Combined De Novo Transcriptome and Metabolome Analysis of Common Bean Response to Fusarium oxysporum f. sp. phaseoli Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. RNA Sequencing and Identification of Differentially Expressed Genes
2.2. Differential Gene Expression Analysis Related to FOP Infection
2.2.1. Structural Defense
2.2.2. Signaling
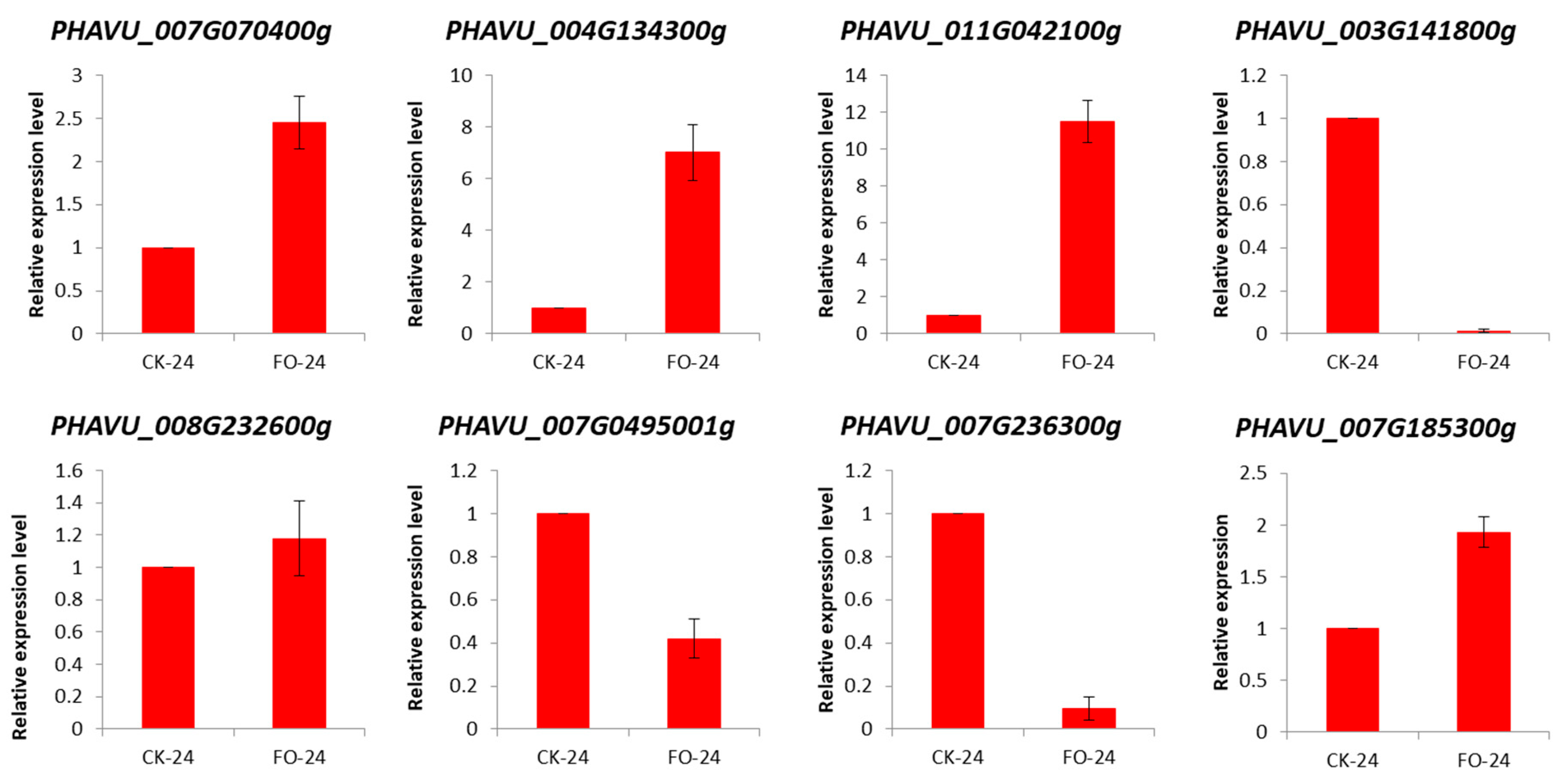
2.3. Validation of DEGs by qRT-PCR
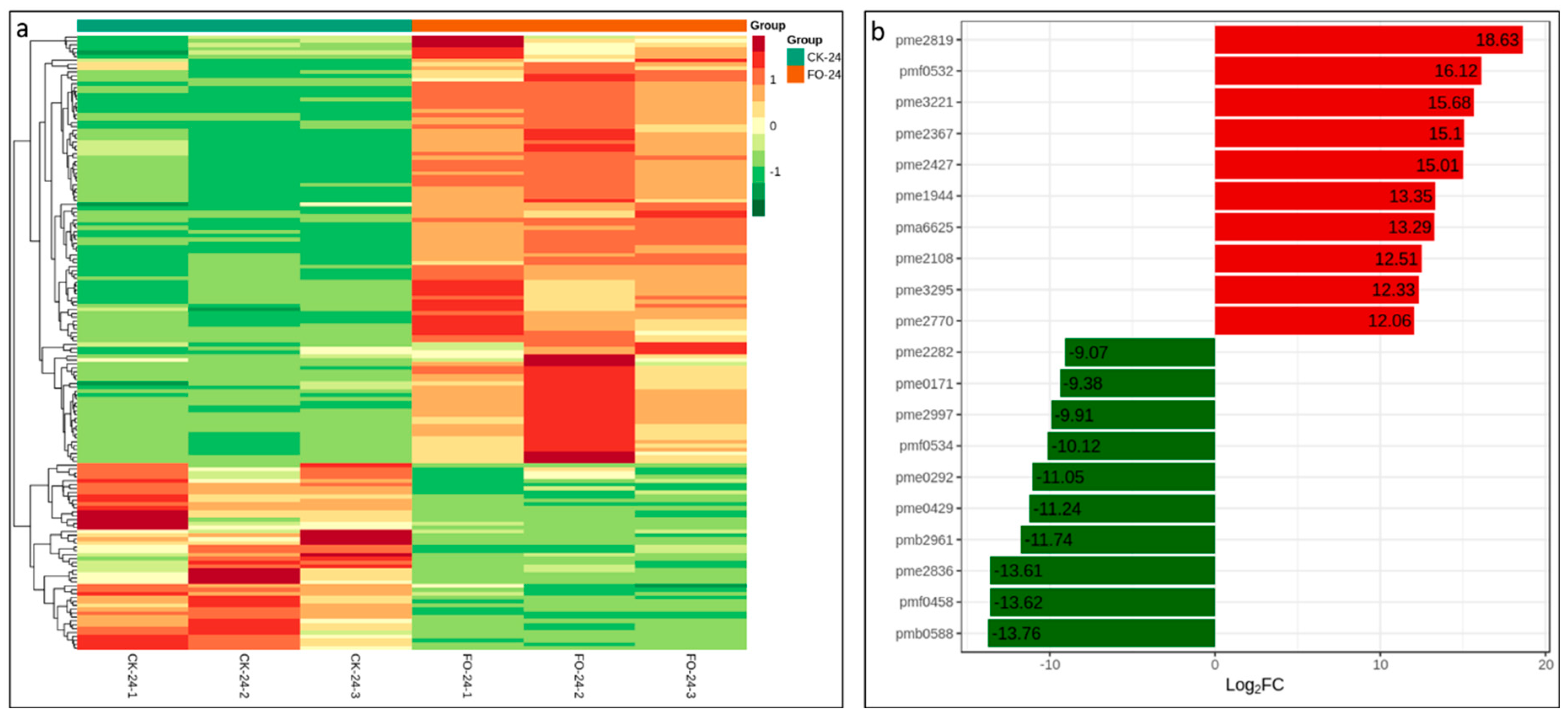
2.4. Metabolite Profile
3. Discussion
3.1. Structural Defense in Response to FOP Infection in Common Bean
3.2. Modulation of Defense Related Proteins in Common Bean
3.3. Crucial Role of Hormones in Signaling Pathways in Common Bean-FOP Pathosystem
3.4. FOP Induced Energy Metabolism and Nitrogen Mobilization
3.5. FOP Resistance in Common Bean is Mediated by Flavonoid Biosynthesis Pathway
4. Materials and Methods
4.1. Plant Growth and In Vivo Inoculations
4.2. RNA Extraction, cDNA Library Construction, and Transcriptome Sequencing
4.3. De Novo Assembly, Functional Annotation, Classification, and Metabolic Pathway Analysis
4.4. Differential Expression and Enrichment Analysis
4.5. Quantitative RT-PCR Analysis
4.6. Widely Targeted Metabolomics
4.7. Sample Preparation
4.8. Chromatographic Mass Spectrometry Acquisition Conditions
4.9. Metabolomics Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Availability of Data
References
- Harter, L. A Fusarium disease of beans. Phytopathology 1929, 19, 1–84. [Google Scholar]
- Buruchara, R.A.; Camacho, L. Common bean reaction to Fusarium oxysporum f. sp. phaseoli, the cause of severe vascular wilt in Central Africa. J. Phytopathol. 2000, 148, 39–45. [Google Scholar] [CrossRef]
- Xue, R.; Wu, J.; Zhu, Z.; Wang, L.; Wang, X.; Wang, S.; Blair, M.W. Differentially expressed genes in resistant and susceptible common bean (Phaseolus vulgaris L.) genotypes in response to Fusarium oxysporum f. sp. phaseoli. PLoS ONE 2015, 10, e0127698. [Google Scholar] [CrossRef] [PubMed]
- Niño-Sánchez, J.; Tello, V.; Casado-del Castillo, V.; Thon, M.R.; Benito, E.P.; Díaz-Mínguez, J.M. Gene expression patterns and dynamics of the colonization of common bean (Phaseolus vulgaris L.) by highly virulent and weakly virulent strains of Fusarium oxysporum. Front. Microbiol. 2015, 6, 234. [Google Scholar] [CrossRef] [Green Version]
- Batista, R.O.; Silva, J.L.O.; Nicoli, A.; Carneiro, P.C.S.; Carneiro, J.E.d.S.; Júnior, P.; Queiroz, M.V.d. Resistance to Fusarium wilt in common bean. Crop Breed. Appl. Biotechnol. 2016, 16, 226–233. [Google Scholar] [CrossRef] [Green Version]
- De Ascensao, A.R.; Dubery, I.A. Panama disease: Cell wall reinforcement in banana roots in response to elicitors from Fusarium oxysporum f. sp. cubense race four. Phytopathology 2000, 90, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cenci, A.; Rouard, M.; Zhang, D.; Wang, Y.; Tang, W.; Zheng, S.-J. Transcriptomic analysis of resistant and susceptible banana corms in response to infection by Fusarium oxysporum f. sp. cubense tropical race 4. Sci. Rep. 2019, 9, 8199. [Google Scholar] [CrossRef]
- Zvirin, T.; Herman, R.; Brotman, Y.; Denisov, Y.; Belausov, E.; Freeman, S.; Perl-Treves, R. Differential colonization and defence responses of resistant and susceptible melon lines infected by Fusarium oxysporum race 1· 2. Plant Pathol. 2010, 59, 576–585. [Google Scholar] [CrossRef]
- Silvia Sebastiani, M.; Bagnaresi, P.; Sestili, S.; Biselli, C.; Zechini, A.; Orrù, L.; Cattivelli, L.; Ficcadenti, N. ranscriptome analysis of the melon-Fusarium oxysporum f. sp. melonis race 1.2 pathosystem in susceptible and resistant plants. Front. Plant Sci. 2017, 8, 362. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Chakraborti, D.; Rangi, R.K.; Basu, D.; Das, S. A molecular insight into the early events of Chickpea (Cicer arietinum) and Fusarium oxysporum f. sp. ciceri (Race 1) interaction through cDNA-AFLP analysis. Phytopathology 2009, 99, 1245–1257. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Chakraborti, D.; Sengupta, A.; Basu, D.; Das, S. Primary metabolism of chickpea is the initial target of wound inducing early sensed Fusarium oxysporum f. sp. ciceri race I. PLoS ONE 2010, 5, e9030. [Google Scholar] [CrossRef] [PubMed]
- Kumar, Y.; Zhang, L.; Panigrahi, P.; Dholakia, B.B.; Dewangan, V.; Chavan, S.G.; Kunjir, S.M.; Wu, X.; Li, N.; Rajmohanan, P.R. Fusarium oxysporum mediates systems metabolic reprogramming of chickpea roots as revealed by a combination of proteomics and metabolomics. Plant Biotechnol. J. 2016, 14, 1589–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowd, C.; Wilson, I.W.; McFadden, H. Gene expression profile changes in cotton root and hypocotyl tissues in response to infection with Fusarium oxysporum f. sp. vasinfectum. Mol. Plant Microbe Interact. 2004, 17, 654–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagopodi, A.L.; Ram, A.F.; Lamers, G.E.; Punt, P.J.; Van den Hondel, C.A.; Lugtenberg, B.J.; Bloemberg, G.V. Novel aspects of tomato root colonization and Infection by Fusarium oxysporum f. sp. radicis-lycopersici revealed by confocal laser scanning microscopic analysis using the green fluorescent protein as a marker. Mol. Plant Microbe Interact. 2002, 15, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrocal-Lobo, M.; Molina, A. Arabidopsis defense response against Fusarium oxysporum. Trends Plant Sci. 2008, 13, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Kidd, B.N.; Kadoo, N.Y.; Dombrecht, B.; Tekeoglu, M.; Gardiner, D.M.; Thatcher, L.F.; Aitken, E.A.; Schenk, P.M.; Manners, J.M.; Kazan, K. Auxin signaling and transport promote susceptibility to the root-infecting fungal pathogen Fusarium oxysporum in Arabidopsis. Mol. Plant Microbe Interact. 2011, 24, 733–748. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Suero, M.; Khanshour, A.; Martinez, Y.; Rickauer, M. A study on the susceptibility of the model legume plant Medicago truncatula to the soil-borne pathogen Fusarium oxysporum. Eur. J. Plant Pathol. 2010, 126, 517–530. [Google Scholar] [CrossRef]
- Li, C.-Y.; Deng, G.-M.; Yang, J.; Viljoen, A.; Jin, Y.; Kuang, R.-b.; Zuo, C.-W.; Lv, Z.-C.; Yang, Q.-S.; Sheng, O. Transcriptome profiling of resistant and susceptible Cavendish banana roots following inoculation with Fusarium oxysporum f. sp. cubense tropical race 4. BMC Genom. 2012, 13, 374. [Google Scholar] [CrossRef] [Green Version]
- Klessig, D.F.; Durner, J.; Shah, J.; Yang, Y. Salicylic acid-mediated signal transduction in plant disease resistance. In Phytochemical Signals and Plant-microbe Interactions; Romeo, J., Downum, K., Verpoorte, R., Eds.; Springer: Boston, MA, USA, 1998; Volume 32, pp. 119–137. [Google Scholar]
- Yang, Y.; Shah, J.; Klessig, D.F. Signal perception and transduction in plant defense responses. Genes Dev. 1997, 11, 1621–1639. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, M.A.; Rehman, H.M.; Imtiaz, M.; Baloch, F.S.; Lee, J.D.; Yang, S.H.; Lee, S.I.; Chung, G. Systems Identification and Characterization of Cell Wall Reassembly and Degradation Related Genes in Glycine max (L.) Merill, a Bioenergy Legume. Sci. Rep. 2017, 7, 10862. [Google Scholar] [CrossRef] [Green Version]
- Bigeard, J.; Colcombet, J.; Hirt, H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol. Plant 2015, 8, 521–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hondo, D.; Hase, S.; Kanayama, Y.; Yoshikawa, N.; Takenaka, S.; Takahashi, H. The LeATL6-associated ubiquitin/proteasome system may contribute to fungal elicitor-activated defense response via the jasmonic acid-dependent signaling pathway in tomato. Mol. Plant Microbe Interact. 2007, 20, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolland, F.; Moore, B.; Sheen, J. Sugar sensing and signaling in plants. Plant Cell 2002, 14, S185–S205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Limones, C.; Hervás, A.; Navas-Cortés, J.A.; Jiménez-Dı́az, R.M.; Tena, M. Induction of an antioxidant enzyme system and other oxidative stress markers associated with compatible and incompatible interactions between chickpea (Cicer arietinum L.) and Fusarium oxysporum f. sp. ciceris. Physiol. Mol. Plant Pathol. 2002, 61, 325–337. [Google Scholar]
- Liu, C.; Hao, F.; Hu, J.; Zhang, W.; Wan, L.; Zhu, L.; Tang, H.; He, G. Revealing different systems responses to brown planthopper infestation for pest susceptible and resistant rice plants with the combined metabonomic and gene-expression analysis. J. Proteome Res. 2010, 9, 6774–6785. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Tzin, V.; Romeis, J.; Peng, Y.; Li, Y. Combined transcriptome and metabolome analyses to understand the dynamic responses of rice plants to attack by the rice stem borer Chilo suppressalis (Lepidoptera: Crambidae). BMC Plant Biol. 2016, 16, 259. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-B.; Wang, S.-M.; Jing, R.-L.; Mao, X.-G. Cloning the PvP5CS gene from common bean (Phaseolus vulgaris) and its expression patterns under abiotic stresses. J. Plant Physiol. 2009, 166, 12–19. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261.e212. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Ying, H.; Pingcuo, G.; Wang, S.; Zhao, F.; Cui, Y.; Shi, J.; Zeng, H.; Zeng, X. Identification of Potential Metabolites Mediating Bird’s Selective Feeding on Prunus mira Flowers. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Zeng, X.; Shi, J.; Xu, Q.; Wang, Y.; Jabu, D.; Sang, Z.; Nyima, T. Time-course comparative metabolite profiling under osmotic stress in tolerant and sensitive tibetan hulless barley. BioMed Res. Int. 2018. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Zeng, X.; Yang, Q.; Xu, Q.; Wang, Y.; Jabu, D.; Sang, Z.; Tashi, N. Gene coexpression network analysis combined with metabonomics reveals the resistance responses to powdery mildew in Tibetan hulless barley. Sci. Rep. 2018, 8, 14928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zeng, X.; Xu, Q.; Mei, X.; Yuan, H.; Jiabu, D.; Sang, Z.; Nyima, T. Metabolite profiling in two contrasting Tibetan hulless barley cultivars revealed the core salt-responsive metabolome and key salt-tolerance biomarkers. AoB Plants 2019, 11, plz021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtasik, W.; Kulma, A.; Dymińska, L.; Hanuza, J.; Czemplik, M.; Szopa, J. Evaluation of the significance of cell wall polymers in flax infected with a pathogenic strain of Fusarium oxysporum. BMC Plant Biol. 2016, 16, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, D.; Khurana, J.P. Role of pathogenesis-related (PR) proteins in plant defense mechanism. In Molecular Aspects of Palnt-Pathogen Interact; Springer: Singapore, 2018. [Google Scholar] [CrossRef]
- Andersen, E.J.; Ali, S.; Byamukama, E.; Yen, Y.; Nepal, M.P. Disease resistance mechanisms in plants. Genes 2018, 9, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K. Glycosylphosphatidylinositol-anchored proteins in Arabidopsis and one of their common roles in signaling transduction. Front. Plant Sci. 2019, 10, 1022. [Google Scholar] [CrossRef] [Green Version]
- Mou, S.; Liu, Z.; Guan, D.; Qiu, A.; Lai, Y.; He, S. Functional analysis and expressional characterization of rice ankyrin repeat-containing protein, OsPIANK1 in basal defense against Magnaporthe oryzae attack. PLoS ONE 2013, 8, e59699. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.P.; Badruzsaufari, E.; Schenk, P.M.; Manners, J.M.; Desmond, O.J.; Ehlert, C.; Maclean, D.J.; Ebert, P.R.; Kazan, K. Antagonistic interaction between abscisic acid and jasmonate-ethylene signaling pathways modulates defense gene expression and disease resistance in Arabidopsis. Plant Cell 2004, 16, 3460–3479. [Google Scholar] [CrossRef] [Green Version]
- Kidd, B.N.; Edgar, C.I.; Kumar, K.K.; Aitken, E.A.; Schenk, P.M.; Manners, J.M.; Kazan, K. The mediator complex subunit PFT1 is a key regulator of jasmonate-dependent defense in Arabidopsis. Plant Cell 2009, 21, 2237–2252. [Google Scholar] [CrossRef] [Green Version]
- Barth, C.; Jander, G. Arabidopsis myrosinases TGG1 and TGG2 have redundant function in glucosinolate breakdown and insect defense. Plant J. 2006, 46, 549–562. [Google Scholar] [CrossRef]
- Fan, J.; Crooks, C.; Creissen, G.; Hill, L.; Fairhurst, S.; Doerner, P.; Lamb, C. Pseudomonas sax genes overcome aliphatic isothiocyanate–mediated non-host resistance in Arabidopsis. Science 2011, 331, 1185–1188. [Google Scholar] [CrossRef]
- Deng, Y.; Li, C.; Li, H.; Lu, S. Identification and characterization of flavonoid biosynthetic enzyme genes in Salvia miltiorrhiza (Lamiaceae). Molecules 2018, 23, 1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farag, M.A.; Huhman, D.V.; Dixon, R.A.; Sumner, L.W. Metabolomics reveals novel pathways and differential mechanistic and elicitor-specific responses in phenylpropanoid and isoflavonoid biosynthesis in Medicago truncatula cell cultures. Plant Physiol. 2008, 146, 387–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.I.; Muthusamy, M.; Nawaz, M.A.; Hong, J.K.; Lim, M.-H.; Kim, J.A.; Jeong, M.-J. Genome-wide analysis of spatiotemporal gene expression patterns during floral organ development in Brassica rapa. Mol. Genet. Genom. 2019, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG Database: A tool for fenome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2015, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Li, J.; Wu, S.; Zhu, Y.; Chen, Y.; He, F. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Profile hidden markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; McCarthy, D.J.; Chen, Y.; Okoniewski, M.; Smyth, G.K.; Huber, W.; Robinson, M.D. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 2013, 8, 1765. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-discovery approach for sample matching of a nerve-agent precursor using liquid chromatography− mass spectrometry, XCMS, and chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Wu, Q.; He, W.; He, T.; Wu, Q.; Miao, Y. Combined De Novo Transcriptome and Metabolome Analysis of Common Bean Response to Fusarium oxysporum f. sp. phaseoli Infection. Int. J. Mol. Sci. 2019, 20, 6278. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246278
Chen L, Wu Q, He W, He T, Wu Q, Miao Y. Combined De Novo Transcriptome and Metabolome Analysis of Common Bean Response to Fusarium oxysporum f. sp. phaseoli Infection. International Journal of Molecular Sciences. 2019; 20(24):6278. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246278
Chicago/Turabian StyleChen, Limin, Quancong Wu, Weimin He, Tianjun He, Qianqian Wu, and Yeminzi Miao. 2019. "Combined De Novo Transcriptome and Metabolome Analysis of Common Bean Response to Fusarium oxysporum f. sp. phaseoli Infection" International Journal of Molecular Sciences 20, no. 24: 6278. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246278