Lercanidipine Synergistically Enhances Bortezomib Cytotoxicity in Cancer Cells via Enhanced Endoplasmic Reticulum Stress and Mitochondrial Ca2+ Overload

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
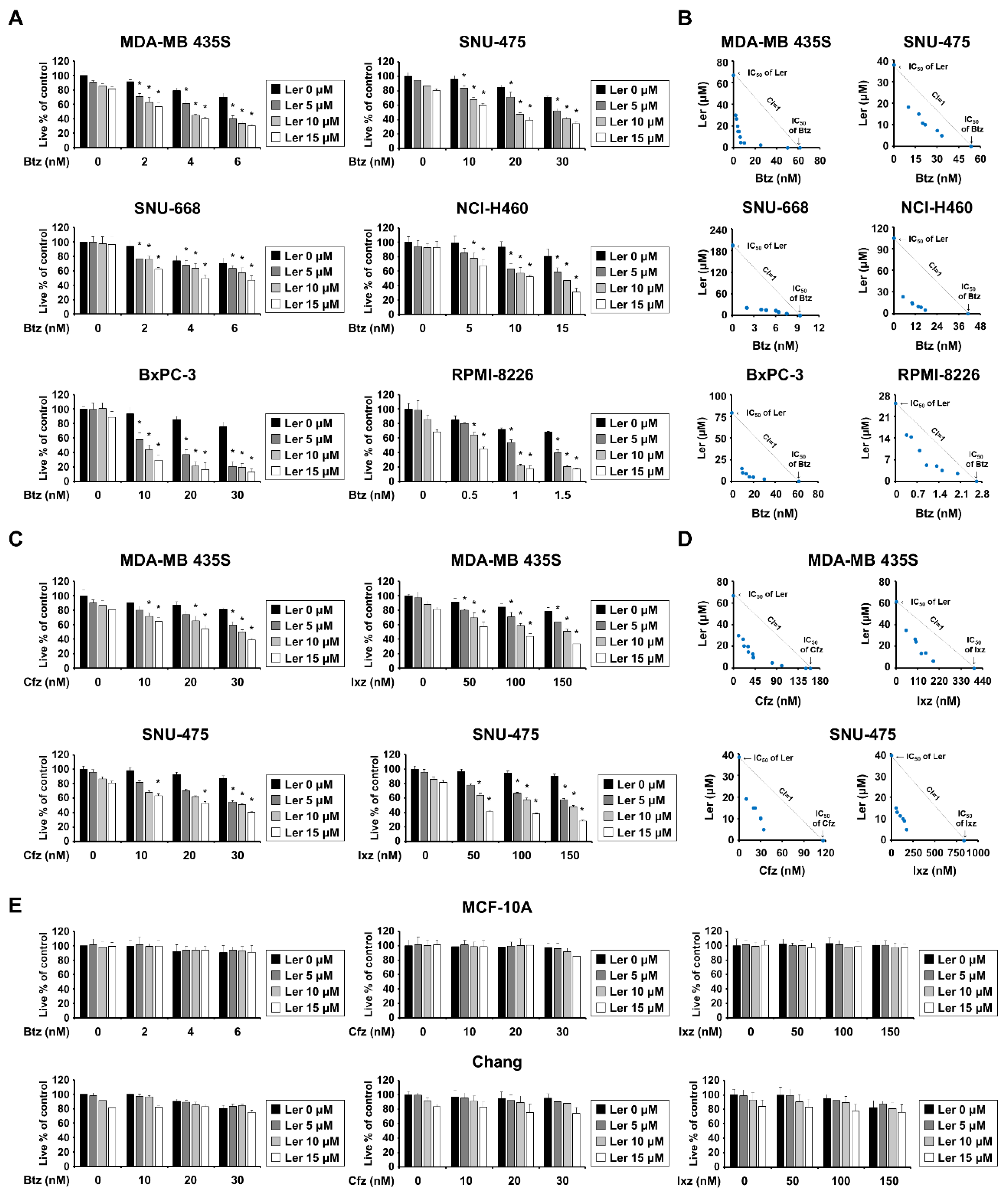
2.1. Lercanidipine Effectively Enhances PI-Mediated Cell Death in Various Cancer Cells
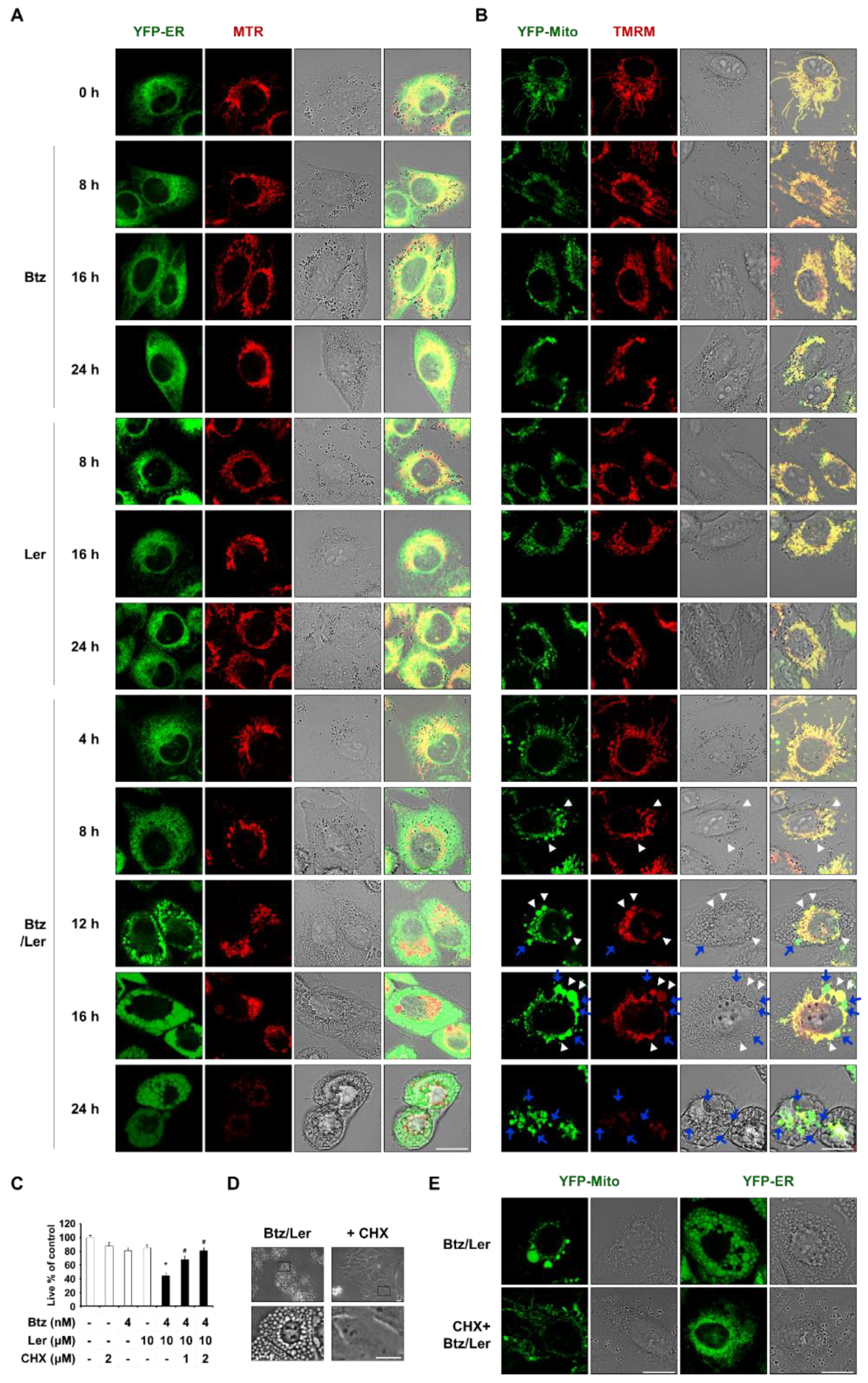
2.2. Combination of Ler and Btz Induces Paraptosis in Cancer Cells
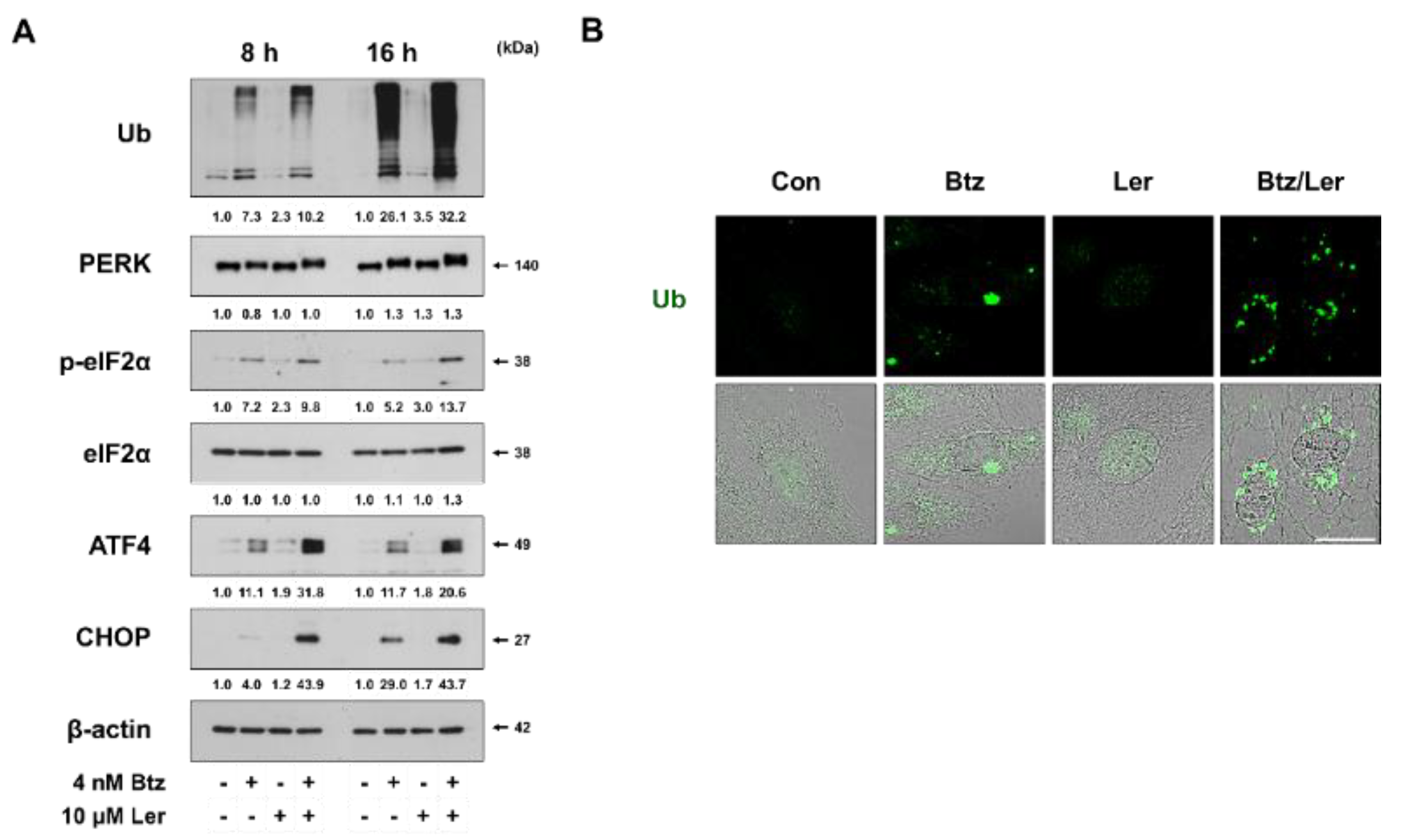
2.3. Combination of Btz and Ler Enhances ER Stress
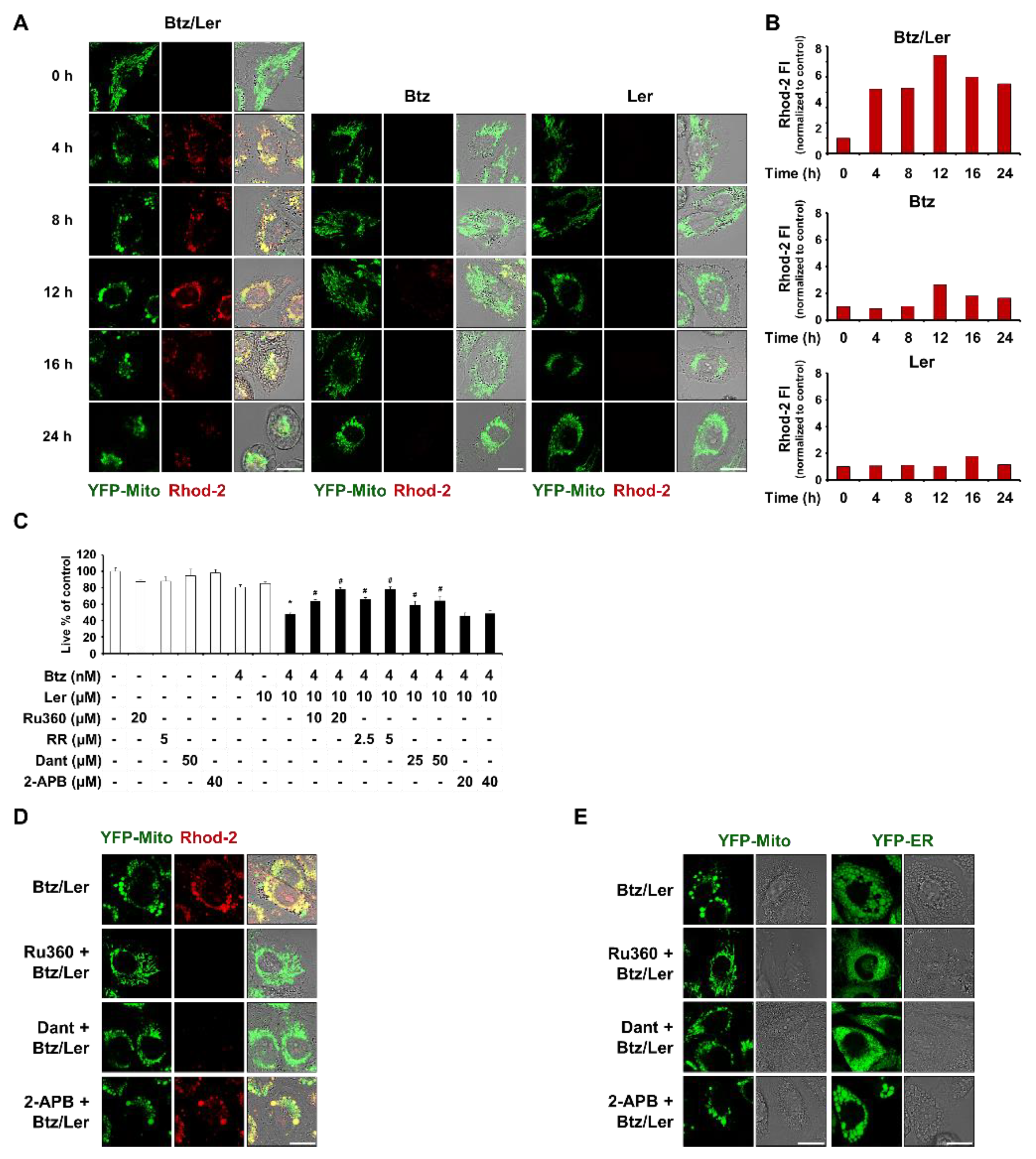
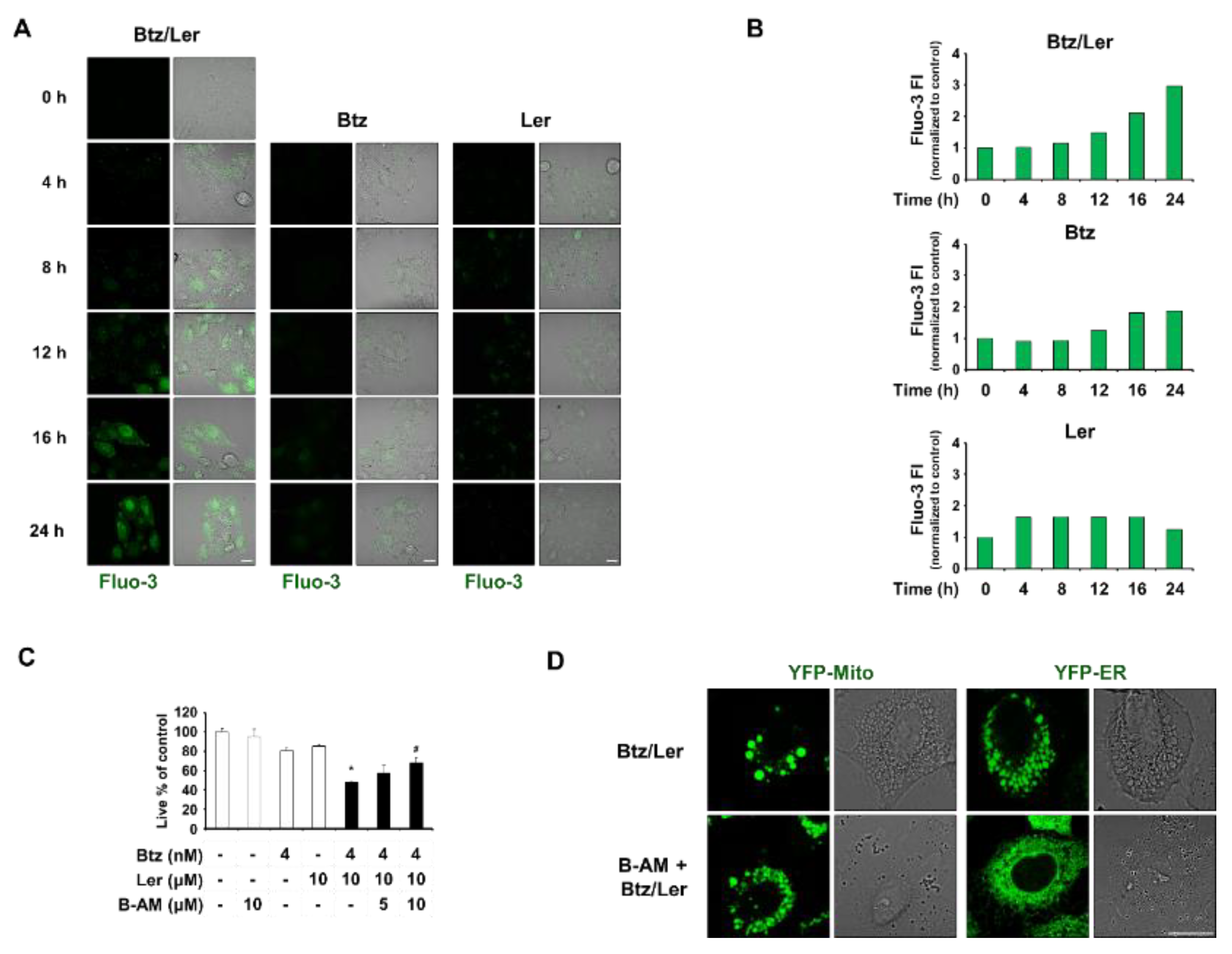
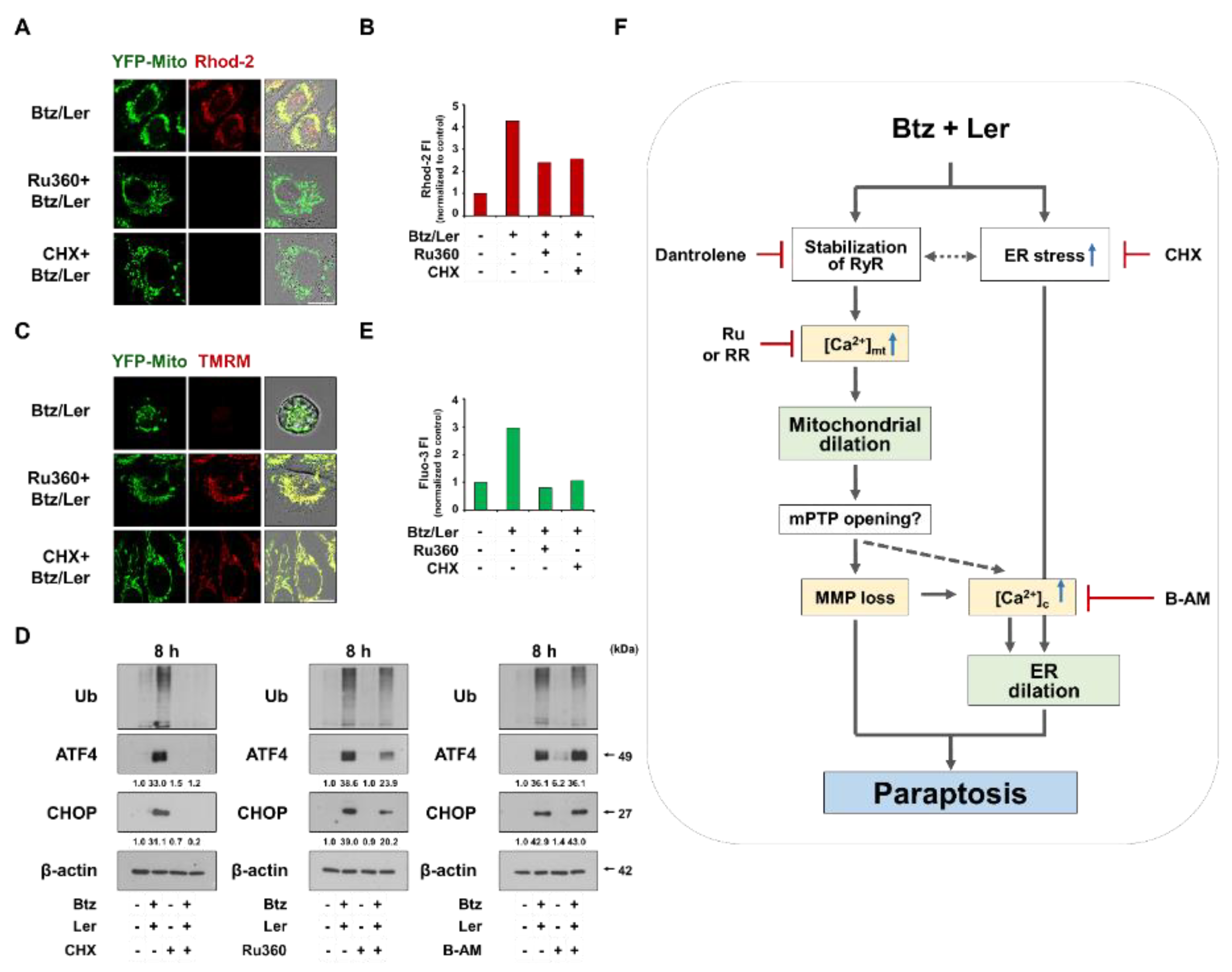
2.4. Mitochondrial Ca2+ Overload Induced by Btz and Ler is Critical for Mitochondrial Dilation during The Progression of Paraptosis
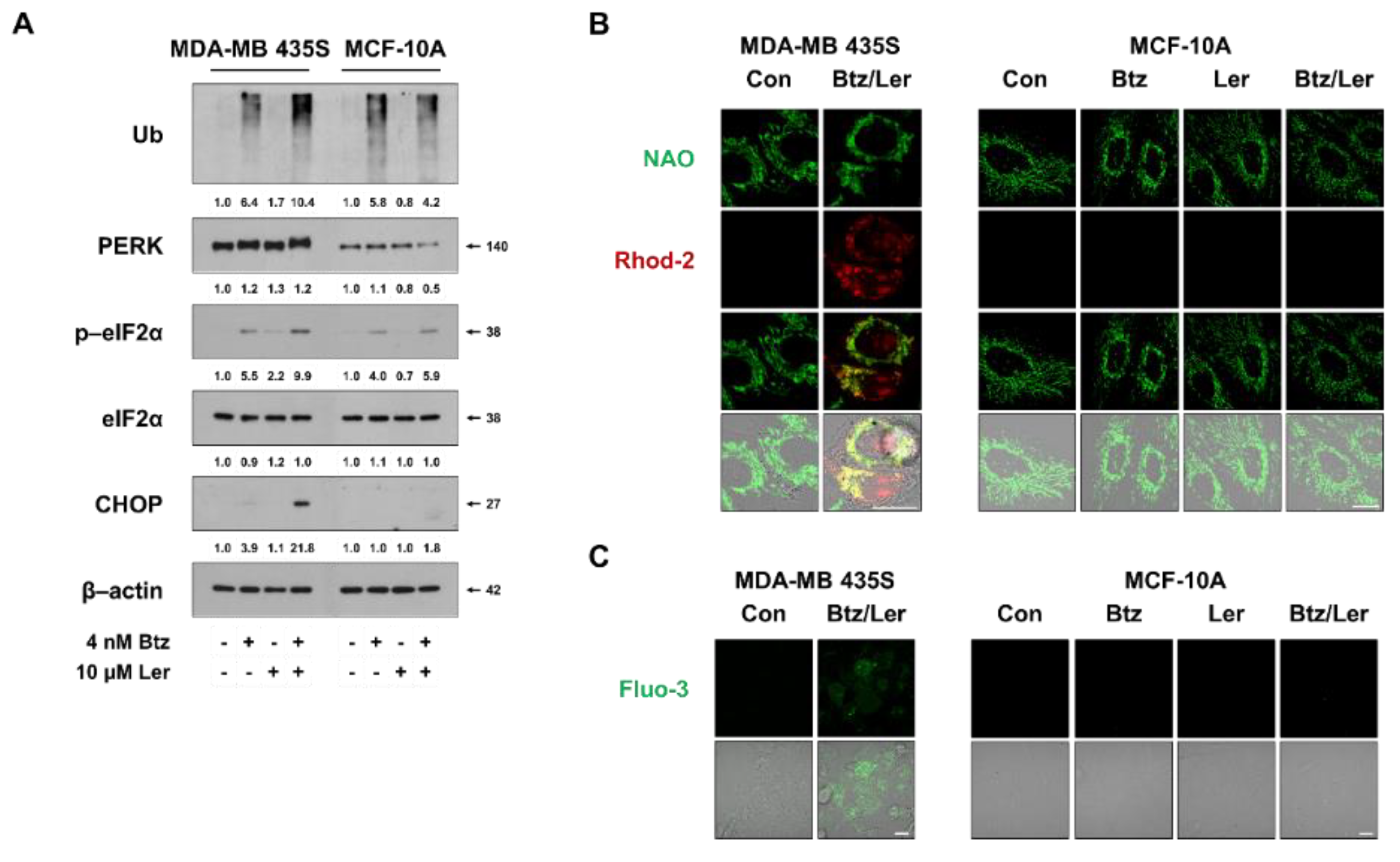
2.5. Combination of Btz and Ler Aggravates ER Stress and Disrupts Ca2+ Homeostasis Selectively in Breast Cancer Cells
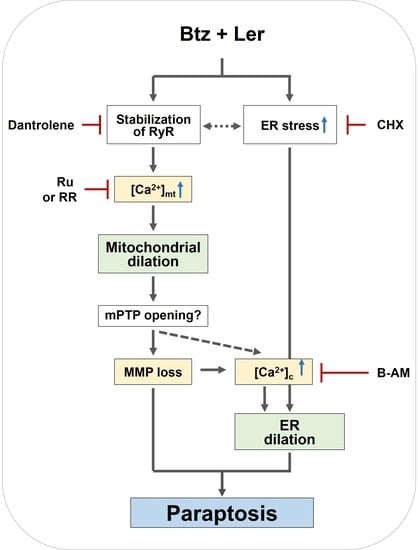
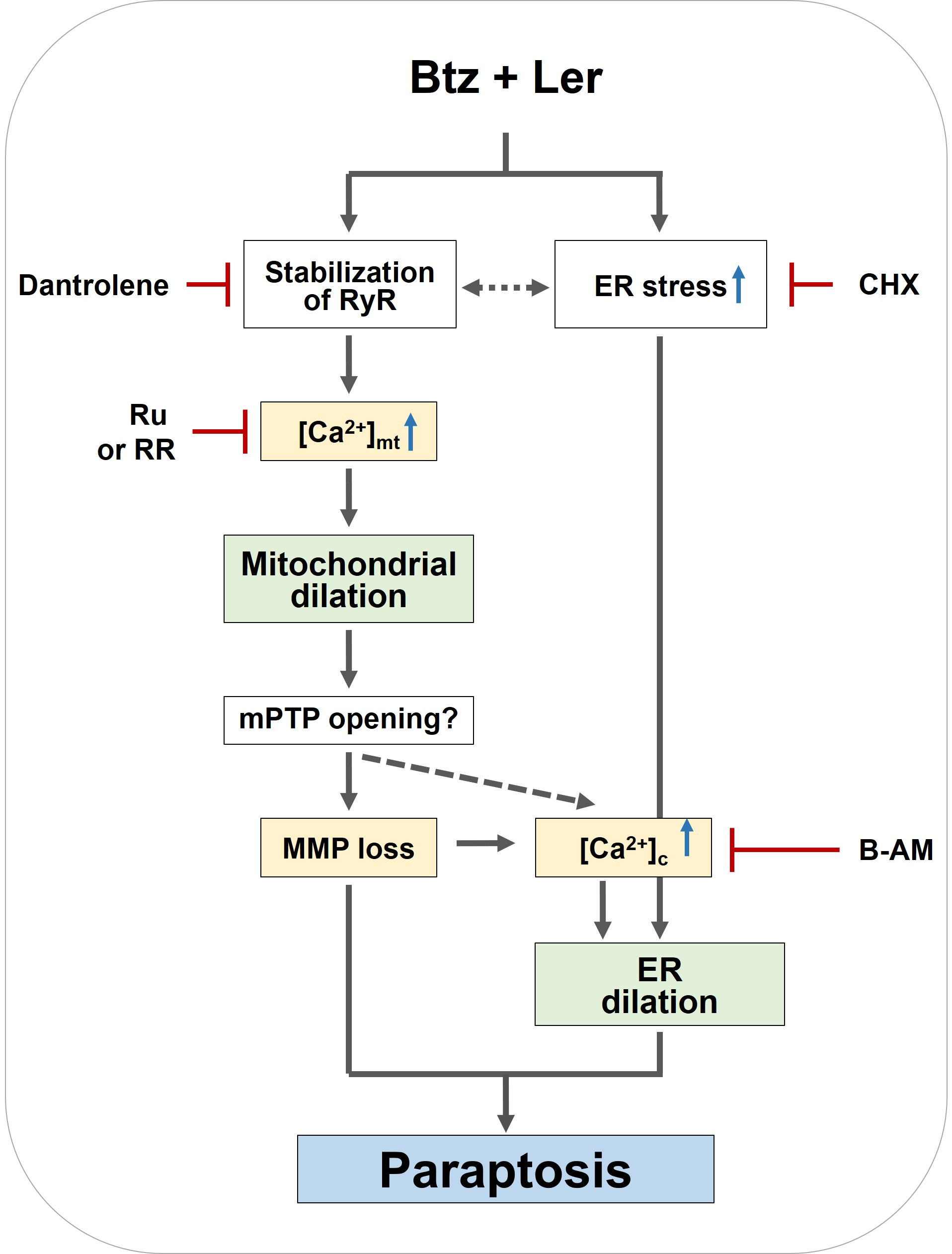
2.6. Mitochondrial Ca2+ Overload Critically Contributes to The Dilation of Both Mitochondria and The ER in Btz/Ler-Induced Paraptosis
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Morphological Examination of The ER and Mitochondria
4.5. Immunoblot Analyses
4.6. Immunofluorescence Microscopy
4.7. Measurement of Cytosolic and Mitochondrial Ca2+ Levels
4.8. Isobologram Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATF4 | Activating transcription factor 4 |
| CHOP | CCAT-enhancer-binding protein homologous protein |
| eIF2α | Eukaryotic translation initiation factor 2 subunit 1 |
| ER | Endoplasmic reticulum |
| MCU | Mitochondrial calcium uniporter |
| PERK | Protein kinase R (PKR)-like endoplasmic reticulum kinase |
| Ub | Ubiquitin |
References
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Miller, Z.; Jun, Y.; Lee, W.; Kim, K.B. Next-generation proteasome inhibitors for cancer therapy. Transl. Res. 2018, 198, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of therapy with bortezomib in solid tumors: A review based on 32 clinical trials. Future Oncol. 2014, 10, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Burris, H.A., 3rd; Gordon, M.; Lee, P.; Sausville, E.A.; Rosen, P.J.; Patnaik, A.; Cutler, R.E., Jr.; Wang, Z.; Lee, S.; et al. A phase I/II study of carfilzomib 2-10-min infusion in patients with advanced solid tumors. Cancer Chemother Pharm. 2013, 72, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Driscoll, J.J.; Minter, A.; Driscoll, D.A.; Burris, J.K. The ubiquitin+proteasome protein degradation pathway as a therapeutic strategy in the treatment of solid tumor malignancies. Anticancer Agents Med. Chem. 2011, 11, 242–246. [Google Scholar] [CrossRef]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother Pharm. 2018, 81, 227–243. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.R.; Stonier, P.D. Repurposing old drugs in oncology: Opportunities with clinical and regulatory challenges ahead. J. Clin. Pharm. 2019, 44, 6–22. [Google Scholar] [CrossRef] [Green Version]
- Burnier, M.; Pruijm, M.; Wuerzner, G. Treatment of essential hypertension with calcium channel blockers: What is the place of lercanidipine? Expert Opin. Drug Metab. Toxicol. 2009, 5, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Bang, L.M.; Chapman, T.M.; Goa, K.L. Lercanidipine: A review of its efficacy in the management of hypertension. Drugs 2003, 63, 2449–2472. [Google Scholar] [CrossRef]
- Pruijm, M.T.; Maillard, M.P.; Burnier, M. Patient adherence and the choice of antihypertensive drugs: Focus on lercanidipine. Vasc. Health Risk Manag. 2008, 4, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
- Piccirillo, J.F.; Tierney, R.M.; Costas, I.; Grove, L.; Spitznagel, E.L., Jr. Prognostic importance of comorbidity in a hospital-based cancer registry. JAMA 2004, 291, 2441–2447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, P.; Thanendrarajan, S.; Paydak, H.; Vallurupalli, S.; Jambhekar, K.; Bhatti, S.; Schinke, C.D.; Davies, F.E.; Mehta, J.L. Cardiovascular complications of multiple myeloma in the elderly. Expert Rev. Cardiovasc. 2017, 15, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Coca, A.; Mazon, P.; Aranda, P.; Redon, J.; Divison, J.A.; Martinez, J.; Calvo, C.; Galceran, J.M.; Barrios, V.; Roca-Cusachs, I.C.A. Role of dihydropyridinic calcium channel blockers in the management of hypertension. Expert Rev. Cardiovasc. 2013, 11, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Shekari, F.; Sadeghpour, H.; Javidnia, K.; Saso, L.; Nazari, F.; Firuzi, O.; Miri, R. Cytotoxic and multidrug resistance reversal activities of novel 1,4-dihydropyridines against human cancer cells. Eur. J. Pharm. 2015, 746, 233–244. [Google Scholar] [CrossRef]
- Viale, M.; Cordazzo, C.; de Totero, D.; Budriesi, R.; Rosano, C.; Leoni, A.; Ioan, P.; Aiello, C.; Croce, M.; Andreani, A.; et al. Inhibition of MDR1 activity and induction of apoptosis by analogues of nifedipine and diltiazem: An in vitro analysis. Investig. New Drugs 2011, 29, 98–109. [Google Scholar] [CrossRef]
- Sperandio, S.; De Belle, I.; Bredesen, D.E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [Green Version]
- Sperandio, S.; Poksay, K.; de Belle, I.; Lafuente, M.J.; Liu, B.; Nasir, J.; Bredesen, D.E. Paraptosis: Mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004, 11, 1066–1075. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Kim, I.Y.; Saha, S.; Choi, K.S. Paraptosis in the anti-cancer arsenal of natural products. Pharmacol. Ther. 2016, 162, 120–133. [Google Scholar] [CrossRef]
- Yoon, M.J.; Kang, Y.J.; Lee, J.A.; Kim, I.Y.; Kim, M.A.; Lee, Y.S.; Park, J.H.; Lee, B.Y.; Kim, I.A.; Kim, H.S.; et al. Stronger proteasomal inhibition and higher CHOP induction are responsible for more effective induction of paraptosis by dimethoxycurcumin than curcumin. Cell Death Dis. 2014, 5, e1112. [Google Scholar] [CrossRef]
- Nedungadi, D.; Binoy, A.; Pandurangan, N.; Pal, S.; Nair, B.G.; Mishra, N. 6-Shogaol induces caspase-independent paraptosis in cancer cells via proteasomal inhibition. Exp. Cell Res. 2018, 364, 243–251. [Google Scholar] [CrossRef]
- Yoon, M.J.; Kim, E.H.; Lim, J.H.; Kwon, T.K.; Choi, K.S. Superoxide anion and proteasomal dysfunction contribute to curcumin-induced paraptosis of malignant breast cancer cells. Free Radic. Biol. Med. 2010, 48, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Kim, E.H.; Kwon, T.K.; Park, S.A.; Choi, K.S. Simultaneous mitochondrial Ca2+ overload and proteasomal inhibition are responsible for the induction of paraptosis in malignant breast cancer cells. Cancer Lett. 2012, 324, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Lee, A.R.; Jeong, S.A.; Kim, Y.S.; Kim, J.Y.; Kwon, Y.J.; Choi, K.S. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget 2014, 5, 6816–6831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.M.; Kim, I.Y.; Seo, M.J.; Kwon, M.R.; Choi, K.S. Nutlin-3 enhances the bortezomib sensitivity of p53-defective cancer cells by inducing paraptosis. Exp. Mol. Med. 2017, 49, e365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Li, R.; Zhao, X.; Ma, C.; Lv, X.; Liu, L.; Liu, P. Morusin induces paraptosis-like cell death through mitochondrial calcium overload and dysfunction in epithelial ovarian cancer. Chem. Biol. Interact. 2018, 283, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Ni, H.M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Rizzuto, R. Molecular control of mitochondrial calcium uptake. Biochem. Biophys Res. Commun. 2014, 449, 373–376. [Google Scholar] [CrossRef]
- De Stefani, D.; Patron, M.; Rizzuto, R. Structure and function of the mitochondrial calcium uniporter complex. Biochim. Biophys Acta 2015, 1853, 2006–2011. [Google Scholar] [CrossRef]
- Parekh, A.B. Mitochondrial regulation of intracellular Ca2+ signaling: More than just simple Ca2+ buffers. News Physiol. Sci. 2003, 18, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Sammels, E.; Parys, J.B.; Missiaen, L.; De Smedt, H.; Bultynck, G. Intracellular Ca2+ storage in health and disease: A dynamic equilibrium. Cell Calcium. 2010, 47, 297–314. [Google Scholar] [CrossRef]
- Meredith, P.A. Lercanidipine: A novel lipophilic dihydropyridine calcium antagonist with long duration of action and high vascular selectivity. Expert. Opin. Investig. Drugs 1999, 8, 1043–1062. [Google Scholar] [CrossRef] [PubMed]
- Angelico, P.; Guarneri, L.; Leonardi, A.; Testa, R. Vascular-selective effect of lercanidipine and other 1,4-dihydropyridines in isolated rabbit tissues. J. Pharm. Pharm. 1999, 51, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Burnier, M.; Gasser, U.E. Efficacy and tolerability of lercanidipine in patients with hypertension: Results of a Phase IV study in general practice. Expert Opin. Pharm. 2007, 8, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Mimnaugh, E.G.; Xu, W.; Vos, M.; Yuan, X.; Neckers, L. Endoplasmic reticulum vacuolization and valosin-containing protein relocalization result from simultaneous hsp90 inhibition by geldanamycin and proteasome inhibition by velcade. Mol. Cancer Res. 2006, 4, 667–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, P.; Von Stockum, S. The permeability transition pore as a Ca2+ release channel: New answers to an old question. Cell Calcium. 2012, 52, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.; Gerasimenko, O.V. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef] [Green Version]
- Deniaud, A.; Sharaf el dein, O.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Gunter, T.E.; Sheu, S.S. Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim. Biophys. Acta 2009, 1787, 1291–1308. [Google Scholar] [CrossRef] [Green Version]
- Smaili, S.S.; Russell, J.T. Permeability transition pore regulates both mitochondrial membrane potential and agonist-evoked Ca2+ signals in oligodendrocyte progenitors. Cell Calcium 1999, 26, 121–130. [Google Scholar] [CrossRef]
- Altschuld, R.A.; Hohl, C.M.; Castillo, L.C.; Garleb, A.A.; Starling, R.C.; Brierley, G.P. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am. J. Physiol. 1992, 262, H1699–H1704. [Google Scholar] [CrossRef]
- Weigl, L.G.; Hohenegger, M.; Kress, H.G. Dihydropyridine-induced Ca2+ release from ryanodine-sensitive Ca2+pools in human skeletal muscle cells. J. Physiol. 2000, 525, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Robin, G.; Allard, B. Dihydropyridine receptors actively control gating of ryanodine receptors in resting mouse skeletal muscle fibres. J. Physiol. 2012, 590, 6027–6036. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, R.T. Bi-directional coupling between dihydropyridine receptors and ryanodine receptors. Front. Biosci. 2002, 7, d659–d670. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, D.B.; Zhorov, B.S. Structural model for dihydropyridine binding to L-type calcium channels. J. Biol. Chem. 2009, 284, 19006–19017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrozo, Z.; Sanchez, G.; Torrealba, N.; Valenzuela, R.; Fernandez, C.; Hidalgo, C.; Lavandero, S.; Donoso, P. Calpains and proteasomes mediate degradation of ryanodine receptors in a model of cardiac ischemic reperfusion. Biochim. Biophys. Acta 2010, 1802, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.R.C.; Abdul-Majeed, S.; Cael, B.; Barta, S.K. Clinical Pharmacokinetics and Pharmacodynamics of Bortezomib. Clin. Pharm. 2019, 58, 157–168. [Google Scholar] [CrossRef]
- Epstein, M. Lercanidipine: A novel dihydropyridine calcium-channel blocker. Heart Dis. 2001, 3, 398–407. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, A.R.; Seo, M.J.; Kim, J.; Lee, D.M.; Kim, I.Y.; Yoon, M.J.; Hoon, H.; Choi, K.S. Lercanidipine Synergistically Enhances Bortezomib Cytotoxicity in Cancer Cells via Enhanced Endoplasmic Reticulum Stress and Mitochondrial Ca2+ Overload. Int. J. Mol. Sci. 2019, 20, 6112. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246112
Lee AR, Seo MJ, Kim J, Lee DM, Kim IY, Yoon MJ, Hoon H, Choi KS. Lercanidipine Synergistically Enhances Bortezomib Cytotoxicity in Cancer Cells via Enhanced Endoplasmic Reticulum Stress and Mitochondrial Ca2+ Overload. International Journal of Molecular Sciences. 2019; 20(24):6112. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246112
Chicago/Turabian StyleLee, A Reum, Min Ji Seo, Jin Kim, Dong Min Lee, In Young Kim, Mi Jin Yoon, Hur Hoon, and Kyeong Sook Choi. 2019. "Lercanidipine Synergistically Enhances Bortezomib Cytotoxicity in Cancer Cells via Enhanced Endoplasmic Reticulum Stress and Mitochondrial Ca2+ Overload" International Journal of Molecular Sciences 20, no. 24: 6112. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246112