-Selective Agonist. Mol. Pharmacol. 2009, 75, 965–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridar, C.; Snider, N.T.; Hollenberg, P.F. Anandamide Oxidation by Wild-Type and Polymorphically Expressed CYP2B6 and CYP2D6. Drug Metab. Dispos. 2011, 39, 782–788. [Google Scholar] [CrossRef] [Green Version]
- McDougle, D.R.; Kambalyal, A.; Meling, D.D.; Das, A. Endocannabinoids Anandamide and 2-Arachidonoylglycerol Are Substrates for Human CYP2J2 Epoxygenase. J. Pharmacol. Exp. Ther. 2014, 351, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-K.; Chen, J.; Imig, J.D.; Wei, S.; Hachey, D.L.; Guthi, J.S.; Falck, J.R.; Capdevila, J.H.; Harris, R.C. Identification of Novel Endogenous Cytochrome P450 Arachidonate Metabolites with High Affinity for Cannabinoid Receptors. J. Biol. Chem. 2008, 283, 24514–24524. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Chen, F.; Thakur, A.; Hong, H. Cysteinyl Leukotrienes and Their Receptors: Emerging Therapeutic Targets in Central Nervous System Disorders. CNS Neurosci. Ther. 2016, 22, 943–951. [Google Scholar] [CrossRef]
- Lenz, Q.F.; Arroyo, D.S.; Temp, F.R.; Poersch, A.B.; Masson, C.J.; Jesse, A.C.; Marafiga, J.R.; Reschke, C.R.; Iribarren, P.; Mello, C.F. Cysteinyl leukotriene receptor (CysLT) antagonists decrease pentylenetetrazol-induced seizures and blood–brain barrier dysfunction. Neuroscience 2014, 277, 859–871. [Google Scholar] [CrossRef]
- Gorter, J.A.; Aronica, E.; van Vliet, E.A. The Roof is Leaking and a Storm is Raging: Repairing the Blood–Brain Barrier in the Fight Against Epilepsy. Epilepsy Curr. 2019, 19, 177–181. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.-Y.; Zhang, X.-Y.; Wang, X.-R.; Xu, D.-M.; Chen, L.; Zhang, L.-H.; Fang, S.-H.; Lu, Y.-B.; Zhang, W.-P.; Wei, E.-Q. Cysteinyl leukotriene receptor 1 mediates LTD4-induced activation of mouse microglial cells in vitro. Acta Pharmacol. Sin. 2014, 35, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, T.; Downs, J.; Olson, H.; Bergin, A.M.; Smith, S.; Leonard, H. Cannabis for refractory epilepsy in children: A review focusing on CDKL5 Deficiency Disorder. Epilepsy Res. 2019, 151, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Maa, E.; Figi, P. The case for medical marijuana in epilepsy. Epilepsia 2014, 55, 783–786. [Google Scholar] [CrossRef]
- Russo, E.B.; Jiang, H.-E.; Li, X.; Sutton, A.; Carboni, A.; del Bianco, F.; Mandolino, G.; Potter, D.J.; Zhao, Y.-X.; Bera, S.; et al. Phytochemical and genetic analyses of ancient cannabis from Central Asia. J. Exp. Bot. 2008, 59, 4171–4182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Academies of Sciences, Engineering, and Medicine. The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research; The National Academies Press: Washington, DC, USA, 2017; p. 486. [Google Scholar] [CrossRef] [Green Version]
- Gloss, D.; Vickrey, B. Cannabinoids for epilepsy. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hall, W.; Stjepanović, D.; Caulkins, J.; Lynskey, M.; Leung, J.; Campbell, G.; Degenhardt, L. Public health implications of legalising the production and sale of cannabis for medicinal and recreational use. Lancet 2019, 394, 1580–1590. [Google Scholar] [CrossRef]
- Hill, K.P.; George, T.P. Cannabis legalisation in Canada: A crucial trial balloon. Lancet Psychiatry 2019, 6, 5–6. [Google Scholar] [CrossRef]
- Destrée, L.; Amiet, D.; Carter, A.; Lee, R.; Lorenzetti, V.; Segrave, R.; Youssef, G.; Solowij, N.; Yücel, M. Exploring the association of legalisation status of cannabis with problematic cannabis use and impulsivity in the USA. Drugs Context 2018, 7, 212541. [Google Scholar] [CrossRef]
- Martin, J.H.; Bonomo, Y.A. Medicinal cannabis in Australia: The missing links. Med. J. Aust. 2016, 204, 371–373. [Google Scholar] [CrossRef]
- Lowrey, T. ACT Legalises Personal Cannabis Use, becoming First Australian Jurisdiction to Do So. Available online: https://www.abc.net.au/news/2019-09-25/act-first-jurisdiction-to-legalise-personal-cannabis-use/11530104 (accessed on 26 September 2019).
- Adams, I.B.; Martin, B.R. Cannabis: Pharmacology and toxicology in animals and humans. Addiction 1996, 91, 1585–1614. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Cilio, M.R.; Cross, H.; Fernandez-Ruiz, J.; French, J.; Hill, C.; Katz, R.; Di Marzo, V.; Jutras-Aswad, D.; Notcutt, W.G.; et al. Cannabidiol: Pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 2014, 55, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, N.A.; Glyn, S.E.; Akiyama, S.; Hill, T.D.M.; Hill, A.J.; Weston, S.E.; Burnett, M.D.A.; Yamasaki, Y.; Stephens, G.J.; Whalley, B.J.; et al. Cannabidiol exerts anti-convulsant effects in animal models of temporal lobe and partial seizures. Seizure 2012, 21, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, N.A.; Hill, A.J.; Smith, I.; Bevan, S.A.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J. Pharmacol. Exp. Ther. 2010, 332, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, P.H.; Barker-Haliski, M.; White, H.S.; Whalley, B.J.; Glyn, S.; Sandhu, H.; Jones, N.; Bazelot, M.; Williams, C.M.; McNeish, A.J. Cannabidiol reduces seizures and associated behavioral comorbidities in a range of animal seizure and epilepsy models. Epilepsia 2019, 60, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Stockings, E.; Zagic, D.; Campbell, G.; Weier, M.; Hall, W.D.; Nielsen, S.; Herkes, G.K.; Farrell, M.; Degenhardt, L. Evidence for cannabis and cannabinoids for epilepsy: A systematic review of controlled and observational evidence. J. Neurol. Neurosurg. Psychiatry 2018, 89, 741–753. [Google Scholar] [CrossRef]
- Huestis, M.A. Human cannabinoid pharmacokinetics. Chem. Biodivers. 2007, 4, 1770–1804. [Google Scholar] [CrossRef] [Green Version]
- Andréasson, S.; Engström, A.; Allebeck, P.; Rydberg, U. Cannabis and Schizophrenia A Longitudinal Study of Swedish Conscripts. Lancet 1987, 330, 1483–1486. [Google Scholar] [CrossRef]
- Friedman, D.; French, J.A.; Maccarrone, M. Safety, efficacy, and mechanisms of action of cannabinoids in neurological disorders. Lancet Neurol. 2019, 18, 504–512. [Google Scholar] [CrossRef]
- Devinsky, O.; Cross, J.H.; Laux, L.; Marsh, E.; Miller, I.; Nabbout, R.; Scheffer, I.E.; Thiele, E.A.; Wright, S. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N. Engl. J. Med. 2017, 376, 2011–2020. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Patel, A.D.; Cross, J.H.; Villanueva, V.; Wirrell, E.C.; Privitera, M.; Greenwood, S.M.; Roberts, C.; Checketts, D.; VanLandingham, K.E.; et al. Effect of Cannabidiol on Drop Seizures in the Lennox–Gastaut Syndrome. N. Engl. J. Med. 2018, 378, 1888–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, E.A.; Marsh, E.D.; French, J.A.; Mazurkiewicz-Beldzinska, M.; Benbadis, S.R.; Joshi, C.; Lyons, P.D.; Taylor, A.; Roberts, C.; Sommerville, K.; et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2018, 391, 1085–1096. [Google Scholar] [CrossRef]
- Perucca, E. Cannabinoids in the Treatment of Epilepsy: Hard Evidence at Last? J. Epilepsy Res. 2017, 7, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Scheffer, I.E.; Sadleir, L.G. Efficacy of cannabinoids in paediatric epilepsy. Dev. Med. Child Neurol. 2019, 61, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Ondřej, Z.; Gabriela, D.; Kristýna, N.; Miroslav, T.; Alexandra, Š.; Lumír, H.; Jan, J. Cannabinoids and Cytochrome P450 Interactions. Curr. Drug Metab. 2016, 17, 206–226. [Google Scholar] [CrossRef]
- Jiang, R.; Yamaori, S.; Okamoto, Y.; Yamamoto, I.; Watanabe, K. Cannabidiol Is a Potent Inhibitor of the Catalytic Activity of Cytochrome P450 2C19. Drug Metab. Pharmacokinet. 2013, 28, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Morrison, G.; Crockett, J.; Blakey, G.; Sommerville, K. A Phase 1, Open-Label, Pharmacokinetic Trial to Investigate Possible Drug-Drug Interactions Between Clobazam, Stiripentol, or Valproate and Cannabidiol in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2019. [Google Scholar] [CrossRef]
- Geffrey, A.L.; Pollack, S.F.; Bruno, P.L.; Thiele, E.A. Drug–drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 2015, 56, 1246–1251. [Google Scholar] [CrossRef]
- Thiele, E.A.; Devinsky, O.; Checketts, D.; Knappertz, V. Cannabidiol Treatment Responder Analysis in Patients with Lennox-Gastaut Syndrome on and off Clobazam [abstract]. In Proceedings of the American Epilepsy Society Annual Meeting, New Orleans, LA, USA, 6–10 December 2019. [Google Scholar]
- Thiele, E.; Marsh, E.; Mazurkiewicz-Beldzinska, M.; Halford, J.J.; Gunning, B.; Devinsky, O.; Checketts, D.; Roberts, C. Cannabidiol in patients with Lennox-Gastaut syndrome: Interim analysis of an open-label extension study. Epilepsia 2019, 60, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Patel, A.D.; Thiele, E.A.; Wong, M.H.; Appleton, R.; Harden, C.L.; Greenwood, S.; Morrison, G.; Sommerville, K.; GWPCARE1 Part A Study Group. Randomized, dose-ranging safety trial of cannabidiol in Dravet syndrome. Neurology 2018, 90, e1204–e1211. [Google Scholar] [CrossRef] [Green Version]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPartland, J.M.; Duncan, M.; Di Marzo, V.; Pertwee, R.G. Are cannabidiol and Δ(9) -tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br. J. Pharmacol. 2015, 172, 737–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perucca, E.; Wiebe, S. Not all that glitters is gold: A guide to the critical interpretation of drug trials in epilepsy. Epilepsia Open 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tham, M.; Yilmaz, O.; Alaverdashvili, M.; Kelly, M.E.M.; Denovan-Wright, E.M.; Laprairie, R.B. Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. Br. J. Pharmacol. 2019, 176, 1455–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.S.; Stella, N.; Catterall, W.A.; Westenbroek, R.E. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 11229–11234. [Google Scholar] [CrossRef] [Green Version]
- Friedman, D.; Devinsky, O. Cannabinoids in the Treatment of Epilepsy. N. Engl. J. Med. 2015, 373, 1048–1058. [Google Scholar] [CrossRef]
- Katona, I. Cannabis and Endocannabinoid Signaling in Epilepsy. In Endocannabinoids; Pertwee, R.G., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 285–316. [Google Scholar]
- Bouron, A. Phyto and endocannabinoids exert complex actions on calcium and zinc signaling in mouse cortical neurons. Biochem. Pharmacol. 2018, 152, 244–251. [Google Scholar] [CrossRef]
- Leweke, F.M.; Piomelli, D.; Pahlisch, F.; Muhl, D.; Gerth, C.W.; Hoyer, C.; Klosterkötter, J.; Hellmich, M.; Koethe, D. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl. Psychiatry 2012, 2, e94. [Google Scholar] [CrossRef] [Green Version]
- Elmes, M.W.; Kaczocha, M.; Berger, W.T.; Leung, K.; Ralph, B.P.; Wang, L.; Sweeney, J.M.; Miyauchi, J.T.; Tsirka, S.E.; Ojima, I.; et al. Fatty Acid Binding Proteins (FABPs) are Intracellular Carriers for Δ9-Tetrahydrocannabinol (THC) and Cannabidiol (CBD). J. Biol. Chem. 2015, 290, 8711–8721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Allarà, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; Di Marzo, V. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 2011, 163, 1479–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVuono, M.V.; Hrelja, K.M.; Sabaziotis, L.; Rajna, A.; Rock, E.M.; Limebeer, C.L.; Mutch, D.M.; Parker, L.A. Conditioned gaping produced by high dose Δ9-tetrahydracannabinol: Dysregulation of the hypothalamic endocannabinoid system. Neuropharmacology 2018, 141, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-H.; Lee, S.T.; Lin, W.-W. Effects of cannabinoids on LPS-stimulated inflammatory mediator release from macrophages: Involvement of eicosanoids. J. Cell. Biochem. 2001, 81, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Ruhaak, L.R.; Felth, J.; Karlsson, P.C.; Rafter, J.J.; Verpoorte, R.; Bohlin, L. Evaluation of the Cyclooxygenase Inhibiting Effects of Six Major Cannabinoids Isolated from Cannabis sativa. Biol. Pharm. Bull. 2011, 34, 774–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornheim, L.M.; Kim, K.Y.; Chen, B.L.; Correia, M.A. The Effect of Cannabidiol on Mouse Hepatic Microsomal Cytochrome P450-Dependent Anandamide Metabolism. Biochem. Biophys. Res. Commun. 1993, 197, 740–746. [Google Scholar] [CrossRef]
- Arnold, W.R.; Weigle, A.T.; Das, A. Cross-talk of cannabinoid and endocannabinoid metabolism is mediated via human cardiac CYP2J2. J. Inorg. Biochem. 2018, 184, 88–99. [Google Scholar] [CrossRef]
- Massi, P.; Valenti, M.; Vaccani, A.; Gasperi, V.; Perletti, G.; Marras, E.; Fezza, F.; Maccarrone, M.; Parolaro, D. 5-Lipoxygenase and anandamide hydrolase (FAAH) mediate the antitumor activity of cannabidiol, a non-psychoactive cannabinoid. J. Neurochem. 2008, 104, 1091–1100. [Google Scholar] [CrossRef]
- Fleck, J.; Temp, F.R.; Marafiga, J.R.; Jesse, A.C.; Milanesi, L.H.; Rambo, L.M.; Mello, C.F. Montelukast reduces seizures in pentylenetetrazol-kindled mice. Braz. J. Med. Biol. Res. 2016, 49. [Google Scholar] [CrossRef] [Green Version]
- Rehni, A.K.; Singh, T.G. Modulation of leukotriene D4 attenuates the development of seizures in mice. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 97–106. [Google Scholar] [CrossRef]
- Takahashi, Y.; Imai, K.; Ikeda, H.; Kubota, Y.; Yamazaki, E.; Susa, F. Open study of pranlukast add-on therapy in intractable partial epilepsy. Brain Dev. 2013, 35, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.-H.; Sørgård, M.; Di Marzo, V.; Julius, D.; Högestätt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.A.; Hill, C.L.; Leo, A.; Alhusaini, A.; Soubrane, C.; Mazzarella, E.; Russo, E.; Whalley, B.J.; Di Marzo, V.; Stephens, G.J. Nonpsychotropic Plant Cannabinoids, Cannabidivarin (CBDV) and Cannabidiol (CBD), Activate and Desensitize Transient Receptor Potential Vanilloid 1 (TRPV1) Channels in Vitro: Potential for the Treatment of Neuronal Hyperexcitability. ACS Chem. Neurosci. 2014, 5, 1131–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, E.B.; Burnett, A.; Hall, B.; Parker, K.K. Agonistic Properties of Cannabidiol at 5-HT1a Receptors. Neurochem. Res. 2005, 30, 1037–1043. [Google Scholar] [CrossRef]
- Limebeer, C.L.; Rock, E.M.; Sharkey, K.A.; Parker, L.A. Nausea-Induced 5-HT Release in the Interoceptive Insular Cortex and Regulation by Monoacylglycerol Lipase (MAGL) Inhibition and Cannabidiol. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarra-Lecue, I.; Mollinedo-Gajate, I.; Meana, J.J.; Callado, L.F.; Diez-Alarcia, R.; Urigüen, L. Chronic cannabis promotes pro-hallucinogenic signaling of 5-HT2A receptors through Akt/mTOR pathway. Neuropsychopharmacology 2018, 43, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Nichol, K.; Stott, C.; Jones, N.; Gray, R.A.; Bazelot, M.; Whalley, B.J. The proposed multimodal mechanism of action of cannabidiol (CBD) in epilepsy: Modulation of intracellular calcium and adenosine-mediated signaling (P5.5-007). Neurology 2019, 92, P5.5-007. [Google Scholar]
- Pandolfo, P.; Silveirinha, V.; Santos-Rodrigues, A.D.; Venance, L.; Ledent, C.; Takahashi, R.N.; Cunha, R.A.; Köfalvi, A. Cannabinoids inhibit the synaptic uptake of adenosine and dopamine in the rat and mouse striatum. Eur. J. Pharmacol. 2011, 655, 38–45. [Google Scholar] [CrossRef]
- Carrier, E.J.; Auchampach, J.A.; Hillard, C.J. Inhibition of an equilibrative nucleoside transporter by cannabidiol: A mechanism of cannabinoid immunosuppression. Proc. Natl. Acad. Sci. USA 2006, 103, 7895–7900. [Google Scholar] [CrossRef] [Green Version]
- Rimmerman, N.; Ben-Hail, D.; Porat, Z.; Juknat, A.; Kozela, E.; Daniels, M.P.; Connelly, P.S.; Leishman, E.; Bradshaw, H.B.; Shoshan-Barmatz, V.; et al. Direct modulation of the outer mitochondrial membrane channel, voltage-dependent anion channel 1 (VDAC1) by cannabidiol: A novel mechanism for cannabinoid-induced cell death. Cell Death Dis. 2013, 4, e949. [Google Scholar] [CrossRef]
- Niesink, R.J.M.; van Laar, M. Does Cannabidiol Protect Against Adverse Psychological Effects of THC? Front. Psychiatry 2013, 4, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, D.T.; Robson, P.; House, H.; Makela, P.; Aram, J. A preliminary controlled study to determine whether whole-plant cannabis extracts can improve intractable neurogenic symptoms. Clin. Rehabil. 2003, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.T.; Makela, P.; Robson, P.; House, H.; Bateman, C. Do cannabis-based medicinal extracts have general or specific effects on symptoms in multiple sclerosis? A double-blind, randomized, placebo-controlled study on 160 patients. Mult. Scler. J. 2004, 10, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Mato, S.; Del Olmo, E.; Pazos, A. Ontogenetic development of cannabinoid receptor expression and signal transduction functionality in the human brain. Eur. J. Neurosci. 2003, 17, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- De Fonseca, F.R.; Ramos, J.A.; Bonnin, A.; Fernández-Ruiz, J.J. Presence of cannabinoid binding sites in the brain from early postnatal ages. NeuroReport 1993, 4, 135–138. [Google Scholar] [CrossRef]
- Verdurand, M.; Nguyen, V.; Stark, D.; Zahra, D.; Gregoire, M.-C.; Greguric, I.; Zavitsanou, K. Comparison of Cannabinoid CB1 Receptor Binding in Adolescent and Adult Rats: A Positron Emission Tomography Study Using [18F]MK-947. Int. J. Mol. Imaging 2011, 2011, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, B.; Sundram, S.; Bradbury, R.; Scarr, E.; Copolov, D. Studies on [3H]CP-55940 binding in the human central nervous system: Regional specific changes in density of cannabinoid-1 receptors associated with schizophrenia and cannabis use. Neuroscience 2001, 103, 9–15. [Google Scholar] [CrossRef]
- Dalton, V.S.; Zavitsanou, K. Cannabinoid effects on CB1 receptor density in the adolescent brain: An autoradiographic study using the synthetic cannabinoid HU210. Synapse 2010, 64, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Rubino, T.; Prini, P.; Piscitelli, F.; Zamberletti, E.; Trusel, M.; Melis, M.; Sagheddu, C.; Ligresti, A.; Tonini, R.; Di Marzo, V.; et al. Adolescent exposure to THC in female rats disrupts developmental changes in the prefrontal cortex. Neurobiol. Dis. 2015, 73, 60–69. [Google Scholar] [CrossRef]
- Abush, H.; Akirav, I. Short- and long-term cognitive effects of chronic cannabinoids administration in late-adolescence rats. PLoS ONE 2012, 7, e31731. [Google Scholar] [CrossRef]
- Boulos, P.K.; Dalwani, M.S.; Tanabe, J.; Mikulich-Gilbertson, S.K.; Banich, M.T.; Crowley, T.J.; Sakai, J.T. Brain Cortical Thickness Differences in Adolescent Females with Substance Use Disorders. PLoS ONE 2016, 11, e0152983. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.; Perez-Iglesias, R.; Roiz-Santiañez, R.; Tordesillas-Gutierrez, D.; Pazos, A.; Gutierrez, A.; Vazquez-Barquero, J.L.; Crespo-Facorro, B. Gyrification brain abnormalities associated with adolescence and early-adulthood cannabis use. Brain Res. 2010, 1317, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chye, Y.; Suo, C.; Lorenzetti, V.; Batalla, A.; Cousijn, J.; Goudriaan, A.E.; Martin-Santos, R.; Whittle, S.; Solowij, N.; Yücel, M. Cortical surface morphology in long-term cannabis users: A multi-site MRI study. Eur. Neuropsychopharmacol. 2019, 29, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Mashhoon, Y.; Sava, S.; Sneider, J.T.; Nickerson, L.D.; Silveri, M.M. Cortical thinness and volume differences associated with marijuana abuse in emerging adults. Drug Alcohol Depend. 2015, 155, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobus, J.; Squeglia, L.M.; Meruelo, A.D.; Castro, N.; Brumback, T.; Giedd, J.N.; Tapert, S.F. Cortical thickness in adolescent marijuana and alcohol users: A three-year prospective study from adolescence to young adulthood. Dev. Cogn. Neurosci. 2015, 16, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khongkow, M.; Yata, T.; Boonrungsiman, S.; Ruktanonchai, U.R.; Graham, D.; Namdee, K. Surface modification of gold nanoparticles with neuron-targeted exosome for enhanced blood–brain barrier penetration. Sci. Rep. 2019, 9, 8278. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654. Available online: https://0-www-nature-com.brum.beds.ac.uk/articles/ncb1596#supplementary-information (accessed on 21 October 2019). [CrossRef] [Green Version]
- Almughlliq, F.B.; Koh, Y.Q.; Peiris, H.N.; Vaswani, K.; Arachchige, B.J.; Reed, S.; Mitchell, M.D. Eicosanoid pathway expression in bovine endometrial epithelial and stromal cells in response to lipopolysaccharide, interleukin 1 beta, and tumor necrosis factor alpha. Reprod. Biol. 2018, 18, 390–396. [Google Scholar] [CrossRef]
- Almughlliq, F.B.; Koh, Y.Q.; Peiris, H.N.; Vaswani, K.; McDougall, S.; Graham, E.M.; Burke, C.R.; Arachchige, B.J.; Reed, S.; Mitchell, M.D. Proteomic content of circulating exosomes in dairy cows with or without uterine infection. Theriogenology 2018, 114, 173–179. [Google Scholar] [CrossRef]
- Almughlliq, F.B.; Koh, Y.Q.; Peiris, H.N.; Vaswani, K.; McDougall, S.; Graham, E.M.; Burke, C.R.; Mitchell, M.D. Effect of exosomes from plasma of dairy cows with or without an infected uterus on prostaglandin production by endometrial cell lines. J. Dairy Sci. 2017, 100, 9143–9152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, Y.Q.; Mitchell, M.D.; Almughlliq, F.B.; Vaswani, K.; Peiris, H.N. Regulation of inflammatory mediator expression in bovine endometrial cells: Effects of lipopolysaccharide, interleukin 1 beta, and tumor necrosis factor alpha. Physiol. Rep. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, B.; Yue, S.; Galli, U.; Rana, S.; Gross, W.; Müller, M.; Giese, N.A.; Kalthoff, H.; Becker, T.; Büchler, M.W.; et al. Combined evaluation of a panel of protein and miRNA serum-exosome biomarkers for pancreatic cancer diagnosis increases sensitivity and specificity. Int. J. Cancer 2015, 136, 2616–2627. [Google Scholar] [CrossRef]
- Kan, A.A.; de Jager, W.; de Wit, M.; Heijnen, C.; van Zuiden, M.; Ferrier, C.; van Rijen, P.; Gosselaar, P.; Hessel, E.; van Nieuwenhuizen, O.; et al. Protein expression profiling of inflammatory mediators in human temporal lobe epilepsy reveals co-activation of multiple chemokines and cytokines. J. Neuroinflamm. 2012, 9, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persike, D.S.; Marques-Carneiro, J.E.; Stein, M.L.d.L.; Yacubian, E.M.T.; Centeno, R.; Canzian, M.; Fernandes, M.J.d.S. Altered Proteins in the Hippocampus of Patients with Mesial Temporal Lobe Epilepsy. Pharmaceuticals 2018, 11, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Tan, L.; Tan, L.; Tian, Y.; Ma, J.; Tan, C.-C.; Wang, H.-F.; Liu, Y.; Tan, M.-S.; Jiang, T.; et al. Circulating microRNAs are promising novel biomarkers for drug-resistant epilepsy. Sci. Rep. 2015, 5, 10201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemiya, T.; Maehara, M.; Matsumura, K.; Yasuda, S.; Sugiura, H.; Yamagata, K. Prostaglandin E2 produced by late induced COX-2 stimulates hippocampal neuron loss after seizure in the CA3 region. Neurosci. Res. 2006, 56, 103–110. [Google Scholar] [CrossRef]
- Wolfe, L.S.; Mamer, O.A. Measurement of prostaglandin F2α levels in human cerebrospinal fluid in normal and pathological conditions. Prostaglandins 1975, 9, 183–192. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, H.; Xie, W.; Meng, F.; Zhang, K.; Jiang, Y.; Zhang, X.; Zhang, J. Altered microRNA profiles in plasma exosomes from mesial temporal lobe epilepsy with hippocampal sclerosis. Oncotarget 2016, 8, 4136–4146. [Google Scholar] [CrossRef]
- Woodcock, E.A.; Hillmer, A.T.; Mason, G.F.; Cosgrove, K.P. Imaging Biomarkers of the Neuroimmune System among Substance Use Disorders: A Systematic Review. Mol. Neuropsychiatry 2019, 5, 125–146. [Google Scholar] [CrossRef]
- Ocque, A.J.; Hagler, C.E.; DiFrancesco, R.; Lombardo, J.; Morse, G.D. Development and validation of an assay to measure cannabidiol and Δ9-tetrahydrocannabinol in human EDTA plasma by UHPLC-MS/MS. J. Chromatogr. B 2019, 1112, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, R.; Pichini, S.; Pellegrini, M.; Tittarelli, R.; Pantano, F.; Mannocchi, G.; Rotolo Maria, C.; Busardò Francesco, P. Determination of cannabinoids in oral fluid and urine of “light cannabis” consumers: A pilot study. Clin. Chem. Lab. Med. 2018, 57, 238. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.; Broecker, S.; Madea, B.; Hess, C. Decarbonylation: A metabolic pathway of cannabidiol in humans. Drug Test. Anal. 2019. [Google Scholar] [CrossRef] [PubMed]
- Giacovazzo, G.; Bisogno, T.; Piscitelli, F.; Verde, R.; Oddi, S.; Maccarrone, M.; Coccurello, R. Different Routes to Inhibit Fatty Acid Amide Hydrolase: Do All Roads Lead to the Same Place? Int. J. Mol. Sci. 2019, 20, 4503. [Google Scholar] [CrossRef] [Green Version]
- Burstein, S.H. Ajulemic acid: Potential treatment for chronic inflammation. Pharmacol. Res. Perspect. 2018, 6, e00394. [Google Scholar] [CrossRef]
- Administration U.S.F.D. FDA Approves First Drug Comprised of an Active Ingredient Derived from Marijuana to Treat Rare, Severe Forms of Epilepsy. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-comprised-active-ingredient-derived-marijuana-treat-rare-severe-forms (accessed on 20 October 2019).
- Pichler, E.-M.; Kawohl, W.; Seifritz, E.; Roser, P. Pure delta-9-tetrahydrocannabinol and its combination with cannabidiol in treatment-resistant Tourette syndrome: A case report. Int. J. Psychiatry Med. 2019, 54, 150–156. [Google Scholar] [CrossRef]
- Tartaglia, N.; Bonn-Miller, M.; Hagerman, R. Treatment of Fragile X Syndrome with Cannabidiol: A Case Series Study and Brief Review of the Literature. Cannabis Cannabinoid Res. 2019, 4, 3–9. [Google Scholar] [CrossRef]
- Zamberletti, E.; Gabaglio, M.; Parolaro, D. The Endocannabinoid System and Autism Spectrum Disorders: Insights from Animal Models. Int. J. Mol. Sci. 2017, 18, 1916. [Google Scholar] [CrossRef]
- Navarro, G.; Varani, K.; Reyes-Resina, I.; Sánchez de Medina, V.; Rivas-Santisteban, R.; Sánchez-Carnerero Callado, C.; Vincenzi, F.; Casano, S.; Ferreiro-Vera, C.; Canela, E.I.; et al. Cannabigerol Action at Cannabinoid CB1 and CB2 Receptors and at CB1–CB2 Heteroreceptor Complexes. Front. Pharmacol. 2018, 9, 632. [Google Scholar] [CrossRef]
- Pharmaceuticals, G. GW Pharmaceuticals Announces Preliminary Results of Phase 2a Study for its Pipeline Compound GWP42006. Available online: https://www.gwpharm.com/about/news/gw-pharmaceuticals-announces-preliminary-results-phase-2a-study-its-pipeline-compound (accessed on 25 November 2019).
- Taylor, L.; Gidal, B.; Blakey, G.; Tayo, B.; Morrison, G. A Phase I, Randomized, Double-Blind, Placebo-Controlled, Single Ascending Dose, Multiple Dose, and Food Effect Trial of the Safety, Tolerability and Pharmacokinetics of Highly Purified Cannabidiol in Healthy Subjects. CNS Drugs 2018, 32, 1053–1067. [Google Scholar] [CrossRef] [Green Version]
- Merrick, J.L.B.; Sebree, T.; Yaksh, T.; O’Neill, C.; Banks, S.L. Identification of Psychoactive Degradants of Cannabidiol in Simulated Gastric and Physiological Fluid. Cannabis Cannabinoid Res. 2016, 1, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Crume, T.L.; Juhl, A.L.; Brooks-Russell, A.; Hall, K.E.; Wymore, E.; Borgelt, L.M. Cannabis Use During the Perinatal Period in a State With Legalized Recreational and Medical Marijuana: The Association Between Maternal Characteristics, Breastfeeding Patterns, and Neonatal Outcomes. J. Pediatr. 2018, 197, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, S.; Rossi, G.; Oddi, S.; Di Nisio, V.; Maccarrone, M. Role of Major Endocannabinoid-Binding Receptors during Mouse Oocyte Maturation. Int. J. Mol. Sci. 2019, 20, 2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, M.D.; Rice, G.E.; Vaswani, K.; Kvaskoff, D.; Peiris, H.N. Differential Regulation of Eicosanoid and Endocannabinoid Production by Inflammatory Mediators in Human Choriodecidua. PLoS ONE 2016, 11, e0148306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bara, A.; Manduca, A.; Bernabeu, A.; Borsoi, M.; Serviado, M.; Lassalle, O.; Murphy, M.; Wager-Miller, J.; Mackie, K.; Pelissier-Alicot, A.-L.; et al. Sex-dependent effects of in utero cannabinoid exposure on cortical function. eLife 2018, 7, e36234. [Google Scholar] [CrossRef] [PubMed]
- Tortoriello, G.; Morris, C.V.; Alpar, A.; Fuzik, J.; Shirran, S.L.; Calvigioni, D.; Keimpema, E.; Botting, C.H.; Reinecke, K.; Herdegen, T.; et al. Miswiring the brain: Δ9-tetrahydrocannabinol disrupts cortical development by inducing an SCG10/stathmin-2 degradation pathway. EMBO J. 2014, 33, 668–685. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwan Cheung, K.A.; Peiris, H.; Wallace, G.; Holland, O.J.; Mitchell, M.D. The Interplay between the Endocannabinoid System, Epilepsy and Cannabinoids. Int. J. Mol. Sci. 2019, 20, 6079. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236079
Kwan Cheung KA, Peiris H, Wallace G, Holland OJ, Mitchell MD. The Interplay between the Endocannabinoid System, Epilepsy and Cannabinoids. International Journal of Molecular Sciences. 2019; 20(23):6079. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236079
Chicago/Turabian StyleKwan Cheung, Keith A., Hassendrini Peiris, Geoffrey Wallace, Olivia J. Holland, and Murray D. Mitchell. 2019. "The Interplay between the Endocannabinoid System, Epilepsy and Cannabinoids" International Journal of Molecular Sciences 20, no. 23: 6079. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236079