Coagulatory Defects in Type-1 and Type-2 Diabetes



Medical and Biological Sciences Building, School of Medicine, University of St Andrews, St Andrews KY16 9TF, UK
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(24), 6345; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246345
Submission received: 14 November 2019
/
Revised: 6 December 2019
/
Accepted: 12 December 2019
/
Published: 16 December 2019
(This article belongs to the Special Issue Molecular and Cellular Basis of Thrombotic Diseases)
Abstract
:Diabetes (both type-1 and type-2) affects millions of individuals worldwide. A major cause of death for individuals with diabetes is cardiovascular diseases, in part since both types of diabetes lead to physiological changes that affect haemostasis. Those changes include altered concentrations of coagulatory proteins, hyper-activation of platelets, changes in metal ion homeostasis, alterations in lipid metabolism (leading to lipotoxicity in the heart and atherosclerosis), the presence of pro-coagulatory microparticles and endothelial dysfunction. In this review, we explore the different mechanisms by which diabetes leads to an increased risk of developing coagulatory disorders and how this differs between type-1 and type-2 diabetes.
Keywords:
atherosclerosis; endothelial dysfunction; magnesium; metal ions; microparticles; platelets; thrombosis; zinc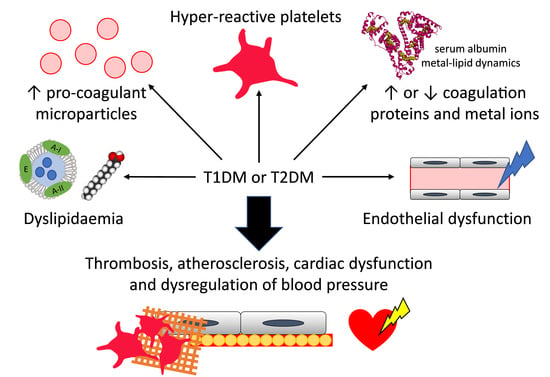
1. Introduction
Diabetes is a term used to describe a group of conditions that impact upon the body’s ability to properly control blood glucose levels. In 2017, combined occurrences of type-1 diabetes mellitus (T1DM) and type-2 diabetes mellitus (T2DM) were estimated at 425 million individuals worldwide [1]. This number is predicted to rise to 629 million by 2045 [1]. All forms of diabetes are characterised by defective signalling of insulin, the peptide hormone responsible for stimulating cellular glucose uptake. In T1DM, the insulin-secreting β-cells in the islets of the pancreas are destroyed, by the immune system in the first subtype of T1DM, idiopathically in the second subtype [2]. This results in a decrease of insulin production. T2DM is a polygenetic disease; it can be divided into two subtypes, with and without obesity, and several genes can predispose individuals to developing the disease. It displays a heterogenous phenotype that is the consequence of resistance to insulin signalling, often due to defects associated with insulin receptors. At the beginning of the disease, insulin secretion is impaired and results in hyperinsulinemia. However, as the disease progresses, β-cells can become damaged, leading to hypoinsulinemia [3].
Both T1DM and T2DM have wide-ranging consequences for the body as glucose levels are associated with many physiological processes. These include lipid metabolism and the regulation of inflammation, vasodilatation, basic cell growth and replication. Unmanaged diabetes and hyperglycaemia can worsen these physiological changes, potentially leading to diabetes-associated complications. In particular, individuals with diabetes are two to three times more likely to develop cardiovascular diseases than those without diabetes [1]. For example, several coagulatory defects are observed in individuals with T1DM or T2DM. Indeed, the vascular endothelium is altered in individuals with both these types of diabetes, and so hypertension, premature atherosclerosis and more extensive vascular diseases can be found in affected individuals compared to the general population, thus also increasing their risk of plaque rupture (in the case of atherosclerosis) and thrombus formation [4,5,6]. Furthermore, in individuals with diabetes, platelets are hyper-reactive, giving rise to increased activation of prothrombotic factors and decreased fibrinolysis which results in an increased risk of thrombosis [4,7]. In addition, the altered lipid profile found in individuals with diabetes affects cardiac function and can cause lipotoxicity in the heart [5]. Due to these factors, up to 80% of individuals with diabetes die as a result of cardiovascular complications [8]. The prognosis following a cardiovascular event remains poor for individuals with diabetes despite intensive research on the subject and the development of new therapies [4,9]. Thus, it is important to better understand the underlying mechanisms that drive the haemostatic changes observed in T1DM and T2DM.
Despite the known increased risk of cardiovascular disease in individuals with diabetes, the pathophysiology underlying this relationship is complex and not completely understood. Nevertheless, among the many physiological changes induced by diabetes that can impact on the cardiovascular system are changes in the concentrations of plasma proteins and metal ions, altered lipid metabolism and lipid composition (resulting in altered metabolic regulation), cardiac lipotoxicity and atherosclerosis, endothelial dysfunction, platelet hyper-activation and the presence of pro-coagulatory particles in the blood. Here, we review the molecular and cellular changes that can lead to the increased thrombotic risk observed in individuals with diabetes.
2. Diabetes and Thrombosis: Abnormal Coagulation Mechanisms
2.1. Alterations of Plasma Protein Concentrations
T1DM and T2DM are associated with changes in blood coagulability, including alterations in clot structure and in the kinetics of clot formation and lysis. The factors responsible for these alterations include changes in the concentration and activity of numerous coagulatory proteins, resulting in defective thrombin generation and changes in the molecular make-up of fibrin clots. Proteins identified as exhibiting an altered concentration in both T1DM and T2DM are summarised in Table 1, while Figure 1 summarises the activity of those proteins in coagulation. Proteins with elevated concentrations in both types of diabetes include von Willebrand factor (vWF) [10,11,12], (pre)kallikrein [13,14], factor V [15], (activated) factor VII [15,16,17], factor VIII [15,18], factor X [15], factor XI [14], prothrombin [15], and fibrinogen [19,20,21] (although a study has also reported its reduction in T1DM [22]). Proteins only elevated in T2DM include: kininogen [23], soluble tissue factor [16,24], factor IX [18], (activated) factor XII [18,25], and factor XIII [26]. In contrast, in T1DM, activated factor XII levels are reduced [27]. Simultaneously to changes in pro-coagulation proteins, several anticoagulation proteins have a reduced plasma concentration in both types of diabetes, including protein C [15,19,28,29] and protein S [30], but thrombomodulin [29,31] has an elevated concentration in both types of diabetes and tissue factor pathway inhibitor levels are elevated in T2DM [16]. A number of reports have examined antithrombin concentration in T2DM. One such study found reduced concentrations [32], whilst two other studies reported elevated concentrations of this protein associated with the disease [18,33]. The cause of this difference is not known; it may be due to a difference in methodology or to the individuals studied being at a different stage of progression of the disease. Antithrombin cofactors, the heparan sulphate glycosaminoglycans in the endothelium surface layer, are largely responsible for the anticoagulant properties of the endothelium [34]; the concentration of these molecules is decreased in the arteries of individuals with T2DM, especially in those with lesions [35]. The pro-fibrinolysis protein, tissue plasminogen activator has an increased concentration in individuals with glucose intolerance [36] and in individuals with T1DM [37] or T2DM [24], but its availability is decreased because of the elevated concentration of plasminogen activator inhibitor 1 (PAI-1) associated with glucose intolerance in non-diabetic individuals [36] and with individuals with T2DM [10,19,20,24,38]. In contrast, in T1DM, the PAI-1 concentration is reduced [39]. The concentration of other inhibitors of fibrinolysis, including thrombin-activatable fibrinolysis inhibitor [40,41] and α2-macroglobulin [32,42], are also elevated in both T1DM and T2DM, while the α2-antiplasmin concentration is elevated in T2DM [19], but in T1DM it has been reported to be elevated in two studies [19,43], but reduced in another [22]. Again, the origin of this difference is not known but could be due to differences in methodology or to different stages of progression of the disease in the individuals studied. Changes in plasma protein concentrations as well as plasma glucose levels lead to an increase in plasma viscosity in T2DM [44] and an increasing trend has been measured in T1DM, especially in individuals with bad glycaemic control [45,46]. Most but not all changes in the protein concentration of coagulatory proteins in plasma are due to uncontrolled glycaemia and so can often be reversed through control of blood glucose levels: for example, protein C, protein S and antithrombin concentrations have been demonstrated to increase in T2DM subjects following improvement in glycaemic control [47]. Some proteomic alterations are influenced by genetic factors. For example, the increase in fibrinogen concentration and factor VII coagulant activity in T2DM are also seen in first degree relatives of individuals without the disease [48]. In addition, chronic inflammation (as is associated with both T1DM and T2DM) leads to activation of both the complement system and the kinin–kallikrein system, resulting in the activation of factor XII and elevated concentrations of several proteins including factor VIII, tissue factor, prothrombin and fibrinogen [7].
The concentration of coagulatory proteins is not the only factor that impacts on coagulation. While elevated concentrations of PAI-1 are found in T2DM [10,19,20,24,38], in T1DM it is the activity of the protein that is reduced [22,52]. Also, alterations in fibrin(ogen) function are also more complex than just a change in concentration: an examination of clots formed from fibrinogen purified from individuals with T2DM and controls found the T2DM-derived samples exhibited denser and less porous clots [53]. This can be explained by elevated glycation of fibrinogen in diabetes and can be abrogated with better glycaemic control [53,54,55]. Fibrinogen is not the only protein affected by poor glycaemic control; plasminogen, the precursor of plasmin, undergoes increased glycation in individuals with T1DM, thereby leading to a reduced fibrinolytic-activity of plasmin [51]. Furthermore, the activity of antithrombin is inhibited by methylglyoxal, a by-product of hyperglycaemia [56]. In addition, in healthy individuals subjected to combined hyperglycaemia and hyperinsulinemia, the tissue factor pathway has been shown to be increasingly activated (compared to an euglycemia–hyperinsulinemia group), as reflected by elevated concentrations of activated factor VII and tissue factor pathway inhibitor, as well as in an increase in factor VII activity [57]. Thus, thrombin generation (followed by measuring levels of the thrombin–antithrombin complex) is increased in individuals with T2DM or T1DM [28,58]. Good glycaemic control is also important for anticoagulant activity, with better glycaemic control in T2DM leading to a reduction of thrombin generation [59] and an increase of the anticoagulant activity of antithrombin, protein C and protein S [47].
Thus, changes in both the concentrations and activities of coagulation proteins have important consequences on fibrin clot formation, clot lysis parameters and fibrin clot ultrastructure. Elevated PAI-1 concentration results in prolonged lysis time of the fibrin clot in individuals with T2DM [10]. The elevated concentration of complement protein C3 found in T1DM results in the protein being increasingly incorporated into fibrin clots formed from fibrinogen purified from blood from those individuals with T1DM, leading to delayed fibrin clot lysis [60]. This has also been observed in T2DM [61]. In T1DM, both lysis time and the concentration of C3 improved with better glycaemic control [60]. Similarly, α2-antiplasmin is also increasingly incorporated into fibrin clots in T1DM and T2DM, which has been shown to increase lysis resistance [22,62]. Fibrin clots formed in individuals with T1DM, like with T2DM, are more compact in correlation with glycaemic control [63]. In both types of diabetes, fibrin clots are more resistant to fibrinolysis [10,53,63]. Diabetes duration also has an impact, with prolonged T2DM duration (more than five years) associated with increased thrombin generation, reduced fibrinolysis and a pro-thrombotic phenotype even with good glycaemic control [64]. PAI-1 and t-PA antigen levels are also higher in prolonged T2DM duration while fibrinogen, plasminogen, soluble thrombomodulin and thrombin-activatable fibrinolysis inhibitor antigen levels are unaffected [64]. In addition, differences in coagulatory protein levels and clot parameters were found between males and females with T1DM or T2DM: indeed, fibrinogen and PAI-1 concentrations are higher in females with T2DM than in males with T2DM, and after correcting for those factors, females still had more compact clots that were resistant to fibrinolysis than males [65], However, another study with fewer individuals found unchanged fibrinogen levels and reduced PAI-1 levels in females with T2DM [66]. The incorporation of α2-antiplasmin in clots was found to be increased in females with T2DM, making them more resistant to fibrinolysis [66]. In T1DM, clot density and fibrinogen concentration in females are the same as in males, while their factor XIII concentration is higher than in males [67]. PAI-1 concentration is also similar between females and males with T1DM [39]. When looking at a younger cohort with T1DM (under 30 years old), females have a prolonged lysis time compared to males, but this is not the case for an older cohort [67]. Thus, in both types of diabetes, these combined changes result in an increase in pro-coagulation mechanism and a decrease in anticoagulation and fibrinolysis, leading to an elevated thrombosis risk.
2.2. Changes in Metal Ion Homeostasis
Metal ions play numerous roles in blood plasma, which include structural and catalytic functions. The plasma concentration of several metal ions is known to be altered in T1DM and T2DM [68]. This is important as many of these are necessary for the normal functioning of proteins involved in coagulation [69,70,71,72,73]. Ca2+ is an important regulator of coagulation. Ca2+ is released by activated platelets and is required for clotting to take place (in particular for tenase and prothrombinase complexes to function). Chelating agents that bind calcium (e.g., citrate and ethylenediaminetetraacetic acid) are common anticoagulants used when taking blood samples. Several studies have found associations between high levels of calcium in the blood and risk of developing T2DM. Such studies include the PREDIMED study [74], the Atherosclerosis Risk in Communities (ARIC) study [75], the Insulin Resistance Atherosclerosis study [76] and the Tromsø study [77]. In addition, elevated total plasma calcium levels are also found in individuals with T2DM compared to healthy controls; no difference was observed between males and females and the duration of diabetes had no influence on calcium levels [78]. Serum calcium levels are unchanged in T1DM [79,80]. The effects of elevated calcium levels on coagulation in T2DM have not been fully characterised. In the general population, a meta-analysis has shown that taking high levels of calcium supplements (>1000 mg/day) increases cardiovascular risk in men but not in women where the benefits of taking the supplements outweighs the risks [81]. In this study, calcium supplementation was found to also increase the risk of coronary artery calcification and atherosclerosis in both sexes [81]. However not all calcium taken in supplements will be absorbed and much of the absorbed excess calcium will be stored in bones. In individuals with T2DM, plasma calcium levels are elevated. Thus, further studies are necessary to determine if part of the hypercoagulability found in individuals with T2DM may be explained by alterations in plasma calcium concentration.
In addition to Ca2+, Zn2+ is also extremely important in regulating coagulation [69]. Like with Ca2+, Zn2+ is also released by activated platelets, as well as damaged epithelial cells and atherosclerotic plaques; it is also contained by neutrophils, lymphocytes and erythrocytes and may therefore be released at sites of injury (although this has yet to be confirmed) [82]. Zn2+ is involved in all steps of coagulation: pro-coagulatory, anti-coagulatory, pro-fibrinolysis and anti-fibrinolysis mechanisms, as well as platelet activation and aggregation [69]. Zinc deficiency causes bleeding and platelet aggregation disorders [69]. In T2DM and T1DM, zinc concentrations are reduced compared to healthy subjects [83,84]. However, in plasma, Zn2+ ions are transported by serum albumin, which is also the main transporter of free fatty acids (FFAs) [85]. When a FFA binds to the high affinity binding site FA2 on serum albumin, the protein conformation changes and the main Zn2+ binding site is disrupted and can no longer bind Zn2+ [85]. Thus, when pathological concentrations of FFAs are present in the blood, such as is the case in T2DM and in some cases of T1DM, Zn2+ handling/buffering by serum albumin is dysregulated and plasma zinc speciation (the molecules to which it is bound) is altered [86]. Recently we have shown using size-exclusion-chromatography-ICP-MS that Zn2+ is redistributed among other plasma proteins in the presence of pathophysiological FFA concentrations [87]. Considering the many coagulatory proteins that are regulated by Zn2+, this altered zinc speciation can dysregulate coagulation, resulting in increased platelet aggregation, increased fibrin clot density, and delayed fibrinolysis, thus potentially participating in the elevated thrombotic risks found in T2DM [86].
Mg2+ is also important for coagulation. Mg2+ can potentiate the activation of factor X by activated factor IX while in the presence of activated factor VIII, phospholipids and Ca2+ [88], the activation of factor IX by activated factor VII-tissue factor complex [88], and the inactivation of activated factor V by activated protein C [89]. Mg2+ also affects clot time by accelerating clotting at low concentrations and slowing or completely preventing fibrin clotting at high concentration (as it competes with Ca2+ for binding to coagulation factors [90,91,92,93]). Furthermore, Mg2+ shortens fibrin clot lysis time, possibly through an inhibition of PAI-1 in the presence of thrombin and vitronectin [92]. Magnesium deficiency in humans and animals has been shown to cause hypercoagulability [94]. Magnesium deficiency has been observed in T1DM in both males and females, although the effect is more pronounced in females [68]. Magnesium deficiency in T1DM has been associated with delayed fibrinolysis and a higher thrombotic risk [39]. Individuals with T2DM are also at risk of magnesium deficiency and may therefore be affected by a similar mechanism [95]. Bad glycaemic control is associated with magnesium deficiency, as it reduces the tubular reabsorption of magnesium [68].
Total plasma copper levels are elevated in T2DM and T1DM [83,96]. The potential effect of copper on coagulation is not well known, despite Cu+ and Cu2+ being essential cofactors of several coagulation proteins (e.g., coagulation factors V and VIII) [72,73]. Elevated dietary levels of copper in rats (which were reflected in copper concentrations in the liver) were found not to affect clot time when clotting was induced by thromboplastin and Ca2+ (when assaying for factor X, thrombin and fibrinogen activation) or Ca2+ and phospholipids (when testing the whole extrinsic pathway of coagulation) [97]. However, more than 95% of copper found in plasma is carried by the protein ceruloplasmin [71]. Levels of ceruloplasmin are elevated in individuals with T1DM, possibly due to inflammatory processes [96]. In T2DM, ceruloplasmin levels can be reduced or elevated; a meta-analysis has shown that globally ceruloplasmin levels are increased but not significantly (p = 0.06) and that sex does not influence this parameter [83]. However, another study compared T2DM individuals without diabetic complications with T2DM individuals with complications and found the latter group to have higher ceruloplasmin levels [98]. In cases where high blood concentrations of ceruloplasmin has been observed, the protein has been shown to bind to activated protein C to reduce its anticoagulant activity and induce acquired activated protein C resistance—a state associated with a higher risk of venous thrombosis [70,71]. No direct study on the link between ceruloplasmin levels in diabetes and coagulation has been performed.
Individuals with T2DM have higher plasma levels of ferritin and higher, but not significantly so, total plasma levels of iron (p = 0.06); sex does not influence either parameter [83]. Individuals with T1DM are deficient in iron; diabetes duration or sex had no influence on iron deficiency, but the menstrual cycle did [99,100]. For those individuals deficient in iron, treatment includes supplementation with Fe3+ salt [101]. This has been shown to impact on coagulation by extending the clotting time of plasma (possibly by competing with calcium in binding coagulation factors), weakening the fibrin clot (by interacting with fibrinogen and fibrin), and inducing the precipitation of plasma proteins to form “insoluble coagulums” resistant to lysis (notably by binding and degrading serum albumin and possibly transferrin), thus increasing the risk of thrombosis [101]. In addition, Fe3+ has been shown to initiate the conversion of fibrinogen into a fibrin-like polymer, parafibrin, that is resistant to proteolysis and so is deposited in blood vessels [102]. The persistent presence of this parafibrin has been argued to cause chronic inflammation [102]. Thus, the altered levels of plasma metal ions in individuals with T1DM and T2DM will impact on coagulation and the risk of developing cardiovascular diseases.
2.3. Changes in Lipid Metabolism at the Origin of Atherosclerosis and Lipotoxicity
Both T1DM and T2DM are associated with changes in lipid metabolism. Plasma cholesterol, low-density lipoprotein (LDL) and triglyceride concentrations are increased and high-density lipoprotein (HDL) concentration is decreased in individuals with T2DM, and in individuals with T1DM and bad glycaemic control [103,104]. Unchanged cholesterol levels in individuals with T1DM and good glycaemic control can be deceptive as lipid profiles and functioning are altered [105,106]. Traditionally, high levels of total cholesterol and LDL have been regarded as a major risk factor of atherosclerosis and cardiovascular disease in the general population. However, a recent review of the literature by Ravnskov et al. has argued that total cholesterol and LDL do not cause those diseases [107]. They explain the difference between this new view and the traditional view as the failure of most meta-analyses to properly account for negative studies [107]. They also argue that the associations between cardiovascular disease and LDL or cholesterol concentrations found in certain cohorts can be explained through different mechanisms. A possible explanation is that infections can cause cardiovascular disease and that LDL participates in immune functioning by adhering to and inactivating microorganisms and their toxic products [107]. Another is that stress also causes cardiovascular diseases as increased production of adrenalin and noradrenaline contribute to hypertension and hyper-coagulation, and that cholesterol is a precursor for cortisol and other steroid stress hormones [107]. However, whether this new view reflects what really happens at the molecular level is unclear as this study looked at the general population and not individuals with T1DM and T2DM, in which lipid-lowering drugs remain an essential treatment to prevent the development of complications, including cardiovascular diseases [104,105].
In diabetes, high LDL levels are associated with cardiovascular diseases, but they are not an accurate predictor of cardiovascular risks in T1DM [105,108,109]. Nevertheless, there are higher levels of “small dense LDL” in T1DM and T2DM and these forms of LDL penetrate more easily in the arterial wall than “large buoyant LDL” [110,111,112]. Small dense LDL are also more susceptible to oxidative stress, have a reduced affinity for LDL receptors and have a prolonged half-life in plasma than large buoyant LDL [110]. In addition, they are more easily glycated as they carry a higher proportion of apolipoprotein B, which is exposed to glucose [113]. Furthermore, oxidized LDL inhibits endothelial nitric oxide production [114] and can more easily be taken up by macrophages as part of atherosclerotic plaque formation [112]. These characteristics are all associated with endothelial dysfunction (see Section 2.4) and cardiovascular diseases [105,112,115].
HDL has long been thought to have protective properties against cardiovascular diseases. However, recent evidence has shown that a low HDL concentration is associated with an increased cardiovascular risk, but that a high HDL concentration does not have a protective effect and could even be dangerous, as has been shown in the general population and in individuals with T1DM [106,116,117]. These unexpected results may be explained by the existence of different HDL subspecies with different functions [118]. Thus, it may be beneficial to directly measure HDL function, such as its role in promoting reverse cholesterol transport (the net movement of cholesterol from peripheral tissues to the liver to be excreted through the bile) [118]. For example, when the macrophages present in artery walls accumulate excess cholesterol, the ATP-binding cassette transporters A1 and G1 are induced and this results in the efflux of cholesterol from the macrophages to the HDL [118]. HDL efflux has been found to be a better predictor of cardiovascular disease than HDL levels [119,120,121,122], although other studies have disputed this [123,124,125,126]. HDL efflux is reduced in individuals with T2DM [127,128] or T1DM [129]. This may be because reactive oxygen species, which are increased in individuals with diabetes, can impact HDL function [130]. Beyond its role in cholesterol efflux, HDL also has an anti-atherosclerotic effect as it promotes nitric oxide production in endothelial cells, an essential feature for endothelial function [118]. HDL also has anti-inflammatory and antioxidant effects [118]. A study comparing the effects of HDL taken from individuals with T2DM and controls showed that in T2DM, HDL failed to stimulate nitric oxide production by endothelial cells and did not promote endothelial repair [131]. In addition, in T2DM, HDL have reduced levels of HDL-associated sphingosine-1-phosphate (S1P), resulting in a reduced ability to activate endothelial nitric oxide synthase [132]. Furthermore, individuals with T1DM have the same levels of S1P and apolipoprotein M in total HDL as controls, but the HDL-associated apolipoprotein M/S1P complex move to a different subset of HDL, from buoyant HDL to dense HDL, where it has reduced anti-inflammatory effects due to altered S1P1 receptor activation [133].
Hypertriglyceridemia is often associated with T1DM and T2DM [134,135,136]. Cholesterol ester transfer protein (CETP) exchanges cholesterol and triglycerides between very low-density lipoprotein and HDL [137]. When the plasma level of triglycerides is too high, the equilibrium is displaced, and HDL are impoverished in cholesterol and enriched in triglycerides [137]. This results in impaired HDL structure, as the hydrophobic core of the triglycerides partially extrude to the HDL surface [138], and therefore impaired HDL function [139], and (eventually) endothelial dysfunction. Contrary to HDL-cholesterol levels, HDL-triglycerides have been shown to be an effective marker of increased cardiovascular risk in the general population [140] and in individuals with T2DM or with metabolic syndrome [137].
FFA levels are also altered in diabetes; total plasma FFA concentrations and the plasma concentrations of most major FFA species are increased in individuals with T2DM [86], while they are reduced in individuals with T1DM and bad glycaemic control [141]. FFAs are important regulators of many physiological processes and their dysregulation can have important consequences [5]. The most obvious one is the adherence of excess FFAs on the endothelial walls of blood vessels and their subsequent accumulation that results in the formation of atherosclerosis plaques [5]. These plaques can make blood vessels narrower, thus facilitating their full blockage, and plaque rupture is a pro-thrombotic event that can trigger thrombosis and embolism [142]. In addition, the fatty acid translocase CD36, which is located on macrophages and platelets, can be activated by FFA to trigger coagulation [143]. Furthermore, elevated plasma FFA concentrations directly affect fibrin clot parameters; the saturated FFA, stearic acid, has been shown to increase the diameter of fibrin fibres in a purified system, while an unsaturated FFA, oleic acid, reduced it [144]. Stearic and oleic acids also both increased clotting time and reduced the mechanical stability of the clot through a decreased rigidity, a higher deformability and a decreased internal resistance to shear stress [144]. In addition, plasma FFA levels also impact on the function of the heart as they increase its susceptibility to oxidative stress and ischemic damage [145]. Indeed, excess FFAs also lead to the formation of toxic lipids (in particular diacylglycerides and ceramide) thus promoting endoplasmic reticulum stress, mitochondrial dysfunction and the generation of reactive oxygen species [5]. This leads to inflammation, insulin resistance and apoptosis of cells [5]. These toxic lipids also activate protein kinase C (PKC) [5]. PKC activation impairs intracellular Ca2+ handling in the heart, affecting cardiac contractibility and promoting cardiac fibrosis and hypertrophy [145,146]. While n-3 unsaturated FFAs have been shown to have anti-arrhythmic and cardioprotective effects, saturated FFAs can lead to electrophysiological remodelling and sustained and fatal arrhythmias [147]. Thus, the changes in lipid levels and lipid metabolism found in T1DM and T2DM can have a strong impact on the risk of developing cardiovascular diseases, as summarised in Figure 2.
2.4. Endothelial Dysfunction
Endothelial dysfunction can be defined as a diminished production and/or availability of nitric oxide, a molecule involved in vascular homeostasis, vasodilation and platelet inhibition, and as an imbalance between vasodilators and vasoconstrictors in the vasculature. Endothelial dysfunction precedes the development of atherosclerosis and increases the risk of cardiovascular diseases. Diabetes is associated with a series of changes in endothelial function caused by several factors including an excess of plasma FFAs in T2DM and alterations in glucose metabolism, impaired insulin signalling, chronic inflammation and oxidative stress in both T1DM and T2DM [148]. Excess plasma FFAs causes a dysregulation of Ca2+ and insulin signalling, resulting in a reduction in the production of nitric oxide, thus leading to increased endothelial permeability [142]. The activation of the NLRP3 inflammasome by excess FFAs also contributes to this increase in permeability [142]. Excess FFAs also impact on the renin–angiotensin system, resulting in the dysregulation of arterial blood pressure [142]. In addition, the activation of the NF-κB inflammation pathway (by saturated FFAs but not polyunsaturated FFAs) leads to an increase in the production of superoxide in the endothelium [142], which itself increases the concentration of a range of enzymes including oxidative enzyme systems such as NADPH oxidase, xanthine oxidase, cyclooxygenases, lipoxygenases, myeloperoxidases, cytochrome P450 monooxygenase, uncoupled nitric oxide synthase, and peroxidases. Collectively, these enzymes inactivate nitric oxide. Furthermore, the combination of oxidative stress and hyperglycaemia seen in diabetes leads to the glycation of plasma proteins and lipids and generates advanced glycation end-products (AGEs) [148]. These AGEs then accumulate in the vessel walls and disrupt cell function, notably by binding to AGE receptors (RAGEs) [148]. Signalling by RAGEs downregulates nitric oxide synthase in endothelial cells and upregulates expression of vascular cell adhesion molecule, intercellular adhesion molecule, E-selectin (three cellular adhesion molecules), monocyte chemoattractant protein-1 (a regulator of migration and infiltration of monocytes and macrophages), endothelin-1 (a vasoconstrictor) and tissue factor [148]. Insulin resistance itself also reduces nitric oxide production and stimulates endothelin-1 secretion [149]. Dysregulation of the aforementioned pathways contributes to the pro-inflammatory and pro-thrombotic properties of the endothelium in diabetes. In addition, diabetes is associated with decreased endothelial synthesis of prostacyclin, a vasodilator and inhibitor of platelet activation [148]. Furthermore, matrix metalloproteinases are zinc-binding proteinases that degrade components of the extracellular matrix and whose production is upregulated notably by hyperglycaemia, pro-inflammatory mediators and reactive oxygen species [150]. Matrix metalloproteinase levels are associated with cardiovascular disease development and all-cause mortality in T1DM [151,152,153], and with cardiovascular organ damage in T2DM [154]. In diabetes, they increase inflammation, endothelial dysfunction, vascular remodelling and thrombus formation [155,156]. The imbalance of matrix metalloproteinases and their inhibitors observed in diabetes is associated with the formation and destabilisation of atherosclerotic plaques [157,158]. Thus, endothelial dysfunction is a major factor contributing to the risk of cardiovascular disease in individuals with T1DM and T2DM, as summarised in Figure 3.
2.5. Platelet Hyper-Activation
Many of the key changes that impact upon the coagulation system in diabetes involve platelets. In normal physiology, platelets are activated in response to exogenous stimuli including thrombin (which binds to the G protein-coupled receptors, PAR1, PAR3 and PAR4), collagen (which binds to the receptor GPVI-αIIβI) and thromboxane A2. The P2Y12 pathway can amplify these stimuli by triggering the secretion of thromboxane A2 and ADP from internal stores. Activation is carried out through intracellular Ca2+ flux and results in changes in the level of expression of surface glycoproteins (including integrins) which can then act as receptors for platelet agonists and for adhesion proteins involved in platelet aggregation. After platelet activation, P-selectin translocates from α-granule membranes to the plasma membrane, and the GPIIb-IIIa complex on the plasma membrane undergoes a conformational change that exposes a fibrinogen binding site. Platelets then secrete the content of their granules (including Ca2+, Zn2+, coagulation factors and growth factors), adhere to subendothelial surface (GPIb-IX-V binds to vWF, GPIIb-IIIa binds to vWf or fibrinogen, and fibrin and other coagulation factors interact with the platelet surface), and aggregate to form a thrombus. Regulation of platelet function occurs through the action of the anti-aggregants prostacyclin and of nitric oxide, both of which are secreted by intact endothelial cells. Insulin inhibits platelet responses to stimuli through the P2Y12 pathway and sensitises platelets to the anti-aggregant effects of nitric oxide and prostacyclin.
The hyperglycaemia found in T1DM and T2DM results in: (1) reduced nitric oxide and prostacyclin production from the endothelium and nitric oxide production by platelets leading to an imbalance in the anti-aggregation mechanism [159]; (2) reduced insulin sensitivity of platelets in T2DM (or reduced insulin levels in T1DM), which leads to reduced inhibition of the P2Y12 pathway, itself resulting in a reduced platelet response threshold to stimuli and so an increased platelet reactivity [160,161]; (3) the glycation of proteins at the surface of platelets, leading to altered activity of, and signalling by, receptor proteins and to reduced platelet membrane fluidity, thus increasing platelet sensitivity to thrombin and platelet adhesion [7,159,162]; (4) increased activation of PKC which increases platelet activation [163]; (5) increased oxidative stress which activates the PKC pathway, but also leads to an increase in intracellular Ca2+ signalling and so to an increased platelet activation and aggregation [163,164]; (6) decreased production of antioxidants like glutathione, which has been linked to increased formation of thromboxane A2, leading to increased platelet activation [159,165]; (7) elevated basal Ca2+ levels in platelets and disturbed Ca2+ homeostasis which directly regulates platelet activation, platelet morphology, and initiation of coagulation [159,166]; (8) increased surface expression of glycoproteins such as GPIb and GPIIb/IIIa and increased activation of GPIIb/IIIa, leading to increased binding to vWF and fibrin(ogen), both resulting in increased platelet aggregation [159,167].
Furthermore, the hyper-activation of platelets in individuals with diabetes means that they are consumed more rapidly such that platelet turnover is faster [159]. This leads to the generation of new platelets that are themselves inherently hyperactive [168]. In addition, in both T1DM and T2DM, platelet counts have been found to be higher [169]. This parameter responds positively to glycaemic control only in individuals with T1DM, while individuals with T2DM have a higher number of large platelets which display increased platelet reactivity [159,168,169]. Thus, in individuals with diabetes, platelets are more active, leading to increased adhesion, activation and aggregation and the increased production of platelet-derived microparticles [6,159]. Collectively, these changes result in an increased triggering of thrombus formation and an increased release of pro-coagulatory molecules by platelets such as Ca2+, Zn2+, fibrinogen, vasoconstrictors and oxidative reactive species which increase coagulation and the atherosclerotic process in both T1DM and T2DM (Figure 4) [159].
2.6. Pro-Coagulatory Microparticles
During cell growth, proliferation, activation and apoptosis, cells communicate through the release of extracellular vesicles. Microparticles are a heterogenous type of these vesicles with a diameter of 0–0.1 µm and whose content includes lipids, proteins and microRNAs depending on their origin [170]. The shedding of microparticles by cells is triggered notably by pro-inflammatory cytokines, AGEs, oxidative stress, LDLs and hyperglycaemia and their size, structure and content differ depending on the cell type and the stimuli triggering their formation [159]. Microparticles are constantly present in the blood, but many cardiovascular diseases are associated with elevated levels of these, especially microparticles derived from platelet and endothelial cells [171]. In T2DM, microparticle levels in the blood are increased; in particular, endothelial-derived microparticles enriched in CD31, CD62E, CD105 and CD106 [172], as well as platelet-derived microparticles enriched in fibrinogen (two-fold increase compared to non-diabetic individuals [173]), tissue factor (three-fold increase compared to non-diabetic individuals [173]) and P-selectin [174]. In addition, a proteomic analysis carried on microparticles taken from individuals with T2DM and controls has shown that in T2DM, proteins involved in platelet activation, cell adhesion, and inflammation are differentially expressed [175]. High levels of platelet-derived microparticles are associated with atherosclerotic progression and arterial thrombosis in individuals with T2DM [170]. Microparticle levels in T2DM are an independent predictor of adverse cardiovascular events (adjusting for age, gender, hyperlipidaemia, smoking and statin use) [170]. Individuals with T1DM also have elevated levels of endothelial- and platelet-derived microparticles and total levels of microparticles enriched with annexin V [176,177,178]. In T1DM, increased pro-coagulant activity was found to be associated with the total number of microparticles enriched with annexin V [177]. The levels of endothelial- and platelet-derived-microparticles as well as procoagulant activity directly correlated with HbA1c levels in individuals with T1DM [176,177,178].
Microparticles formed from platelets are extremely pro-thrombotic and facilitate thrombin generation [171]. These microparticles are enriched in tissue factor, constituting a “blood-borne” reservoir of this protein [171]. While tissue factor exposed at the blood vessel wall during coagulation initiates thrombus formation, the “blood-borne” tissue factor is involved in the propagation of coagulation [171]. Thus, the increase of “blood-borne” tissue factor found in T2DM contributes to the pro-thrombotic phenotype of the disease [170]. Such platelet-derived microparticles can be trapped in the developing thrombus through interactions of CD15, CD18 and tissue factor with the thrombus [170]. Furthermore, smooth muscle cell-derived highly pro-thrombotic microparticles enriched in tissue factor can be trapped inside atherosclerosis plaques and released during plaque erosion or rupture [179]. Microparticles then create a binding surface for further platelet recruitment and for fibrin after plaque disruption [170]. Microparticles derived from activated platelets can also activate other platelets by releasing arachidonic acid [180]. In addition to tissue factor, microparticles can induce thrombin generation in a factor XII-dependent manner (for erythrocyte- and platelet-derived microparticles) and in a factor XI-dependent manner (for erythrocyte-derived microparticles) [179,181]. Thus, the elevated levels of pro-coagulatory microparticles present in individuals with T1DM and T2DM participate in the high thrombotic risk associated with those diseases (Figure 4).
3. Differences in Thrombotic Risks Between T1DM and T2DM
Despite the different aetiologies of the diseases, numerous similarities exist in the hyper-coagulatory state found in individuals with T1DM and T2DM, as well as differences. Both diseases are characterised by hyperglycaemia, altered insulin metabolism, dyslipidaemia, endothelial dysfunction, oxidative stress and inflammation. However, individuals with T2DM have elevated FFA levels that are not found in T1DM, which can impact on the regulation of many physiological processes, including alterations in fibrin clot parameters, endothelial dysfunction, atherosclerotic plaque formation and cardiac lipotoxicity. In addition, insulin levels are often elevated in T2DM and accompanied with insulin resistance, while they are lowered in T1DM (although insulin resistance can also appear in T1DM [182]). In T2DM, β-cells can become damaged, leading to reduced insulin production and increased platelet reactivity [3]. Furthermore, both diseases are associated with differentially altered plasma concentrations of metal ions, leading to dysregulation of coagulation via distinct mechanisms. Finally, PAI-1 levels are elevated in T2DM but reduced in T1DM, even though both these diabetes types are associated with prolonged fibrin clot lysis times. These distinctions are very important and need to be taken into account during treatment of each disease. The similarities and differences in how T1DM and T2DM impact on coagulation are summarised in Table 2.
Few studies have looked at whether subtypes of T1DM and T2DM have an influence on these pro-thrombotic factors. Obese individuals with T2DM can be expected to have elevated plasma FFA levels compared to non-obese individuals with T2DM and we have already discussed the pro-thrombotic effects of FFAs. In addition, obese individuals with T2DM have higher risks of thrombosis than non-obese individuals with T2DM as they have delayed fibrinolysis, higher plasma concentrations of vWF and fibrinogen and higher levels of factor VII and factor VIII activity [183]. Individuals with T2DM who also have a genetic predisposition to T2DM are likely to be associated with some additional pro-thrombotic factors compared to individuals with T2DM without genetic predisposition; indeed, individuals with some genetic predisposition to T2DM but who have not developed T2DM have an elevated risk of cardiovascular disease [184,185]. Among individuals with T2DM, those who have a genetic predisposition to T2DM are also at higher risk of developing cardiovascular diseases [186]. Those “at risk” genes include genes involved in lipid oxidation (e.g., paraoxonase), antioxidation (e.g., superoxide dismutase) and anti-inflammation (e.g., adiponectin) [187]. When comparing non-fulminant T1DM and fulminant T1DM (a subtype of idiopathic T1DM defined by a short time period between the advent of symptoms and the onset, resulting from the rapid and complete breakdown of pancreatic β cells) over five years, one study found no difference in the development of microangiopathy complications [188], while another study which followed a higher number of individuals found higher microangiopathy incidence in the fulminant T1DM group compared to autoimmune T1DM [189]. Individuals with idiopathic T1DM were also found to have higher body mass index and LDL levels, a higher visceral adiposity index (an indicator of low-grade inflammation and cardiovascular risk) higher levels of obesity (and so presumably higher levels of plasma FFA and of the dysregulations they cause) and hypercholesterolemia and lower HDL levels compared to individuals with autoimmune T1DM [190]. The levels of micro and macrovascular complications were the same between the two groups [190]. Thus, more studies are needed to understand how the different subtypes of T1DM and T2DM differ in terms of cardiovascular biomarkers and thrombosis risk.
4. Current Treatment and Future Perspectives
Due to differences in how T1DM and T2DM impact upon coagulatory pathways, individuals with these diseases respond differently to drugs designed to reduce thrombotic risk and so special care needs to be taken in identifying the most suitable treatment regimen for an individual. The effects of anti-platelet drugs, lipid-lowering drugs and hypoglycaemic drugs on the coagulation system of individuals with diabetes has been reviewed in more depth by Alzahrani and Ajjan [4]. The current treatment for diabetes focuses on lifestyle changes to control glycaemia [191,192,193]. As shown in this review, hyperglycaemia is the origin of many changes in coagulation and good glycemia control has been shown to greatly reduce many of the symptoms, including normalising the plasma concentration of many coagulatory proteins, reducing platelet hyper-activation and aggregation, endothelial dysfunction and plasma levels of microparticles. If lifestyle changes are not sufficient, drugs can be prescribed. Many drugs help control blood glucose levels such as metformin or glipizide (a sulfonylureas type of glucose-lowering drug), which have been shown to reduce plasma levels of PAI-1 in monotherapy and to a greater degree when used in combination [194]. Lowering blood glucose levels reduces the risk of developing cardiovascular diseases in diabetes, but only up to a certain level, as hypoglycaemia has been shown to be pro-thrombotic too [63]. On its own, metformin has beneficial effects including weight loss (and so presumably a reduction in plasma FFA levels), improvements in haemostatic function (more efficient fibrinolysis and reduced clot formation tendency), reduced inflammation and oxidative stress, improved endothelial function and reduced atherosclerotic plaque formation. The latter includes inhibition of the conversion of monocytes to macrophages, reduced invasion of the arterial wall by inflammatory cells and reduced lipid uptake by activated macrophages within the atherosclerotic plaque [195]. Whether metformin influences cardiovascular disease risk is unclear. Two meta-analyses have shown that when metformin is used alone, it does not have a significant effect on cardiovascular disease risk in T2DM; however, these analyses mostly included short-term studies [196,197], while another meta-analysis and a study that followed patients over a longer period (the UK Prospective Diabetes Study) have shown beneficial effects [195,198,199]. Other classes of glucose-lowering drugs, such as sulfonylureas and dipeptidyl peptidase-4 (DPP-4) inhibitors, have also been found to have no beneficial effect on cardiovascular risk in T2DM [200,201,202]. DPP-4 inhibitors have been shown to inhibit platelet aggregation by interfering with tyrosine phosphorylation of the platelet plasma membrane Ca2+-ATPase channel, thus limiting the accumulation of intracellular Ca2+ [203]. They also improve endothelial nitric oxide signalling in the vasculature, thereby reducing endothelial dysfunction, and also reduce inflammation, atherosclerotic plaque formation and oxidative stress [204,205]. Another class of glucose-lowering drug, thiazolidinediones, has been shown to have some beneficial effects on cardiovascular disease risk but also increases the risks of congestive heart failure [200]. Recently, two new classes of glucose-lowering drugs, sodium–glucose cotransporter 2 (SGLT2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists, have been shown to have beneficial effects on cardiovascular disease risk in individuals with T2DM and established cardiovascular disease [200,201]. GLP-1 receptor agonists inhibit platelet aggregation and thrombus formation through enhanced nitric oxide production by activating endothelial nitric oxide synthase, thereby also reducing endothelial dysfunction. [206,207]. They also lower postprandial dyslipidaemia [207]. SGLT2 inhibitors are mostly beneficial in decreasing heart failure as they enhance cardiac cell metabolism, reduce cardiac fibrosis, inhibit Na+/H+ exchange in myocardial cells, modulate adipokine and cytokine production, improve ventricular loading conditions and decrease blood pressure [208,209].
Lipid-lowering drugs such as statins are also useful and used mainly to lower total cholesterol and LDL levels. In addition to their effects on LDL, lipid-lowering drugs used in the treatment of diabetes can have additional beneficial effects. Indeed, many of these drugs, including statins and fenofibrate, also affect FFA levels as we have reviewed before [5]. This is an important effect as FFAs regulate many physiological processes, including endothelial function. Furthermore, lipid lowering treatment with statins can limit the level of circulating microparticles by reducing thrombin generation and the expression of tissue factor, GPIIIa and P-selectin on platelet-derived microparticles in patients with peripheral vascular diseases [170,210]. They also increase the levels and anticoagulant activity of protein C, protein S and antithrombin, reduce the levels of pro-coagulatory proteins prothrombin, factor V, factor VII, factor VIII, factor IX and factor X, reduce the levels of anti-fibrinolytic PAI-1 and reduce the expression of tissue factor by endothelial cells [4,47,211]. However, statins have been reported to both increase [47] and decrease [211] the levels of antithrombin. Statins have been shown to reduce endogenous thrombin potential and thrombosis risk in T2DM and they reduce atherosclerotic risk through a reduction in inflammation and endothelial dysfunction [4]. Statins taken in combination with other LDL-lowering drugs such as ezetimibe or proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors showed beneficial effects on cardiovascular disease risk [212]. In addition, drugs designed to increase HDL-cholesterol levels such as niacin, fenofibrate and CETP inhibitors have been designed. It is important to highlight though that these drugs fail to further decrease cardiovascular risk when administered to individuals already taking statins [137]. Nevertheless, fenofibrates also have some effect on the coagulation system, including decreasing levels of fibrinogen, tissue factor, factor VII and PAI-1 [4].
If therapeutics targeting glycaemia and lipid levels are insufficient at reducing cardiovascular risk markers in diabetes, anti-platelet drugs can also be prescribed [191,192,193]. It is well known that individuals with diabetes are less responsive to anti-platelet drugs, but nevertheless dual treatment with aspirin and the P2Y12 inhibitor clopidogrel is still advised [9]. Other strategies are being examined to lower thrombotic risk in individuals with diabetes, such as directly targeting the hypo-fibrinolysis found in diabetes, as reviewed by Kearney et al. [63]. These new drugs would inhibit thrombin activatable fibrinolysis inhibitor and PAI-1 and decrease the incorporation of α2-antiplasmin and C3 protein into fibrin clots.
As reviewed here, diabetes is a complex disease that strongly impacts on haemostasis and the risk of developing cardiovascular disease in multiple ways. These include alterations in the plasma levels of coagulatory proteins, metal ions and pro-coagulatory particles, lipid metabolism and composition, endothelial function and platelet reactivity. It is important to understand these mechanisms and how they differ between T1DM and T2DM in order to appropriately treat these diseases and to reduce thrombotic risk in affected individuals.
Author Contributions
Conceptualization, A.I.S.S. and A.J.S.; writing—original draft preparation, A.I.S.S. and A.J.S..; writing—review and editing, A.I.S.S. and A.J.S.
Funding
This research was funded by the British Heart Foundation, grant numbers PG/15/9/31270 and FS/15/42/31556.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AGE | Advanced glycation end-products |
| CETP | Cholesterol ester transfer protein |
| DPP-4 | Dipeptidyl peptidase-4 |
| GLP-1 | Glucagon-like peptide-1 |
| HDL | High-density lipoprotein |
| LDL | Low-density lipoprotein |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PKC | Protein kinase C |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| RAGE | Advanced glycation end-product receptors |
| S1P | HDL-associated sphingosine-1-phosphate |
| SGLT2 | Sodium–glucose cotransporter 2 |
| T1DM | Type-1 diabetes mellitus |
| T2DM | Type-2 diabetes mellitus |
| vWF | von Willebrand factor |
References
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed.; International Diabetes Federation: Brussels, Belgium, 2017. [Google Scholar]
- Al Alawi, A.M.; Majoni, S.W.; Falhammar, H. Magnesium and Human Health: Perspectives and Research Directions. Int. J. Endocrinol. 2018, 2018, 9041694. [Google Scholar] [CrossRef] [Green Version]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Alzahrani, S.H.; Ajjan, R.A. Coagulation and fibrinolysis in diabetes. Diab. Vasc. Dis. Res. 2010, 7, 260–273. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Blindauer, C.A.; Stewart, A.J. Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes. Nutrients 2019, 11, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Adjedj, J.; Varenne, O. Diabetes Mellitus, a prothrombotic disease. Ann. Cardiol. Angeiol. 2017, 66, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Lehto, S.; Ronnemaa, T.; Pyorala, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Rivas Rios, J.R.; Franchi, F.; Rollini, F.; Angiolillo, D.J. Diabetes and antiplatelet therapy: From bench to bedside. Cardiovasc. Diagn. Ther. 2018, 8, 594–609. [Google Scholar] [CrossRef]
- Verkleij, C.J.; Bruijn, R.E.; Meesters, E.W.; Gerdes, V.E.; Meijers, J.C.; Marx, P.F. The hemostatic system in patients with type 2 diabetes with and without cardiovascular disease. Clin. Appl. Thromb. Hemost. 2011, 17, E57–E63. [Google Scholar] [CrossRef] [Green Version]
- Galajda, P.; Martinka, E.; Mokan, M.; Kubisz, P. Endothelial markers in diabetes mellitus. Thromb. Res. 1997, 85, 63–65. [Google Scholar] [CrossRef]
- De Mattia, G.; Bravi, M.C.; Laurenti, O.; Moretti, A.; Cipriani, R.; Gatti, A.; Mandosi, E.; Morano, S. Endothelial dysfunction and oxidative stress in type 1 and type 2 diabetic patients without clinical macrovascular complications. Diabetes Res. Clin. Pract. 2008, 79, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, K.; Ciechanowski, K.; Golembiewska, E.; Safranow, K.; Ciechanowicz, A.; Domanski, L.; Myslak, M.; Rozanski, J. Plasma prekallikrein as a risk factor for diabetic retinopathy. Arch. Med. Res. 2005, 36, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Patrassi, G.M.; Vettor, R.; Padovan, D.; Girolami, A. Contact phase of blood coagulation in diabetes mellitus. Eur. J. Clin. Invest. 1982, 12, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Kim, J.E.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S. High coagulation factor levels and low protein C levels contribute to enhanced thrombin generation in patients with diabetes who do not have macrovascular complications. J. Diabetes Complicat. 2014, 28, 365–369. [Google Scholar] [CrossRef] [PubMed]
- El-Hagracy, R.S.; Kamal, G.M.; Sabry, I.M.; Saad, A.A.; Abou El Ezz, N.F.; Nasr, H.A. Tissue Factor, Tissue Factor Pathway Inhibitor and Factor VII Activity in Cardiovascular Complicated Type 2 Diabetes Mellitus. Oman Med. J. 2010, 25, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Boden, G.; Homko, C.; Gunawardana, J.; Rao, A.K. Whole-blood tissue factor procoagulant activity is elevated in type 1 diabetes: Effects of hyperglycemia and hyperinsulinemia. Diabetes Care 2012, 35, 1322–1327. [Google Scholar] [CrossRef] [Green Version]
- Barillari, G.; Fabbro, E.; Pasca, S.; Bigotto, E. Coagulation and oxidative stress plasmatic levels in a type 2 diabetes population. Blood Coagul. Fibrinolysis 2009, 20, 290–296. [Google Scholar] [CrossRef]
- Fattah, M.A.; Shaheen, M.H.; Mahfouz, M.H. Disturbances of haemostasis in diabetes mellitus. Dis. Markers 2003, 19, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Madan, R.; Gupt, B.; Saluja, S.; Kansra, U.C.; Tripathi, B.K.; Guliani, B.P. Coagulation profile in diabetes and its association with diabetic microvascular complications. J. Assoc. Physicians India 2010, 58, 481–484. [Google Scholar]
- Klein, R.L.; Hunter, S.J.; Jenkins, A.J.; Zheng, D.; Semler, A.J.; Clore, J.; Garvey, W.T.; Dcct/Ecic Study, G. Fibrinogen is a marker for nephropathy and peripheral vascular disease in type 1 diabetes: Studies of plasma fibrinogen and fibrinogen gene polymorphism in the DCCT/EDIC cohort. Diabetes Care 2003, 26, 1439–1448. [Google Scholar] [CrossRef] [Green Version]
- Agren, A.; Jorneskog, G.; Elgue, G.; Henriksson, P.; Wallen, H.; Wiman, B. Increased incorporation of antiplasmin into the fibrin network in patients with type 1 diabetes. Diabetes Care 2014, 37, 2007–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothschild, A.M.; Reis, M.L.; Melo, V.L.; Foss, M.C.; Gallo, L., Jr. Increased kininogen levels observed in plasma of diabetic patients are corrected by the administration of insulin. Horm. Metab. Res. 1999, 31, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Sommeijer, D.W.; Hansen, H.R.; van Oerle, R.; Hamulyak, K.; van Zanten, A.P.; Meesters, E.; Spronk, H.M.; ten Cate, H. Soluble tissue factor is a candidate marker for progression of microvascular disease in patients with Type 2 diabetes. J. Thromb. Haemost. 2006, 4, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Song, D.Y.; Gu, J.Y.; Yoo, H.J.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Activation of Factor XII and Kallikrein-kinin System Combined with Neutrophil Extracellular Trap Formation in Diabetic Retinopathy. Exp. Clin. Endocrinol. Diabetes 2019. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, M.W.; Kohler, H.P.; Ariens, R.A.; McCormack, L.J.; Grant, P.J. Circulating levels of coagulation factor XIII in subjects with type 2 diabetes and in their first-degree relatives. Diabetes Care 2000, 23, 703–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colhoun, H.M.; Zito, F.; Norman Chan, N.; Rubens, M.B.; Fuller, J.H.; Humphries, S.E. Activated factor XII levels and factor XII 46C>T genotype in relation to coronary artery calcification in patients with type 1 diabetes and healthy subjects. Atherosclerosis 2002, 163, 363–369. [Google Scholar] [CrossRef]
- Reverter, J.L.; Reverter, J.C.; Tassies, D.; Rius, F.; Monteagudo, J.; Rubies-Prat, J.; Escolar, G.; Ordinas, A.; Sanmarti, A. Thrombomodulin and induced tissue factor expression on monocytes as markers of diabetic microangiopathy: A prospective study on hemostasis and lipoproteins in insulin-dependent diabetes mellitus. Am. J. Hematol. 1997, 56, 93–99. [Google Scholar] [CrossRef]
- Aslan, B.; Eren, N.; Ciğerli, Ş.; Müldür, F.; Yücel, N. Evaluation of Plasma Protein C Antigen, Protein C Activity and Thrombomodulin Levels in Type 2 Diabetic Patients. Turk. J. Med. Sci. 2005, 35, 305–310. [Google Scholar]
- Yasuma, T.; Yano, Y.; D’Alessandro-Gabazza, C.N.; Toda, M.; Gil-Bernabe, P.; Kobayashi, T.; Nishihama, K.; Hinneh, J.A.; Mifuji-Moroka, R.; Roeen, Z.; et al. Amelioration of Diabetes by Protein S. Diabetes 2016, 65, 1940–1951. [Google Scholar] [CrossRef] [Green Version]
- Rigla, M.; Mateo, J.; Fontcuberta, J.; Souto, J.C.; de Leiva, A.; Perez, A. Normalisation of tissue factor pathway inhibitor activity after glycaemic control optimisation in type 1 diabetic patients. Thromb. Haemost. 2000, 84, 223–227. [Google Scholar] [CrossRef]
- Ceriello, A.; Giugliano, D.; Quatraro, A.; Stante, A.; Dello Russo, P.; Torella, R. Increased alpha 2-macroglobulin in diabetes: A hyperglycemia related phenomenon associated with reduced antithrombin III activity. Acta Diabetol. Lat. 1989, 26, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Borsey, D.Q.; Prowse, C.V.; Gray, R.S.; Dawes, J.; James, K.; Elton, R.A.; Clarke, B.F. Platelet and coagulation factors in proliferative diabetic retinopathy. J. Clin. Pathol. 1984, 37, 659–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobczak, A.I.S.; Pitt, S.J.; Stewart, A.J. Glycosaminoglycan Neutralization in Coagulation Control. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasty, F.; Alavi, M.Z.; Moore, S. Distribution of glycosaminoglycans in the intima of human aortas: Changes in atherosclerosis and diabetes mellitus. Diabetologia 1993, 36, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Meigs, J.B.; Mittleman, M.A.; Nathan, D.M.; Tofler, G.H.; Singer, D.E.; Murphy-Sheehy, P.M.; Lipinska, I.; D’Agostino, R.B.; Wilson, P.W. Hyperinsulinemia, hyperglycemia, and impaired hemostasis: The Framingham Offspring Study. JAMA 2000, 283, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Kubisz, P.; Stanciakova, L.; Stasko, J.; Galajda, P.; Mokan, M. Endothelial and platelet markers in diabetes mellitus type 2. World J. Diabetes 2015, 6, 423–431. [Google Scholar] [CrossRef]
- Juhan-Vague, I.; Roul, C.; Alessi, M.C.; Ardissone, J.P.; Heim, M.; Vague, P. Increased plasminogen activator inhibitor activity in non insulin dependent diabetic patients—Relationship with plasma insulin. Thromb. Haemost. 1989, 61, 370–373. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Phoenix, F.A.; Pitt, S.J.; Ajjan, R.A.; Stewart, A.J. Plasma magnesium levels associate with increased fibrin clot density and lysis time in individuals with type-1 diabetes and controls. Thromb. Haemost. (in press).
- Hori, Y.; Gabazza, E.C.; Yano, Y.; Katsuki, A.; Suzuki, K.; Adachi, Y.; Sumida, Y. Insulin resistance is associated with increased circulating level of thrombin-activatable fibrinolysis inhibitor in type 2 diabetic patients. J. Clin. Endocrinol. Metab 2002, 87, 660–665. [Google Scholar] [CrossRef]
- Sherif, E.M.; Elbarbary, N.S.; Abd Al Aziz, M.M.; Mohamed, S.F. Plasma thrombin-activatable fibrinolysis inhibitor levels in children and adolescents with type 1 diabetes mellitus: Possible relation to diabetic microvascular complications. Blood Coagul. Fibrinolysis 2014, 25, 451–457. [Google Scholar] [CrossRef]
- Yoshino, S.; Fujimoto, K.; Takada, T.; Kawamura, S.; Ogawa, J.; Kamata, Y.; Kodera, Y.; Shichiri, M. Molecular form and concentration of serum alpha2-macroglobulin in diabetes. Sci. Rep. 2019, 9, 12927. [Google Scholar] [CrossRef] [PubMed]
- Polat, S.B.; Ugurlu, N.; Yulek, F.; Simavli, H.; Ersoy, R.; Cakir, B.; Erel, O. Evaluation of serum fibrinogen, plasminogen, alpha2-anti-plasmin, and plasminogen activator inhibitor levels (PAI) and their correlation with presence of retinopathy in patients with type 1 DM. J. Diabetes Res. 2014, 2014, 317292. [Google Scholar] [CrossRef] [PubMed]
- Jax, T.W.; Peters, A.J.; Plehn, G.; Schoebel, F.C. Hemostatic risk factors in patients with coronary artery disease and type 2 diabetes—A two year follow-up of 243 patients. Cardiovasc. Diabetol. 2009, 8, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, B.Y.; Vazquez, M.A.; Jaquez, M.G.; Huemoeller, A.H.; Intaglietta, M.; Cabrales, P. Blood pressure directly correlates with blood viscosity in diabetes type 1 children but not in normals. Clin. Hemorheol. Microcirc. 2010, 44, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Singh, N. Blood viscosity, lipid profile, and lipid peroxidation in type-1 diabetic patients with good and poor glycemic control. N. Am. J. Med. Sci. 2013, 5, 562–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addai-Mensah, O.; Annani-Akollor, M.E.; Nsafoah, F.O.; Fondjo, L.A.; Owiredu, E.W.; Danquah, K.O.; Duneeh, R.V.; Amponsah, F.A. Effect of poor glycaemic control on plasma levels and activity of protein C, protein S, and antithrombin III in type 2 diabetes mellitus. PLoS ONE 2019, 14, e0223171. [Google Scholar] [CrossRef]
- Mansfield, M.W.; Heywood, D.M.; Grant, P.J. Circulating levels of factor VII, fibrinogen, and von Willebrand factor and features of insulin resistance in first-degree relatives of patients with NIDDM. Circulation 1996, 94, 2171–2176. [Google Scholar] [CrossRef]
- Leurs, P.B.; van Oerle, R.; Wolffenbuttel, B.H.; Hamulyak, K. Increased tissue factor pathway inhibitor (TFPI) and coagulation in patients with insulin-dependent diabetes mellitus. Thromb. Haemost. 1997, 77, 472–476. [Google Scholar] [CrossRef]
- Ceriello, A.; Quatraro, A.; Marchi, E.; Barbanti, M.; Dello Russo, P.; Lefebvre, P.; Giugliano, D. The role of hyperglycaemia-induced alterations of antithrombin III and factor X activation in the thrombin hyperactivity of diabetes mellitus. Diabet. Med. 1990, 7, 343–348. [Google Scholar] [CrossRef]
- Ajjan, R.A.; Gamlen, T.; Standeven, K.F.; Mughal, S.; Hess, K.; Smith, K.A.; Dunn, E.J.; Anwar, M.M.; Rabbani, N.; Thornalley, P.J.; et al. Diabetes is associated with posttranslational modifications in plasminogen resulting in reduced plasmin generation and enzyme-specific activity. Blood 2013, 122, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Walmsley, D.; Hampton, K.K.; Grant, P.J. Contrasting fibrinolytic responses in type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes. Diabet. Med. 1991, 8, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Dunn, E.J.; Ariens, R.A.; Grant, P.J. The influence of type 2 diabetes on fibrin structure and function. Diabetologia 2005, 48, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Pieters, M.; van Zyl, D.G.; Rheeder, P.; Jerling, J.C.; Loots du, T.; van der Westhuizen, F.H.; Gottsche, L.T.; Weisel, J.W. Glycation of fibrinogen in uncontrolled diabetic patients and the effects of glycaemic control on fibrinogen glycation. Thromb. Res. 2007, 120, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Pieters, M.; Covic, N.; van der Westhuizen, F.H.; Nagaswami, C.; Baras, Y.; Toit Loots, D.; Jerling, J.C.; Elgar, D.; Edmondson, K.S.; van Zyl, D.G.; et al. Glycaemic control improves fibrin network characteristics in type 2 diabetes - a purified fibrinogen model. Thromb. Haemost. 2008, 99, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, R.; Mignemi, N.; Rose, K.; O’Rear, L.; Sarilla, S.; Hamm, H.E.; Barnett, J.V.; Verhamme, I.M.; Schoenecker, J. The hyperglycemic byproduct methylglyoxal impairs anticoagulant activity through covalent adduction of antithrombin III. Thromb. Res. 2014, 134, 1350–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, A.K.; Chouhan, V.; Chen, X.; Sun, L.; Boden, G. Activation of the tissue factor pathway of blood coagulation during prolonged hyperglycemia in young healthy men. Diabetes 1999, 48, 1156–1161. [Google Scholar] [CrossRef]
- Davi, G.; Gennaro, F.; Spatola, A.; Catalano, I.; Averna, M.; Montalto, G.; Amato, S.; Notarbartolo, A. Thrombin-antithrombin III complexes in type II diabetes mellitus. J. Diabetes Complicat. 1992, 6, 7–11. [Google Scholar] [CrossRef]
- Osende, J.I.; Badimon, J.J.; Fuster, V.; Herson, P.; Rabito, P.; Vidhun, R.; Zaman, A.; Rodriguez, O.J.; Lev, E.I.; Rauch, U.; et al. Blood thrombogenicity in type 2 diabetes mellitus patients is associated with glycemic control. J. Am. Coll. Cardiol. 2001, 38, 1307–1312. [Google Scholar] [CrossRef] [Green Version]
- Hess, K.; Alzahrani, S.H.; Mathai, M.; Schroeder, V.; Carter, A.M.; Howell, G.; Koko, T.; Strachan, M.W.; Price, J.F.; Smith, K.A.; et al. A novel mechanism for hypofibrinolysis in diabetes: The role of complement C3. Diabetologia 2012, 55, 1103–1113. [Google Scholar] [CrossRef]
- Hess, K.; Alzahrani, S.H.; Price, J.F.; Strachan, M.W.; Oxley, N.; King, R.; Gamlen, T.; Schroeder, V.; Baxter, P.D.; Ajjan, R.A. Hypofibrinolysis in type 2 diabetes: The role of the inflammatory pathway and complement C3. Diabetologia 2014, 57, 1737–1741. [Google Scholar] [CrossRef]
- Dunn, E.J.; Philippou, H.; Ariens, R.A.; Grant, P.J. Molecular mechanisms involved in the resistance of fibrin to clot lysis by plasmin in subjects with type 2 diabetes mellitus. Diabetologia 2006, 49, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearney, K.; Tomlinson, D.; Smith, K.; Ajjan, R. Hypofibrinolysis in diabetes: A therapeutic target for the reduction of cardiovascular risk. Cardiovasc. Diabetol. 2017, 16, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konieczynska, M.; Fil, K.; Bazanek, M.; Undas, A. Prolonged duration of type 2 diabetes is associated with increased thrombin generation, prothrombotic fibrin clot phenotype and impaired fibrinolysis. Thromb. Haemost. 2014, 111, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzahrani, S.H.; Hess, K.; Price, J.F.; Strachan, M.; Baxter, P.D.; Cubbon, R.; Phoenix, F.; Gamlen, T.; Ariens, R.A.; Grant, P.J.; et al. Gender-specific alterations in fibrin structure function in type 2 diabetes: Associations with cardiometabolic and vascular markers. J. Clin. Endocrinol. Metab. 2012, 97, E2282–E2287. [Google Scholar] [CrossRef] [PubMed]
- Bryk, A.H.; Siudut, J.; Broniatowska, E.; Bagoly, Z.; Barath, B.; Katona, E.; Undas, A. Sex-specific alteration to alpha2-antiplasmin incorporation in patients with type 2 diabetes. Thromb. Res. 2019, 185, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tehrani, S.; Jorneskog, G.; Agren, A.; Lins, P.E.; Wallen, H.; Antovic, A. Fibrin clot properties and haemostatic function in men and women with type 1 diabetes. Thromb. Haemost. 2015, 113, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Sobczak, A.I.S.; Stefanowicz, F.; Pitt, S.J.; Ajjan, R.A.; Stewart, A.J. Total plasma magnesium, zinc, copper and selenium concentrations in type-I and type-II diabetes. Biometals 2019. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.T.; Fredenburgh, J.C.; Weitz, J.I. Zinc, an important cofactor in haemostasis and thrombosis. Thomb. Haemost. 2013, 109, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Ripoll, L.; Mazoyer, E.; Baudin, B.; Lacam, S.; Varsat, B.; Drouet, L.O. Can high plasma ceruloplasmin levels cause acquired activated protein C resistance? Thromb. Haemost. 1998, 79, 449–450. [Google Scholar]
- Vasilyev, V.B. Looking for a partner: Ceruloplasmin in protein-protein interactions. Biometals 2019, 32, 195–210. [Google Scholar] [CrossRef]
- Mann, K.G.; Lawler, C.M.; Vehar, G.A.; Church, W.R. Coagulation Factor V contains copper ion. J. Biol. Chem. 1984, 259, 12949–12951. [Google Scholar] [PubMed]
- Tagliavacca, L.; Moon, N.; Dunham, W.R.; Kaufman, R.J. Identification and functional requirement of Cu(I) and its ligands within coagulation factor VIII. J. Biol. Chem. 1997, 272, 27428–27434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becerra-Tomas, N.; Estruch, R.; Bullo, M.; Casas, R.; Diaz-Lopez, A.; Basora, J.; Fito, M.; Serra-Majem, L.; Salas-Salvado, J. Increased serum calcium levels and risk of type 2 diabetes in individuals at high cardiovascular risk. Diabetes Care 2014, 37, 3084–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, M.R.; Pankow, J.S.; Sibley, S.D.; Selvin, E.; Reis, J.P.; Michos, E.D.; Lutsey, P.L. Serum calcium and incident type 2 diabetes: The Atherosclerosis Risk in Communities (ARIC) study. Am. J. Clin. Nutr. 2016, 104, 1023–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo, C.; Hanley, A.J.; Rewers, M.J.; Haffner, S.M. Calcium and phosphate concentrations and future development of type 2 diabetes: The Insulin Resistance Atherosclerosis Study. Diabetologia 2014, 57, 1366–1374. [Google Scholar] [CrossRef] [Green Version]
- Jorde, R.; Schirmer, H.; Njolstad, I.; Lochen, M.L.; Bogeberg Mathiesen, E.; Kamycheva, E.; Figenschau, Y.; Grimnes, G. Serum calcium and the calcium-sensing receptor polymorphism rs17251221 in relation to coronary heart disease, type 2 diabetes, cancer and mortality: The Tromso Study. Eur. J. Epidemiol. 2013, 28, 569–578. [Google Scholar] [CrossRef]
- Levy, J.; Stern, Z.; Gutman, A.; Naparstek, Y.; Gavin, J.R., 3rd; Avioli, L.V. Plasma calcium and phosphate levels in an adult noninsulin-dependent diabetic population. Calcif. Tissue Int. 1986, 39, 316–318. [Google Scholar] [CrossRef]
- Singh, D.K.; Winocour, P.; Summerhayes, B.; Viljoen, A.; Sivakumar, G.; Farrington, K. Low serum osteoprotegerin levels in normoalbuminuric type 1 diabetes mellitus. Acta Diabetol. 2010, 47 (Suppl. 1), 105–110. [Google Scholar] [CrossRef]
- Wierzbicka, E.; Szalecki, M.; Pludowski, P.; Jaworski, M.; Brzozowska, A. Vitamin D status, body composition and glycemic control in Polish adolescents with type 1 diabetes. Minerva Endocrinol. 2016, 41, 445–455. [Google Scholar]
- Tankeu, A.T.; Ndip Agbor, V.; Noubiap, J.J. Calcium supplementation and cardiovascular risk: A rising concern. J. Clin. Hypertens. 2017, 19, 640–646. [Google Scholar] [CrossRef] [Green Version]
- Sobczak, A.I.S.; Pitt, S.J.; Stewart, A.J. Influence of zinc on glycosaminoglycan neutralisation during coagulation. Metallomics 2018, 10, 1180–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjeevi, N.; Freeland-Graves, J.; Beretvas, S.N.; Sachdev, P.K. Trace element status in type 2 diabetes: A meta-analysis. J. Clin. Diagn. Res. 2018, 12, OE01–OE08. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, C.; Li, L.; Shao, M.; Wang, L.; Lv, X.; Gao, C.; Niu, H.; Li, B. The Study on the Correlation Between Six Kinds of Mineral Elements and Diabetes. Biol. Trace Elem. Res. 2018, 183, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Coverdale, J.P.C.; Khazaipoul, S.; Arya, S.; Stewart, A.J.; Blindauer, C.A. Crosstalk between zinc and free fatty acids in plasma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, A.I.S.; Katundu, K.G.H.; Phoenix, F.A.; Khazaipoul, S.; Yu, R.; Lampiao, F.; Stefanowicz, F.; Blindauer, C.A.; Pitt, S.J.; Smith, T.K.; et al. Plasma non-esterified fatty acids contribute to increased coagulability in type-2 diabetes through altered plasma zinc speciation (preprint). bioRxiv 2019. [Google Scholar] [CrossRef]
- Coverdale, J.P.C.; Barnett, J.P.; Adamu, A.H.; Griffiths, E.J.; Stewart, A.J.; Blindauer, C.A. A metalloproteomic analysis of interactions between plasma proteins and zinc: Elevated fatty acid levels affect zinc distribution. Metallomics 2019. [Google Scholar] [CrossRef] [Green Version]
- Sekiya, F.; Yoshida, M.; Yamashita, T.; Morita, T. Magnesium(II) is a crucial constituent of the blood coagulation cascade. Potentiation of coagulant activities of factor IX by Mg2+ ions. J. Biol. Chem. 1996, 271, 8541–8544. [Google Scholar] [CrossRef] [Green Version]
- Vadivel, K.; Agah, S.; Messer, A.S.; Cascio, D.; Bajaj, M.S.; Krishnaswamy, S.; Esmon, C.T.; Padmanabhan, K.; Bajaj, S.P. Structural and functional studies of gamma-carboxyglutamic acid domains of factor VIIa and activated Protein C: Role of magnesium at physiological calcium. J. Mol. Biol. 2013, 425, 1961–1981. [Google Scholar] [CrossRef] [Green Version]
- Tokutake, T.; Baba, H.; Shimada, Y.; Takeda, W.; Sato, K.; Hiroshima, Y.; Kirihara, T.; Shimizu, I.; Nakazawa, H.; Kobayashi, H.; et al. Exogenous Magnesium Chloride Reduces the Activated Partial Thromboplastin Times of Lupus Anticoagulant-Positive Patients. PLoS ONE 2016, 11, e0157835. [Google Scholar] [CrossRef]
- Van den Besselaar, A.M.; Witteveen, E.; Meeuwisse-Braun, J.; van der Meer, F.J. The influence of exogenous magnesium chloride on the apparent INR determined with human, rabbit, and bovine thromboplastin reagents. Thromb. Haemost. 2003, 89, 43–47. [Google Scholar]
- Jankun, J.; Skrzypczak-Jankun, E.; Lipinski, B. Complex function of magnesium in blood clot formation and lysis. Cent. Eur. J. Immunol. 2013, 38, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Anstall, H.B.; Hayward, G.H.; Huntsman, R.G.; Weitzman, D.; Lehmann, H. The effect of magnesium on blood coagulation in human subjects. Lancet 1959, 1, 814–815. [Google Scholar] [CrossRef]
- Efstratiadis, G.; Sarigianni, M.; Gougourelas, I. Hypomagnesemia and cardiovascular system. Hippokratia 2006, 10, 147–152. [Google Scholar] [PubMed]
- Barbagallo, M.; Dominguez, L.J. Magnesium and type 2 diabetes. World J. Diabetes 2015, 6, 1152–1157. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Negrouk, V.; Perera, M.; Llabre, M.M.; Ricordi, C.; Rongioletti, M.C.A.; Mendez, A.J. Serum copper profile in patients with type 1 diabetes in comparison to other metals. J. Trace Elem. Med. Biol. 2019, 56, 156–161. [Google Scholar] [CrossRef]
- Keen, C.L.; Feldman, B.F.; Knight, J.; O’Neill, S.; Ferrell, F.; Hurley, L.S. The influence of high concentrations of dietary copper on vitamin K-dependent coagulation factors. Proc. Soc. Exp. Biol. Med. 1982, 170, 471–475. [Google Scholar] [CrossRef]
- Memisogullari, R.; Bakan, E. Levels of ceruloplasmin, transferrin, and lipid peroxidation in the serum of patients with Type 2 diabetes mellitus. J. Diabetes Complicat. 2004, 18, 193–197. [Google Scholar] [CrossRef]
- Bergis, D.; Tessmer, L.; Badenhoop, K. Iron deficiency in long standing type 1 diabetes mellitus and its association with depression and impaired quality of life. Diabetes Res. Clin. Pract. 2019, 151, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Akkermans, M.D.; Mieke Houdijk, E.C.A.; Bakker, B.; Boers, A.C.; van der Kaay, D.C.M.; de Vries, M.C.; Claire Woltering, M.; Mul, D.; van Goudoever, J.B.; Brus, F. Iron status and its association with HbA1c levels in Dutch children with diabetes mellitus type 1. Eur. J. Pediatr. 2018, 177, 603–610. [Google Scholar] [CrossRef]
- Jankun, J.; Landeta, P.; Pretorius, E.; Skrzypczak-Jankun, E.; Lipinski, B. Unusual clotting dynamics of plasma supplemented with iron(III). Int. J. Mol. Med. 2014, 33, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, B.; Pretorius, E. Iron-induced fibrin in cardiovascular disease. Curr. Neurovasc. Res. 2013, 10, 269–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ferranti, S.D.; de Boer, I.H.; Fonseca, V.; Fox, C.S.; Golden, S.H.; Lavie, C.J.; Magge, S.N.; Marx, N.; McGuire, D.K.; Orchard, T.J.; et al. Type 1 diabetes mellitus and cardiovascular disease: A scientific statement from the American Heart Association and American Diabetes Association. Circulation 2014, 130, 1110–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarte, J.; Hegele, R.A. Dyslipidemia Management in Adults With Diabetes. Can. J. Diabetes 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, J.; Ho, J.; Soran, H. Cardiovascular Risk in Type 1 Diabetes Mellitus. Diabetes Ther. 2019, 10, 773–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costacou, T.; Evans, R.W.; Orchard, T.J. High-density lipoprotein cholesterol in diabetes: Is higher always better? J. Clin. Lipidol. 2011, 5, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravnskov, U.; de Lorgeril, M.; Diamond, D.M.; Hama, R.; Hamazaki, T.; Hammarskjold, B.; Hynes, N.; Kendrick, M.; Langsjoen, P.H.; Mascitelli, L.; et al. LDL-C does not cause cardiovascular disease: A comprehensive review of the current literature. Expert Rev. Clin. Pharmacol. 2018, 11, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Hero, C.; Svensson, A.M.; Gidlund, P.; Gudbjornsdottir, S.; Eliasson, B.; Eeg-Olofsson, K. LDL cholesterol is not a good marker of cardiovascular risk in Type 1 diabetes. Diabet. Med. 2016, 33, 316–323. [Google Scholar] [CrossRef]
- Soedamah-Muthu, S.S.; Chaturvedi, N.; Toeller, M.; Ferriss, B.; Reboldi, P.; Michel, G.; Manes, C.; Fuller, J.H.; Group, E.P.C.S. Risk factors for coronary heart disease in type 1 diabetic patients in Europe: The EURODIAB Prospective Complications Study. Diabetes Care 2004, 27, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res. 2002, 43, 1363–1379. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, A.J.; Best, J.D.; Klein, R.L.; Lyons, T.J. ‘Lipoproteins, glycoxidation and diabetic angiopathy’. Diabetes Metab Res. Rev. 2004, 20, 349–368. [Google Scholar] [CrossRef]
- Taskinen, M.R. LDL-cholesterol, HDL-cholesterol or triglycerides--which is the culprit? Diabetes Res. Clin. Pract 2003, 61 (Suppl. 1), S19–S26. [Google Scholar] [CrossRef]
- Soran, H.; Durrington, P.N. Susceptibility of LDL and its subfractions to glycation. Curr. Opin. Lipidol. 2011, 22, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Deckert, V.; Brunet, A.; Lantoine, F.; Lizard, G.; Millanvoye-van Brussel, E.; Monier, S.; Lagrost, L.; David-Dufilho, M.; Gambert, P.; Devynck, M.A. Inhibition by cholesterol oxides of NO release from human vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1054–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbrecher, U.P.; Zhang, H.F.; Lougheed, M. Role of oxidatively modified LDL in atherosclerosis. Free Radic. Biol. Med. 1990, 9, 155–168. [Google Scholar] [CrossRef]
- Li, X.; Guan, B.; Wang, Y.; Tse, G.; Zou, F.; Khalid, B.W.; Xia, Y.; Wu, S.; Sun, J. Association between high-density lipoprotein cholesterol and all-cause mortality in the general population of northern China. Sci. Rep. 2019, 9, 14426. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Trigatti, B.L.; Mineo, C.; Knaack, D.; Wilkins, J.T.; Sahoo, D.; Asztalos, B.F.; Mora, S.; Cuchel, M.; Pownall, H.J.; et al. Proceedings of the Ninth HDL (High-Density Lipoprotein) Workshop: Focus on Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Kothari, V.; Bornfeldt, K.E. High-Density Lipoprotein Function in Cardiovascular Disease and Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e10–e16. [Google Scholar] [CrossRef] [Green Version]
- Shea, S.; Stein, J.H.; Jorgensen, N.W.; McClelland, R.L.; Tascau, L.; Shrager, S.; Heinecke, J.W.; Yvan-Charvet, L.; Tall, A.R. Cholesterol Mass Efflux Capacity, Incident Cardiovascular Disease, and Progression of Carotid Plaque. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Ogura, M.; Hori, M.; Harada-Shiba, M. Association Between Cholesterol Efflux Capacity and Atherosclerotic Cardiovascular Disease in Patients With Familial Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [Green Version]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis From the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef] [PubMed]
- Cahill, L.E.; Sacks, F.M.; Rimm, E.B.; Jensen, M.K. Cholesterol efflux capacity, HDL cholesterol, and risk of coronary heart disease: A nested case-control study in men. J. Lipid Res. 2019, 60, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- El Khoudary, S.R.; Ceponiene, I.; Samargandy, S.; Stein, J.H.; Li, D.; Tattersall, M.C.; Budoff, M.J. HDL (High-Density Lipoprotein) Metrics and Atherosclerotic Risk in Women. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2236–2244. [Google Scholar] [CrossRef] [PubMed]
- Zimetti, F.; Freitas, W.M.; Campos, A.M.; Daher, M.; Adorni, M.P.; Bernini, F.; Sposito, A.C.; Zanotti, I.; Brazilian Study on Healthy, A. Cholesterol efflux capacity does not associate with coronary calcium, plaque vulnerability, and telomere length in healthy octogenarians. J. Lipid Res. 2018, 59, 714–721. [Google Scholar] [CrossRef] [Green Version]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Apro, J.; Tietge, U.J.; Dikkers, A.; Parini, P.; Angelin, B.; Rudling, M. Impaired Cholesterol Efflux Capacity of High-Density Lipoprotein Isolated From Interstitial Fluid in Type 2 Diabetes Mellitus-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 787–791. [Google Scholar] [CrossRef] [Green Version]
- Gourgari, E.; Playford, M.P.; Campia, U.; Dey, A.K.; Cogen, F.; Gubb-Weiser, S.; Mete, M.; Desale, S.; Sampson, M.; Taylor, A.; et al. Low cholesterol efflux capacity and abnormal lipoprotein particles in youth with type 1 diabetes: A case control study. Cardiovasc. Diabetol. 2018, 17, 158. [Google Scholar] [CrossRef]
- Mehta, N.U.; Grijalva, V.; Hama, S.; Wagner, A.; Navab, M.; Fogelman, A.M.; Reddy, S.T. Apolipoprotein E-/- Mice Lacking Hemopexin Develop Increased Atherosclerosis via Mechanisms That Include Oxidative Stress and Altered Macrophage Function. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1152–1163. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horvath, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Brinck, J.W.; Thomas, A.; Lauer, E.; Jornayvaz, F.R.; Brulhart-Meynet, M.C.; Prost, J.C.; Pataky, Z.; Lofgren, P.; Hoffstedt, J.; Eriksson, M.; et al. Diabetes Mellitus Is Associated With Reduced High-Density Lipoprotein Sphingosine-1-Phosphate Content and Impaired High-Density Lipoprotein Cardiac Cell Protection. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 817–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frej, C.; Mendez, A.J.; Ruiz, M.; Castillo, M.; Hughes, T.A.; Dahlback, B.; Goldberg, R.B. A Shift in ApoM/S1P Between HDL-Particles in Women With Type 1 Diabetes Mellitus Is Associated With Impaired Anti-Inflammatory Effects of the ApoM/S1P Complex. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1194–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartz, J.C.; de Ferranti, S.; Gidding, S. Hypertriglyceridemia in Diabetes Mellitus: Implications for Pediatric Care. J. Endocr. Soc. 2018, 2, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Selim, S. Frequency and pattern of chronic complications of diabetes and their association with glycemic control. Diabetes Metab. Syndr. 2017, 11 (Suppl. 1), S311–S314. [Google Scholar] [CrossRef]
- Kushner, P.A.; Cobble, M.E. Hypertriglyceridemia: The importance of identifying patients at risk. Postgrad. Med. 2016, 128, 848–858. [Google Scholar] [CrossRef]
- Girona, J.; Amigo, N.; Ibarretxe, D.; Plana, N.; Rodriguez-Borjabad, C.; Heras, M.; Ferre, R.; Gil, M.; Correig, X.; Masana, L. HDL Triglycerides: A New Marker of Metabolic and Cardiovascular Risk. Int. J. Mol. Sci. 2019, 20, 3151. [Google Scholar] [CrossRef] [Green Version]
- Amigo, N.; Mallol, R.; Heras, M.; Martinez-Hervas, S.; Blanco Vaca, F.; Escola-Gil, J.C.; Plana, N.; Yanes, O.; Masana, L.; Correig, X. Lipoprotein hydrophobic core lipids are partially extruded to surface in smaller HDL: “Herniated” HDL, a common feature in diabetes. Sci. Rep. 2016, 6, 19249. [Google Scholar] [CrossRef] [Green Version]
- Verges, B. Lipid modification in type 2 diabetes: The role of LDL and HDL. Fundam. Clin. Pharmacol. 2009, 23, 681–685. [Google Scholar] [CrossRef]
- Holmes, M.V.; Millwood, I.Y.; Kartsonaki, C.; Hill, M.R.; Bennett, D.A.; Boxall, R.; Guo, Y.; Xu, X.; Bian, Z.; Hu, R.; et al. Lipids, Lipoproteins, and Metabolites and Risk of Myocardial Infarction and Stroke. J. Am. Coll. Cardiol. 2018, 71, 620–632. [Google Scholar] [CrossRef]
- Dutta, T.; Kudva, Y.C.; Persson, X.M.; Schenck, L.A.; Ford, G.C.; Singh, R.J.; Carter, R.; Nair, K.S. Impact of Long-Term Poor and Good Glycemic Control on Metabolomics Alterations in Type 1 Diabetic People. J. Clin. Endocrinol. Metab. 2016, 101, 1023–1033. [Google Scholar] [CrossRef]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 206–220. [Google Scholar] [PubMed]
- Tanka-Salamon, A.; Komorowicz, E.; Szabo, L.; Tenekedjiev, K.; Kolev, K. Free Fatty Acids Modulate Thrombin Mediated Fibrin Generation Resulting in Less Stable Clots. PLoS ONE 2016, 11, e0167806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentier, A.C. Abnormal myocardial dietary fatty acid metabolism and diabetic cardiomyopathy. Can. J. Cardiol. 2018, 34, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Salvado, L.; Barroso, E.; Vazquez-Carrera, M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int. J. Cardiol. 2013, 168, 3160–3172. [Google Scholar] [CrossRef]
- Ali, A.; Boutjdir, M.; Aromolaran, A.S. Cardiolipotoxicity, inflammation, and arrhythmias: Role for Interleukin-6 molecular mechanisms. Front. Physiol. 2018, 9, 1866. [Google Scholar] [CrossRef]
- Westein, E.; Hoefer, T.; Calkin, A.C. Thrombosis in diabetes: A shear flow effect? Clin. Sci. 2017, 131, 1245–1260. [Google Scholar] [CrossRef]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular actions of insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef]
- Chen, Q.; Jin, M.; Yang, F.; Zhu, J.; Xiao, Q.; Zhang, L. Matrix metalloproteinases: Inflammatory regulators of cell behaviors in vascular formation and remodeling. Mediators Inflamm. 2013, 2013, 928315. [Google Scholar] [CrossRef] [Green Version]
- Peeters, S.A.; Engelen, L.; Buijs, J.; Jorsal, A.; Parving, H.H.; Tarnow, L.; Rossing, P.; Schalkwijk, C.G.; Stehouwer, C.D.A. Plasma matrix metalloproteinases are associated with incident cardiovascular disease and all-cause mortality in patients with type 1 diabetes: A 12-year follow-up study. Cardiovasc. Diabetol. 2017, 16, 55. [Google Scholar] [CrossRef]
- Peeters, S.A.; Engelen, L.; Buijs, J.; Chaturvedi, N.; Fuller, J.H.; Schalkwijk, C.G.; Stehouwer, C.D.; Group, E.P.C.S. Plasma levels of matrix metalloproteinase-2, -3, -10, and tissue inhibitor of metalloproteinase-1 are associated with vascular complications in patients with type 1 diabetes: The EURODIAB Prospective Complications Study. Cardiovasc. Diabetol. 2015, 14, 31. [Google Scholar] [CrossRef] [Green Version]
- Peeters, S.A.; Engelen, L.; Buijs, J.; Chaturvedi, N.; Fuller, J.H.; Jorsal, A.; Parving, H.H.; Tarnow, L.; Theilade, S.; Rossing, P.; et al. Circulating matrix metalloproteinases are associated with arterial stiffness in patients with type 1 diabetes: Pooled analysis of three cohort studies. Cardiovasc. Diabetol. 2017, 16, 139. [Google Scholar] [CrossRef] [Green Version]
- Kozakova, M.; Morizzo, C.; Goncalves, I.; Natali, A.; Nilsson, J.; Palombo, C. Cardiovascular organ damage in type 2 diabetes mellitus: The role of lipids and inflammation. Cardiovasc. Diabetol. 2019, 18, 61. [Google Scholar] [CrossRef]
- Chase, A.J.; Newby, A.C. Regulation of matrix metalloproteinase (matrixin) genes in blood vessels: A multi-step recruitment model for pathological remodelling. J. Vasc. Res. 2003, 40, 329–343. [Google Scholar] [CrossRef]
- Giebel, S.J.; Menicucci, G.; McGuire, P.G.; Das, A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab. Invest. 2005, 85, 597–607. [Google Scholar] [CrossRef]
- Shah, P.K. Plaque disruption and thrombosis: Potential role of inflammation and infection. Cardiol. Rev. 2000, 8, 31–39. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Daskalopoulou, S.S.; Perrea, D.; Liapis, C.D. Matrix metalloproteinases and diabetic vascular complications. Angiology 2005, 56, 173–189. [Google Scholar] [CrossRef]
- Morel, O.; Jesel, L.; Abbas, M.; Morel, N. Prothrombotic changes in diabetes mellitus. Semin. Thromb. Hemost. 2013, 39, 477–488. [Google Scholar] [CrossRef]
- Trovati, M.; Anfossi, G.; Massucco, P.; Mattiello, L.; Costamagna, C.; Piretto, V.; Mularoni, E.; Cavalot, F.; Bosia, A.; Ghigo, D. Insulin stimulates nitric oxide synthesis in human platelets and, through nitric oxide, increases platelet concentrations of both guanosine-3′, 5′-cyclic monophosphate and adenosine-3′, 5′-cyclic monophosphate. Diabetes 1997, 46, 742–749. [Google Scholar] [CrossRef]
- Westerbacka, J.; Yki-Jarvinen, H.; Turpeinen, A.; Rissanen, A.; Vehkavaara, S.; Syrjala, M.; Lassila, R. Inhibition of platelet-collagen interaction: An in vivo action of insulin abolished by insulin resistance in obesity. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Winocour, P.D.; Watala, C.; Perry, D.W.; Kinlough-Rathbone, R.L. Decreased platelet membrane fluidity due to glycation or acetylation of membrane proteins. Thromb. Haemost. 1992, 68, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Assert, R.; Scherk, G.; Bumbure, A.; Pirags, V.; Schatz, H.; Pfeiffer, A.F. Regulation of protein kinase C by short term hyperglycaemia in human platelets in vivo and in vitro. Diabetologia 2001, 44, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, G.; Wascher, T.C.; Kostner, G.M.; Graier, W.F. Alterations in platelet Ca2+ signalling in diabetic patients is due to increased formation of superoxide anions and reduced nitric oxide production. Diabetologia 1999, 42, 167–176. [Google Scholar] [CrossRef]
- Thomas, G.; Skrinska, V.; Lucas, F.V.; Schumacher, O.P. Platelet glutathione and thromboxane synthesis in diabetes. Diabetes 1985, 34, 951–954. [Google Scholar] [CrossRef]
- Randriamboavonjy, V.; Pistrosch, F.; Bolck, B.; Schwinger, R.H.; Dixit, M.; Badenhoop, K.; Cohen, R.A.; Busse, R.; Fleming, I. Platelet sarcoplasmic endoplasmic reticulum Ca2+-ATPase and mu-calpain activity are altered in type 2 diabetes mellitus and restored by rosiglitazone. Circulation 2008, 117, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, F.K.; Sobel, B.E.; Schneider, D.J. Effects of increased concentrations of glucose on platelet reactivity in healthy subjects and in patients with and without diabetes mellitus. Am. J. Cardiol. 2003, 92, 1362–1365. [Google Scholar] [CrossRef]
- Tschoepe, D.; Roesen, P.; Esser, J.; Schwippert, B.; Nieuwenhuis, H.K.; Kehrel, B.; Gries, F.A. Large platelets circulate in an activated state in diabetes mellitus. Semin. Thromb. Hemost. 1991, 17, 433–438. [Google Scholar] [CrossRef]
- Zaccardi, F.; Rocca, B.; Rizzi, A.; Ciminello, A.; Teofili, L.; Ghirlanda, G.; De Stefano, V.; Pitocco, D. Platelet indices and glucose control in type 1 and type 2 diabetes mellitus: A case-control study. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 902–909. [Google Scholar] [CrossRef]
- Santilli, F.; Marchisio, M.; Lanuti, P.; Boccatonda, A.; Miscia, S.; Davi, G. Microparticles as new markers of cardiovascular risk in diabetes and beyond. Thromb. Haemost. 2016, 116, 220–234. [Google Scholar] [CrossRef]
- Zara, M.; Guidetti, G.F.; Camera, M.; Canobbio, I.; Amadio, P.; Torti, M.; Tremoli, E.; Barbieri, S.S. Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. Int. J. Mol. Sci. 2019, 20, 2840. [Google Scholar] [CrossRef] [Green Version]
- Tramontano, A.F.; Lyubarova, R.; Tsiakos, J.; Palaia, T.; Deleon, J.R.; Ragolia, L. Circulating endothelial microparticles in diabetes mellitus. Mediators Inflamm. 2010, 2010, 250476. [Google Scholar] [CrossRef] [PubMed]
- Stec, J.J.; Silbershatz, H.; Tofler, G.H.; Matheney, T.H.; Sutherland, P.; Lipinska, I.; Massaro, J.M.; Wilson, P.F.; Muller, J.E.; D’Agostino, R.B., Sr. Association of fibrinogen with cardiovascular risk factors and cardiovascular disease in the Framingham Offspring Population. Circulation 2000, 102, 1634–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; McGeoch, S.C.; Johnstone, A.M.; Holtrop, G.; Sneddon, A.A.; MacRury, S.M.; Megson, I.L.; Pearson, D.W.; Abraham, P.; De Roos, B.; et al. Platelet-derived microparticle count and surface molecule expression differ between subjects with and without type 2 diabetes, independently of obesity status. J. Thromb. Thrombolysis 2014, 37, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.D.; Wu, X.Z.; Zhou, Y.; Xue, Y.; Zhang, K.Q. Proteomic characteristics of circulating microparticles in patients with newly-diagnosed type 2 diabetes. Am. J. Transl. Res. 2016, 8, 209–220. [Google Scholar] [PubMed]
- Zahran, A.M.; Mohamed, I.L.; El Asheer, O.M.; Tamer, D.M.; Abo, E.M.G.M.; Abdel-Rahim, M.H.; El-Badawy, O.H.B.; Elsayh, K.I. Circulating Endothelial Cells, Circulating Endothelial Progenitor Cells, and Circulating Microparticles in Type 1 Diabetes Mellitus. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029618825311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatier, F.; Darmon, P.; Hugel, B.; Combes, V.; Sanmarco, M.; Velut, J.G.; Arnoux, D.; Charpiot, P.; Freyssinet, J.M.; Oliver, C.; et al. Type 1 and type 2 diabetic patients display different patterns of cellular microparticles. Diabetes 2002, 51, 2840–2845. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.A.; Adly, A.A.; Ismail, E.A.; Darwish, Y.W.; Kamel, H.A. Platelets microparticles as a link between micro- and macro-angiopathy in young patients with type 1 diabetes. Platelets 2015, 26, 682–688. [Google Scholar] [CrossRef]
- Van Beers, E.J.; Schaap, M.C.; Berckmans, R.J.; Nieuwland, R.; Sturk, A.; van Doormaal, F.F.; Meijers, J.C.; Biemond, B.J.; group, C.s. Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica 2009, 94, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Invest. 1997, 99, 2118–2127. [Google Scholar] [CrossRef] [Green Version]
- Van Der Meijden, P.E.; Van Schilfgaarde, M.; Van Oerle, R.; Renne, T.; ten Cate, H.; Spronk, H.M. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J. Thromb. Haemost. 2012, 10, 1355–1362. [Google Scholar] [CrossRef]
- Donga, E.; Dekkers, O.M.; Corssmit, E.P.; Romijn, J.A. Insulin resistance in patients with type 1 diabetes assessed by glucose clamp studies: Systematic review and meta-analysis. Eur. J. Endocrinol. 2015, 173, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cucuianu, M.; Coca, M. Thrombotic tendency in diabetes mellitus. Revisiting and revising a study initiated 30 years ago. Rom. J. Intern. Med. 2012, 50, 107–115. [Google Scholar] [PubMed]
- Zheng, Q.; Jiang, J.; Huo, Y.; Chen, D. Genetic predisposition to type 2 diabetes is associated with severity of coronary artery disease in patients with acute coronary syndromes. Cardiovasc. Diabetol. 2019, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Pfister, R.; Barnes, D.; Luben, R.N.; Khaw, K.T.; Wareham, N.J.; Langenberg, C. Individual and cumulative effect of type 2 diabetes genetic susceptibility variants on risk of coronary heart disease. Diabetologia 2011, 54, 2283–2287. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Meigs, J.B.; Rexrode, K.M.; Hu, F.B.; Qi, L. Diabetes genetic predisposition score and cardiovascular complications among patients with type 2 diabetes. Diabetes Care 2013, 36, 737–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rosa, S.; Arcidiacono, B.; Chiefari, E.; Brunetti, A.; Indolfi, C.; Foti, D.P. Type 2 Diabetes Mellitus and Cardiovascular Disease: Genetic and Epigenetic Links. Front. Endocrinol. 2018, 9, 2. [Google Scholar] [CrossRef]
- Takaike, H.; Uchigata, Y.; Nakagami, T.; Iwamoto, Y. Incidence and development of diabetic microangiopathy of fulminant type 1 diabetes—Comparison with non-fulminant type 1 diabetes. Intern. Med. 2010, 49, 1079–1083. [Google Scholar] [CrossRef] [Green Version]
- Murase, Y.; Imagawa, A.; Hanafusa, T.; Iwahashi, H.; Uchigata, Y.; Kanatsuka, A.; Kawasaki, E.; Kobayashi, T.; Shimada, A.; Shimizu, I.; et al. Fulminant type 1 diabetes as a high risk group for diabetic microangiopathy—A nationwide 5-year-study in Japan. Diabetologia 2007, 50, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Guarnotta, V.; Vigneri, E.; Pillitteri, G.; Ciresi, A.; Pizzolanti, G.; Giordano, C. Higher cardiometabolic risk in idiopathic versus autoimmune type 1 diabetes: A retrospective analysis. Diabetol. Metab. Syndr. 2018, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- National Institute for Health and Clinical Excellence. Type 1 Diabetes in Adults: Diagnosis and Management, NICE Guideline (NG17). Available online: https://www.nice.org.uk/guidance/ng17 (accessed on 4 September 2019).
- National Institute for Health and Clinical Excellence. Diabetes (Type 1 and Type 2) in Children and Young People: Diagnosis and Management, NICE Guideline (NG18). Available online: https://www.nice.org.uk/guidance/ng18 (accessed on 4 September 2019).
- National Institute for Health and Clinical Excellence. National Institute for Health and Clinical Excellence. Type 2 Diabetes in Adults: Management, NICE Guideline (NG28). Available online: https://www.nice.org.uk/guidance/ng28 (accessed on 31 October 2019).
- Cefalu, W.T.; Schneider, D.J.; Carlson, H.E.; Migdal, P.; Gan Lim, L.; Izon, M.P.; Kapoor, A.; Bell-Farrow, A.; Terry, J.G.; Sobel, B.E. Effect of combination glipizide GITS/metformin on fibrinolytic and metabolic parameters in poorly controlled type 2 diabetic subjects. Diabetes Care 2002, 25, 2123–2128. [Google Scholar] [CrossRef] [Green Version]
- Zilov, A.V.; Abdelaziz, S.I.; AlShammary, A.; Al Zahrani, A.; Amir, A.; Assaad Khalil, S.H.; Brand, K.; Elkafrawy, N.; Hassoun, A.A.K.; Jahed, A.; et al. Mechanisms of action of metformin with special reference to cardiovascular protection. Diabetes Metab. Res. Rev. 2019, 35, e3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamanna, C.; Monami, M.; Marchionni, N.; Mannucci, E. Effect of metformin on cardiovascular events and mortality: A meta-analysis of randomized clinical trials. Diabetes Obes. Metab. 2011, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.J.; Leaver, J.K.; Irving, G.J. Impact of metformin on cardiovascular disease: A meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia 2017, 60, 1620–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.M.; Bellman, S.M.; Stephenson, M.D.; Lisy, K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: A systematic review and meta-analysis. Ageing Res. Rev. 2017, 40, 31–44. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Davies, M.J.; D‘Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2018, 41, 2669–2701. [Google Scholar] [CrossRef] [Green Version]
- Fei, Y.; Tsoi, M.F.; Cheung, B.M.Y. Cardiovascular outcomes in trials of new antidiabetic drug classes: A network meta-analysis. Cardiovasc. Diabetol. 2019, 18, 112. [Google Scholar] [CrossRef] [Green Version]
- Khunti, K.; Chatterjee, S.; Gerstein, H.C.; Zoungas, S.; Davies, M.J. Do sulphonylureas still have a place in clinical practice? Lancet Diabetes Endocrinol. 2018, 6, 821–832. [Google Scholar] [CrossRef]
- Gupta, A.K.; Verma, A.K.; Kailashiya, J.; Singh, S.K.; Kumar, N. Sitagliptin: Anti-platelet effect in diabetes and healthy volunteers. Platelets 2012, 23, 565–570. [Google Scholar] [CrossRef]
- Aroor, A.R.; Manrique-Acevedo, C.; DeMarco, V.G. The role of dipeptidylpeptidase-4 inhibitors in management of cardiovascular disease in diabetes; focus on linagliptin. Cardiovasc. Diabetol. 2018, 17, 59. [Google Scholar] [CrossRef]
- Aroor, A.R.; Sowers, J.R.; Jia, G.; DeMarco, V.G. Pleiotropic effects of the dipeptidylpeptidase-4 inhibitors on the cardiovascular system. Am. J. Physiol. Heart. Circ. Physiol. 2014, 307, H477–H492. [Google Scholar] [CrossRef] [PubMed]
- Cameron-Vendrig, A.; Reheman, A.; Siraj, M.A.; Xu, X.R.; Wang, Y.; Lei, X.; Afroze, T.; Shikatani, E.; El-Mounayri, O.; Noyan, H.; et al. Glucagon-Like Peptide 1 Receptor Activation Attenuates Platelet Aggregation and Thrombosis. Diabetes 2016, 65, 1714–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sposito, A.C.; Berwanger, O.; de Carvalho, L.S.F.; Saraiva, J.F.K. GLP-1RAs in type 2 diabetes: Mechanisms that underlie cardiovascular effects and overview of cardiovascular outcome data. Cardiovasc. Diabetol. 2018, 17, 157. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.K.; Strong, J. The Pleiotropic Effects of Sodium-Glucose Cotransporter-2 Inhibitors: Beyond the Glycemic Benefit. Diabetes. Ther. 2019, 10, 1771–1792. [Google Scholar] [CrossRef] [Green Version]
- Mobarrez, F.; He, S.; Broijersen, A.; Wiklund, B.; Antovic, A.; Antovic, J.; Egberg, N.; Jorneskog, G.; Wallen, H. Atorvastatin reduces thrombin generation and expression of tissue factor, P-selectin and GPIIIa on platelet-derived microparticles in patients with peripheral arterial occlusive disease. Thromb. Haemost. 2011, 106, 344–352. [Google Scholar] [CrossRef]
- Park, H.S.; Gu, J.Y.; Yoo, H.J.; Han, S.E.; Park, C.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Thrombin Generation Assay Detects Moderate-Intensity Statin-Induced Reduction of Hypercoagulability in Diabetes. Clin. Appl. Thromb. Hemost. 2018, 24, 1095–1101. [Google Scholar] [CrossRef]
- American Diabetes, A. 9. Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018, 41, S86–S104. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Simplified schema of coagulation–activation, anticoagulation, fibrinolysis and anti-fibrinolysis by various mechanisms. (A) Platelet activation. (B) Extrinsic pathway of coagulation. (C) Intrinsic pathway of coagulation. (D) Fibrinolysis. Anticoagulation and anti-fibrinolytic activities are indicated in red. All proteins inhibited by antithrombin are indicated in green. Abbreviations used: F, coagulation factor; HMWK, high molecular weight kininogen; K, kallikrein; PAI-1, plasminogen activator inhibitor-1; PK, pre-kallikrein; TAFI, thrombin-activatable fibrinolysis inhibitor; TF, tissue factor; TRPI, tissue factor pathway inhibitor; tPA, tissue plasminogen activator; vWF, von Willebrand factor. Activated coagulation factors are indicated with an “a”.
Figure 1.
Simplified schema of coagulation–activation, anticoagulation, fibrinolysis and anti-fibrinolysis by various mechanisms. (A) Platelet activation. (B) Extrinsic pathway of coagulation. (C) Intrinsic pathway of coagulation. (D) Fibrinolysis. Anticoagulation and anti-fibrinolytic activities are indicated in red. All proteins inhibited by antithrombin are indicated in green. Abbreviations used: F, coagulation factor; HMWK, high molecular weight kininogen; K, kallikrein; PAI-1, plasminogen activator inhibitor-1; PK, pre-kallikrein; TAFI, thrombin-activatable fibrinolysis inhibitor; TF, tissue factor; TRPI, tissue factor pathway inhibitor; tPA, tissue plasminogen activator; vWF, von Willebrand factor. Activated coagulation factors are indicated with an “a”.

Figure 2.
An altered lipid profile increases the risk of cardiovascular disease in T1DM and T2DM. Abbreviations used: FFA, free fatty acid; HDL, high-density lipoprotein; LDL, low-density lipoprotein; T2DM, type-2 diabetes mellitus.
Figure 2.
An altered lipid profile increases the risk of cardiovascular disease in T1DM and T2DM. Abbreviations used: FFA, free fatty acid; HDL, high-density lipoprotein; LDL, low-density lipoprotein; T2DM, type-2 diabetes mellitus.

Figure 3.
The changes in inflammation, oxidative stress and blood levels of glucose, insulin and lipids associated with T1DM and T2DM cause endothelial dysfunction, which itself results in an increase in the risk of cardiovascular disease. Abbreviations used: AGE, advanced glycation end-products; CAM, cell adhesion molecules; FFA, free fatty acid; T2DM, type-2 diabetes mellitus; RAGE, advanced glycation end-product receptors, T1DM, type-1 diabetes mellitus; T2DM, type-2 diabetes mellitus.
Figure 3.
The changes in inflammation, oxidative stress and blood levels of glucose, insulin and lipids associated with T1DM and T2DM cause endothelial dysfunction, which itself results in an increase in the risk of cardiovascular disease. Abbreviations used: AGE, advanced glycation end-products; CAM, cell adhesion molecules; FFA, free fatty acid; T2DM, type-2 diabetes mellitus; RAGE, advanced glycation end-product receptors, T1DM, type-1 diabetes mellitus; T2DM, type-2 diabetes mellitus.

Figure 4.
Mechanisms resulting in the hyper-reactivity, hyper-activation, aggregation and adhesion of platelets in T1DM and T2DM. Abbreviations used: NO, nitric oxide; PGI2, prostacyclin; PKC, protein kinase C; T2DM, type-2 diabetes mellitus.
Figure 4.
Mechanisms resulting in the hyper-reactivity, hyper-activation, aggregation and adhesion of platelets in T1DM and T2DM. Abbreviations used: NO, nitric oxide; PGI2, prostacyclin; PKC, protein kinase C; T2DM, type-2 diabetes mellitus.


{kind=link}
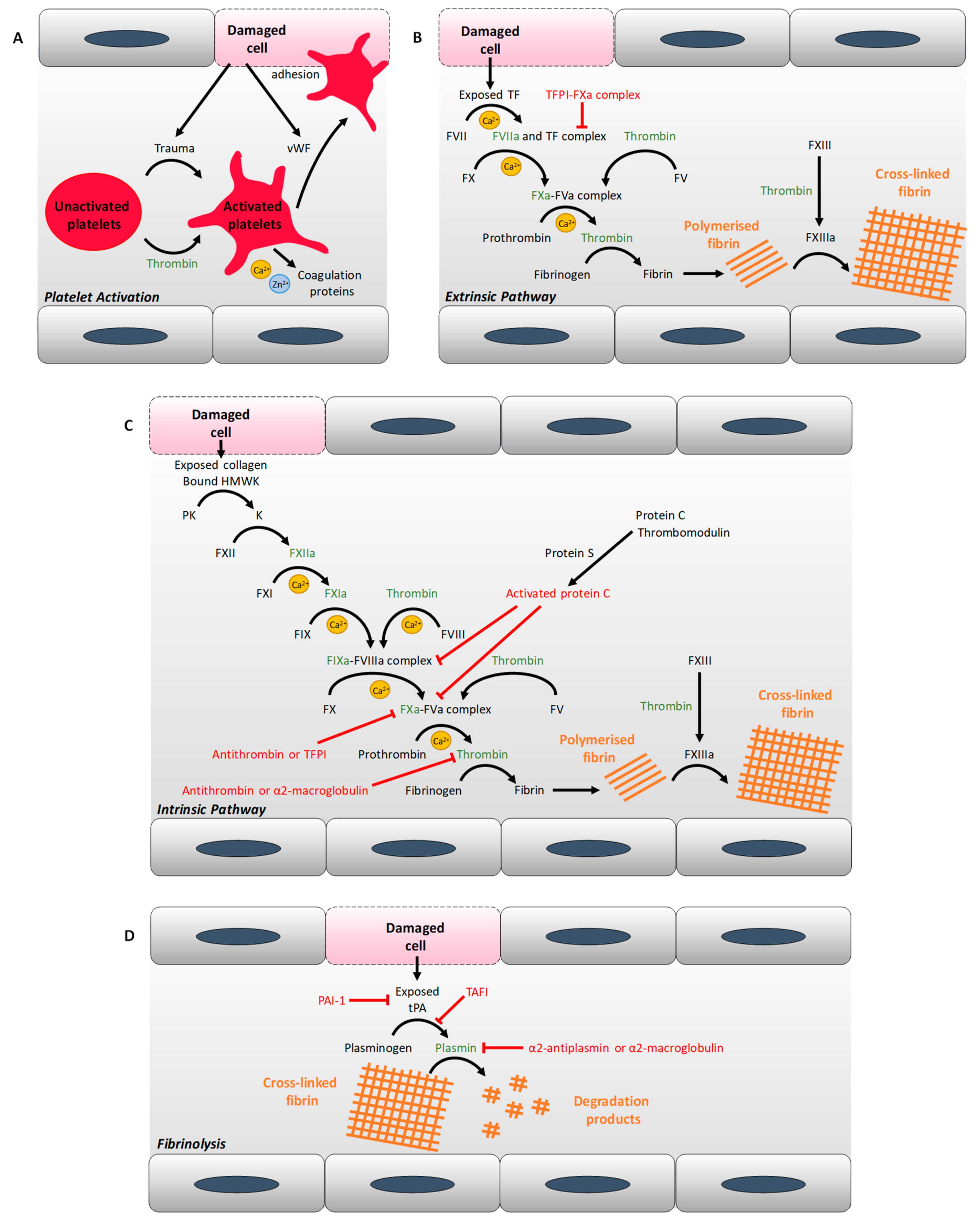{kind=link}
{kind=link}
{kind=link}
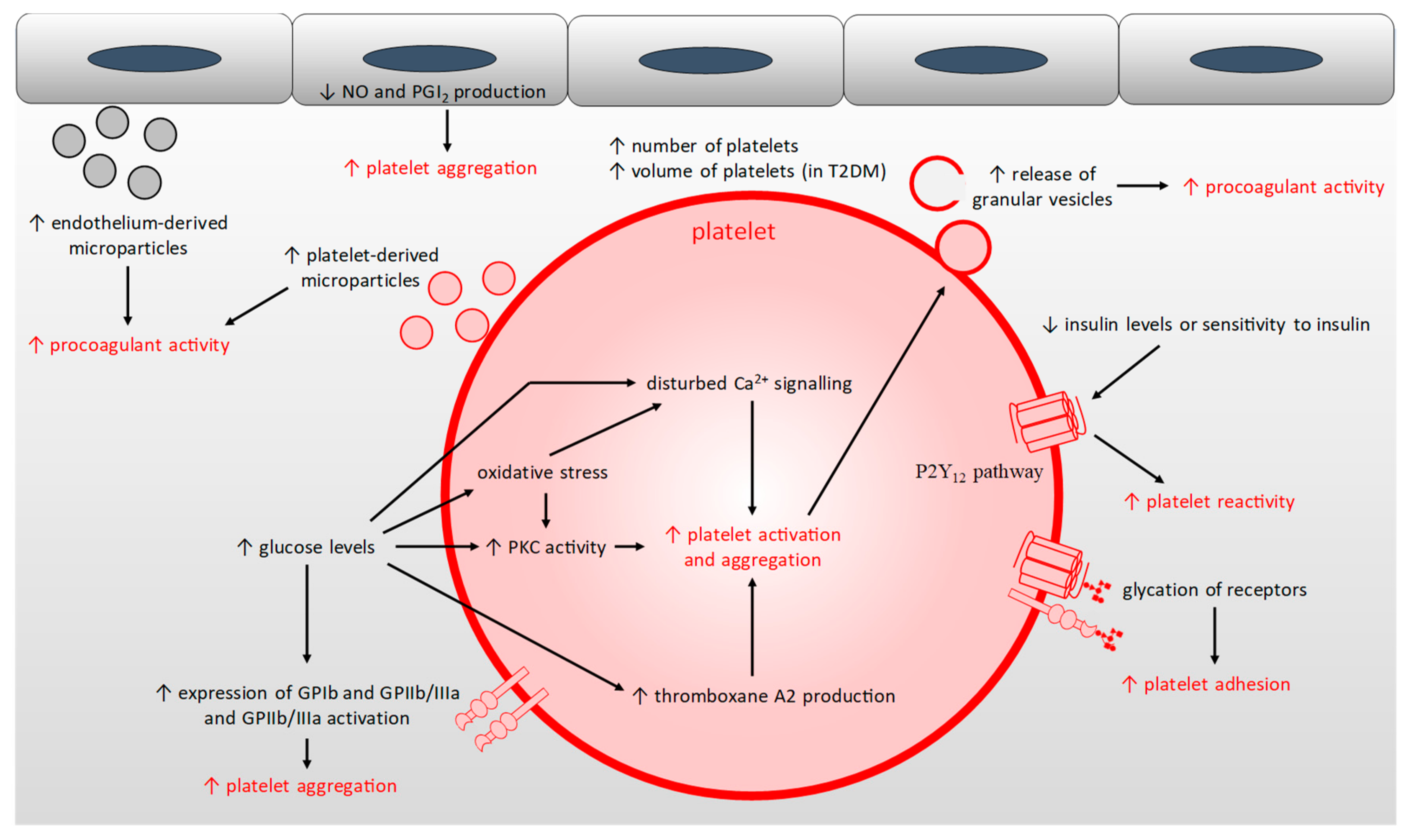{kind=link}
Table 1.
Summary of proteins that have exhibited an altered concentration or activity in individuals with type-1 diabetes mellitus (T1DM) or T2DM. Abbreviations used: PAI-1, plasminogen activator inhibitor-1; vWF, von Willebrand factor.
Table 1.
Summary of proteins that have exhibited an altered concentration or activity in individuals with type-1 diabetes mellitus (T1DM) or T2DM. Abbreviations used: PAI-1, plasminogen activator inhibitor-1; vWF, von Willebrand factor.
| T1DM | T2DM | |
|---|---|---|
| Pro-coagulant proteins | ↑ vWF [12] | ↑ vWF [10,11,12] |
| ↑ prekallikrein [13] | ↑ kininogen [23] | |
| ↑ tissue factor procoagulant activity [17] | ↑ kallikrein [14] | |
| ↑ factor V [15] | ↑ soluble tissue factor [16,24] | |
| ↑ (activated) factor VII [15,17] | ↑ factor V [15] | |
| ↑ factor VIII [15] | ↑ (activated) factor VII [15,16] | |
| ↑ factor X [15] | ↑ factor VIII [15,18] | |
| ↑ factor XI [14] | ↑ factor IX [18] | |
| ↓ activated factor XII [27] | ↑ factor X [15] | |
| ↑ prothrombin [15] | ↑ factor XI [14] | |
| ↓ fibrinogen [22], ↑ fibrinogen in diabetic complications [21] | ↑ (activated) factor XII [18,25] | |
| ↑ factor XIII [26] | ||
| ↑ prothrombin [15] | ||
| ↑ fibrinogen [19,20] | ||
| Anticoagulant proteins | ↓ antithrombin activity [49,50] | ↑ antithrombin [18,33], ↓ antithrombin [32], ↓ antithrombin activity with bad glycaemic control [47] |
| ↓ protein C [15,19,28] | ↓ protein C [15,19,29], ↓ protein C activity with bad glycaemic control [47] | |
| ↓ protein S [30] | ↓ protein S [30], ↓ protein S activity with bad glycaemic control [47] | |
| ↑ tissue factor pathway inhibitor activity [49] | ↑ tissue factor pathway inhibitor [16] | |
| ↑ thrombomodulin [31] | ↑ thrombomodulin [29] | |
| Pro-fibrinolytic proteins | ↑ tissue plasminogen activator in diabetic complications [37] | ↑ tissue plasminogen activator [24] |
| ↓ plasmin activity [51] | ||
| Anti-fibrinolytic proteins | ↓ PAI-1 [39], ↓ PAI-1 activity [22,52] | ↑ PAI-1 [10,19,20,24,38] |
| ↑ α2-antiplasmin [19,43], ↓ α2-antiplasmin [22] | ↑ α2-antiplasmin [19] | |
| ↑ thrombin-activatable fibrinolysis inhibitor [41] | ↑ thrombin-activatable fibrinolysis inhibitor [40] | |
| ↑ α2-maroglobulin [42] | ↑ α2-maroglobulin [32,42] |
↑ indicates an increase in concentration or activity, while ↓ indicates a decrease.
Table 2.
Summary of the similarities and differences in how T1DM and T2DM impact on coagulation. Abbreviations used: AGE, Advanced glycation end-products; FFA, free fatty acid; HDL, high-density lipoprotein; LDL, low-density lipoprotein; PAI-1, plasminogen activator inhibitor; T1DM, type-1 diabetes mellitus; T2DM, type-2 diabetes mellitus.
Table 2.
Summary of the similarities and differences in how T1DM and T2DM impact on coagulation. Abbreviations used: AGE, Advanced glycation end-products; FFA, free fatty acid; HDL, high-density lipoprotein; LDL, low-density lipoprotein; PAI-1, plasminogen activator inhibitor; T1DM, type-1 diabetes mellitus; T2DM, type-2 diabetes mellitus.
| In T1DM | In T2DM | In both T1DM and T2DM | |
|---|---|---|---|
| Coagulation | Reduced PAI-1 levels | Increased levels of anti-fibrinolysis proteins, including PAI-1 | Increased levels of pro-coagulatory proteins |
| Reduced anticoagulant activity | |||
| Denser fibrin fibres, less porous fibrin clot, fibrin clot more resistant to fibrinolysis | |||
| Metal ions | Dysregulation of coagulation by Mg2+ deficiency | Possible dysregulation of coagulation by elevated Ca2+ levels | Possible dysregulation of coagulation by elevated ceruloplasmin levels |
| Possible dysregulation of coagulation by Fe3+ supplements | Dysregulation of coagulation by altered zinc speciation | ||
| Possible dysregulation of coagulation by elevated iron levels | |||
| Lipids | Unchanged or reduced HDL levels | Reduced HDL levels | Elevated levels of small dense LDL that favoured atherosclerotic plaque formation and endothelial dysfunction |
| Reduced plasma FFA levels | Elevated plasma FFA levels causing the destabilisation of fibrin clot, metabolism dysregulation, atherosclerotic plaques and cardiac lipotoxicity | HDL dysfunction causing reduced HDL efflux, reduced anti-inflammatory effects and endothelial dysfunction | |
| Hypertriglyceridemia causing HDL and endothelial dysfunction | |||
| Endothelial dysfunction | Excess FFA levels causing endothelial dysfunction | Endothelial dysfunction causes reduced nitric oxide production, dysregulation of vasodilators and vasoconstrictors | |
| Formation of AGEs dysregulating nitric oxide synthase and protein synthesis by the endothelium, causing endothelial dysfunction | |||
| Matrix metalloproteinases upregulation, causing inflammation, endothelial dysfunction, vascular remodelling, thrombus formation and atherosclerotic plaque formation and destabilisation | |||
| Platelets | Platelets have unchanged volume | Larger platelets | Higher platelet count |
| Hyper-activation, adherence and aggregation of platelets | |||
| Microparticles | Elevated levels of endothelial- and platelet-derived microparticles correlated with HbA1c and associated with pro-coagulatory activity | Elevated levels of endothelial- and platelet-derived microparticles enriched in coagulation proteins and associated with atherosclerosis and thrombosis | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sobczak, A.I.S.; Stewart, A.J. Coagulatory Defects in Type-1 and Type-2 Diabetes. Int. J. Mol. Sci. 2019, 20, 6345. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246345
AMA Style
Sobczak AIS, Stewart AJ. Coagulatory Defects in Type-1 and Type-2 Diabetes. International Journal of Molecular Sciences. 2019; 20(24):6345. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246345
Chicago/Turabian StyleSobczak, Amélie I. S., and Alan J. Stewart. 2019. "Coagulatory Defects in Type-1 and Type-2 Diabetes" International Journal of Molecular Sciences 20, no. 24: 6345. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246345
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.