Transcriptome Analysis of Watermelon Leaves Reveals Candidate Genes Responsive to Cucumber green mottle mosaic virus Infection


Abstract
:
1. Introduction
2. Results
2.1. Phenotypes and Confirmation of CGMMV in Watermelon Post Inoculation
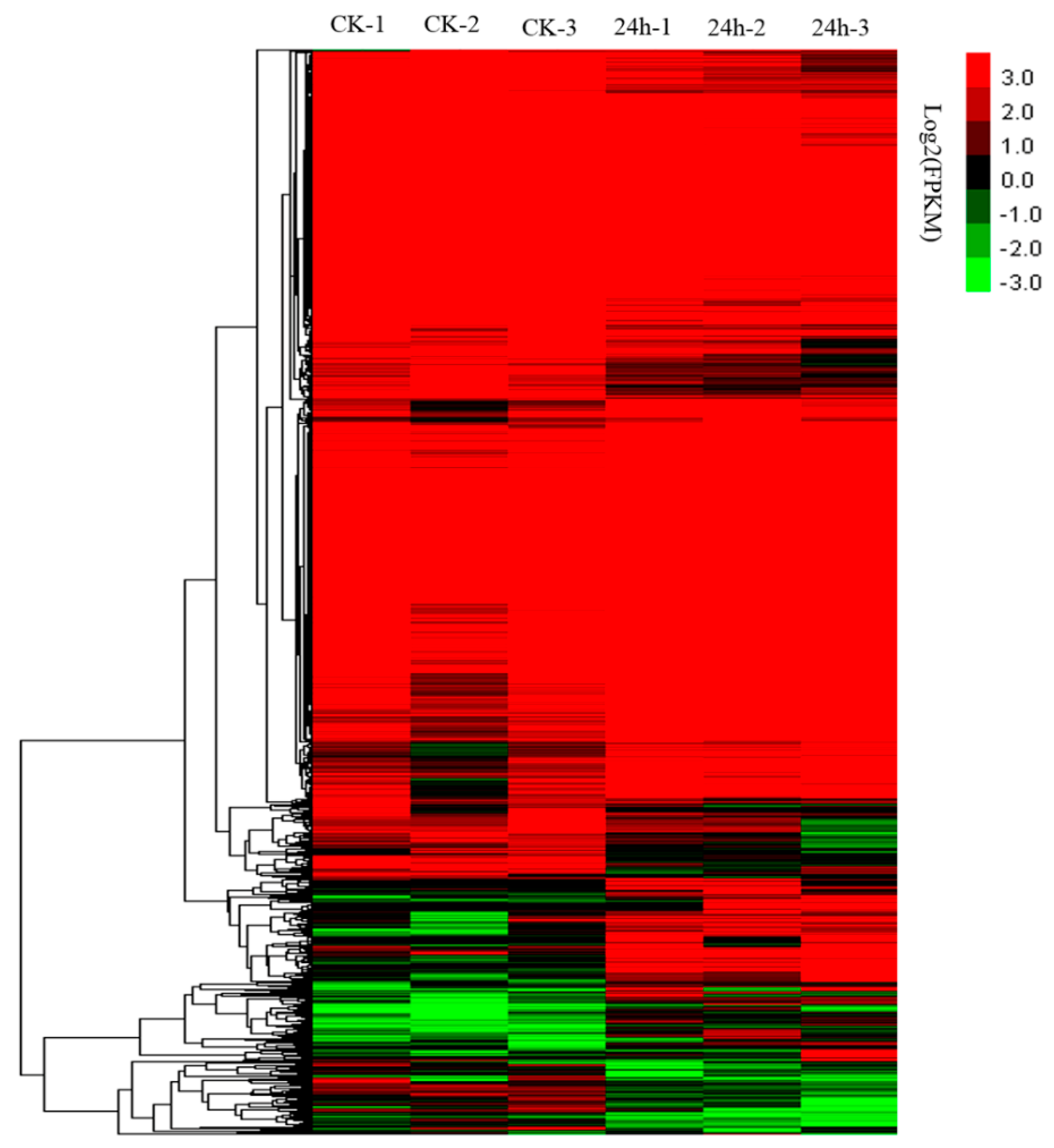
2.2. Overview of the RNA-Seq Results
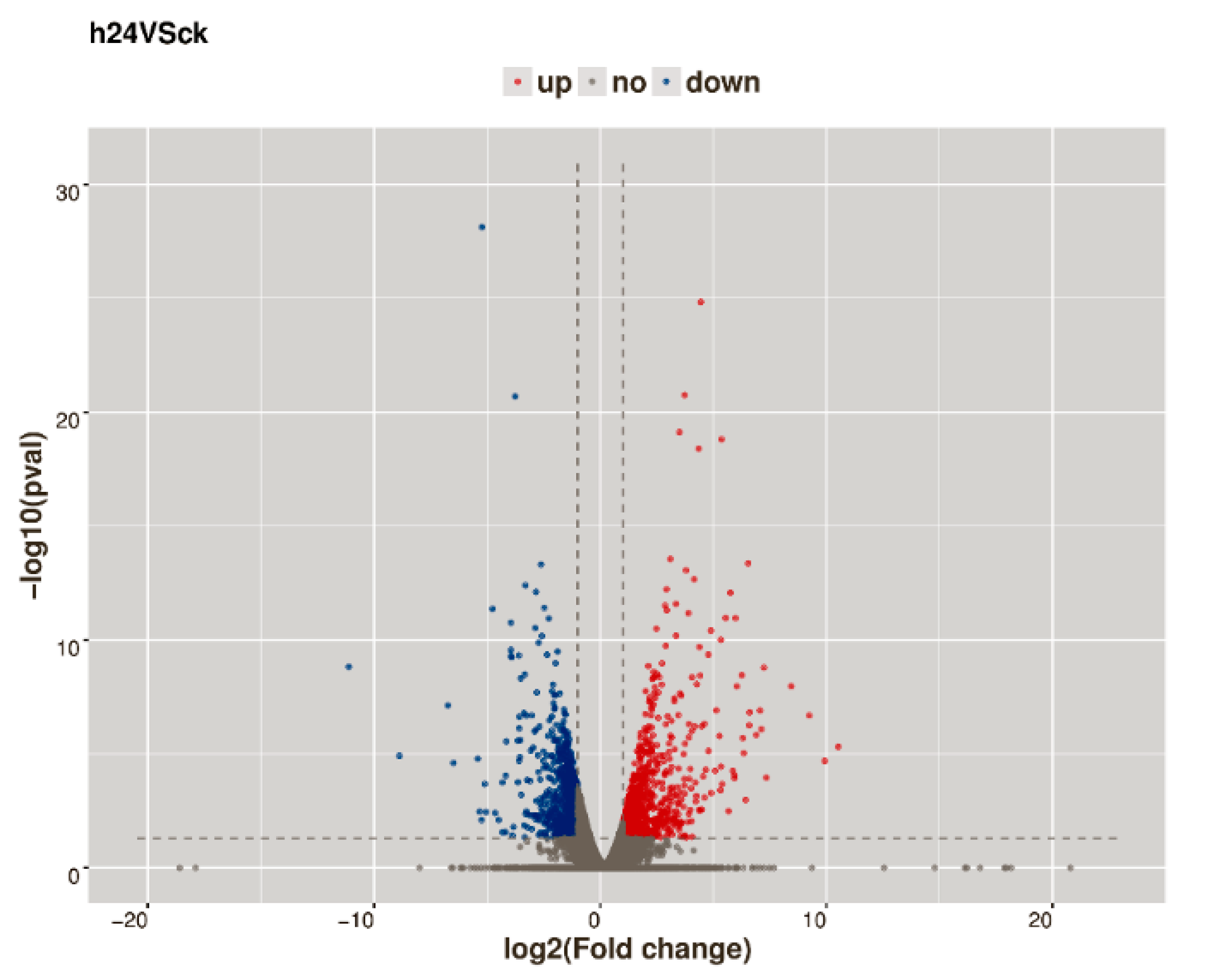
2.3. Analysis of DEGs in Response to CGMMV-Induced Stress
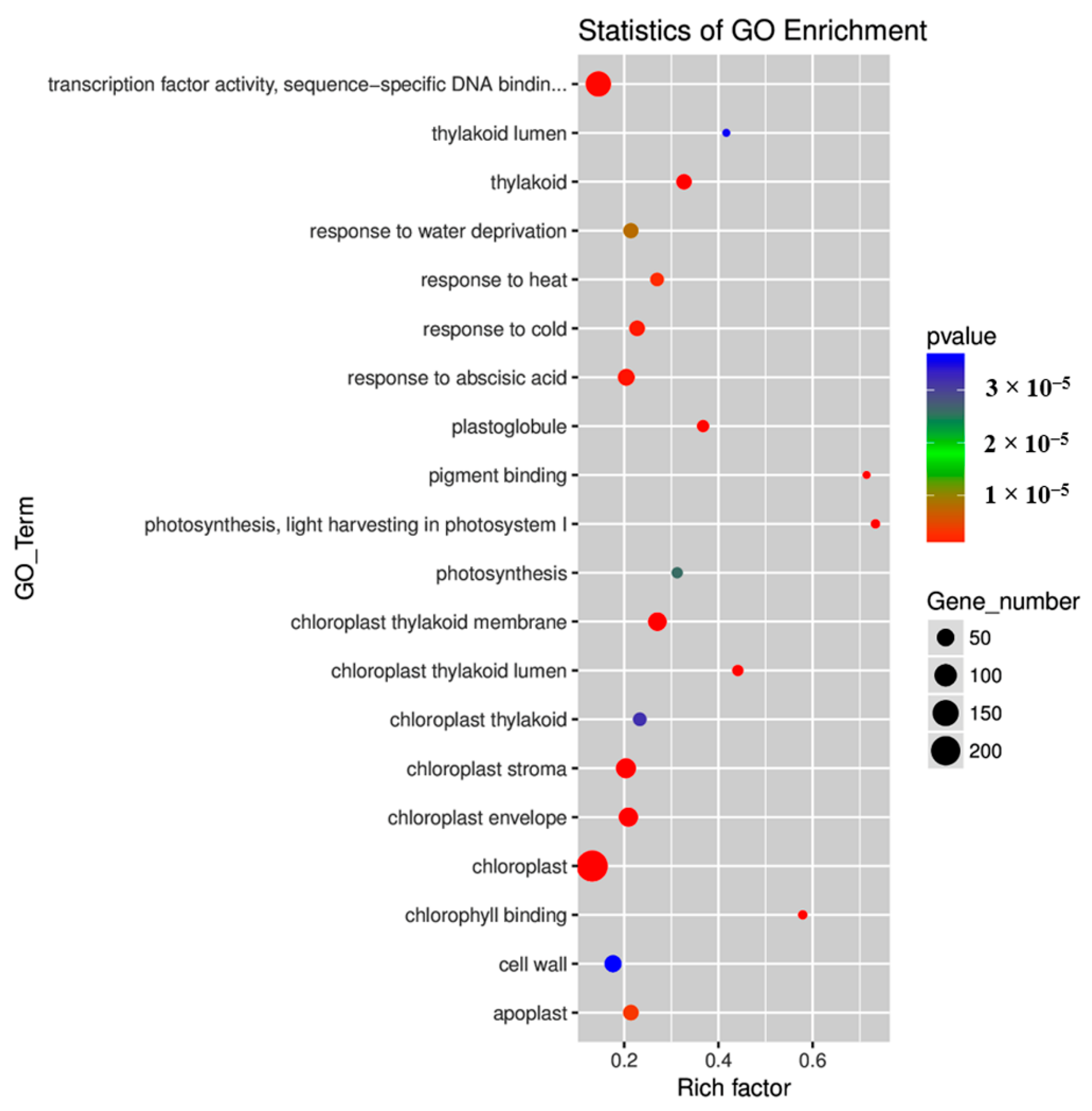
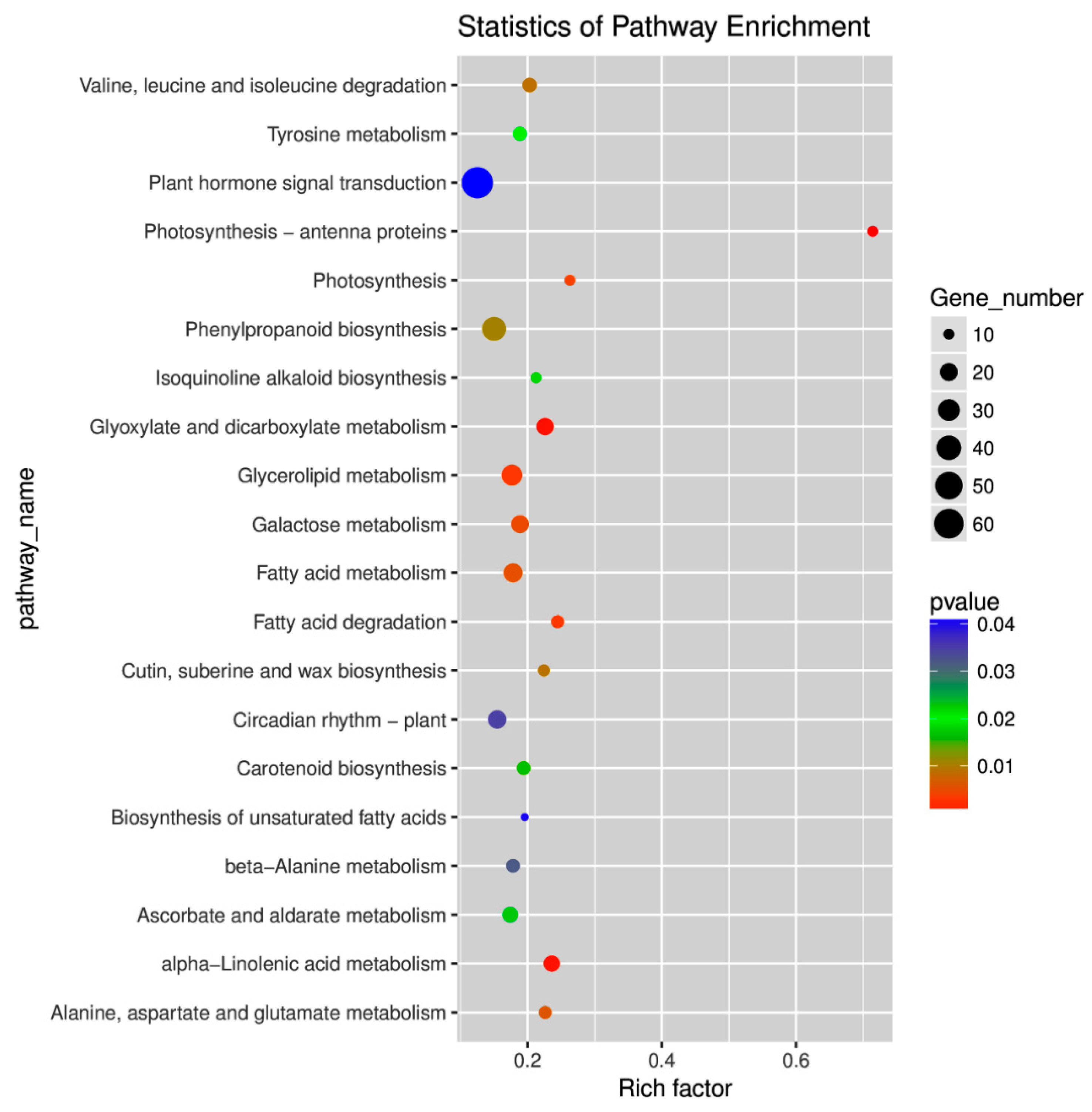
2.4. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analyses of DEGs
2.5. DEGs Involved in Photosynthesis
2.6. DEGs Involved in Plant–Pathogen Interactions
2.7. DEGs Involved in Secondary Metabolism
2.8. DEGs Involved in Plant Hormone Signal Transduction
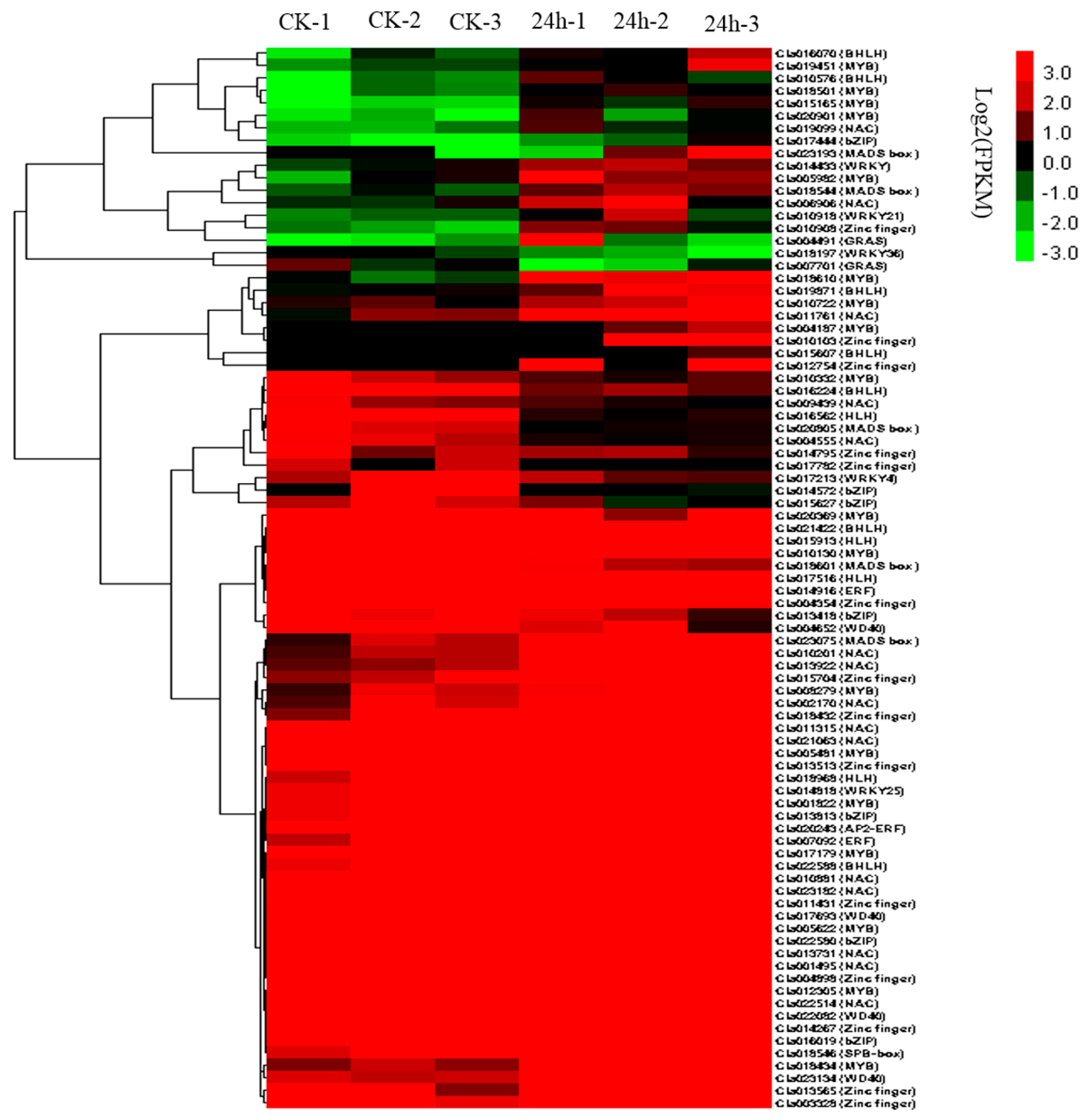
2.9. TFs Involved in CGMMV Stress Response
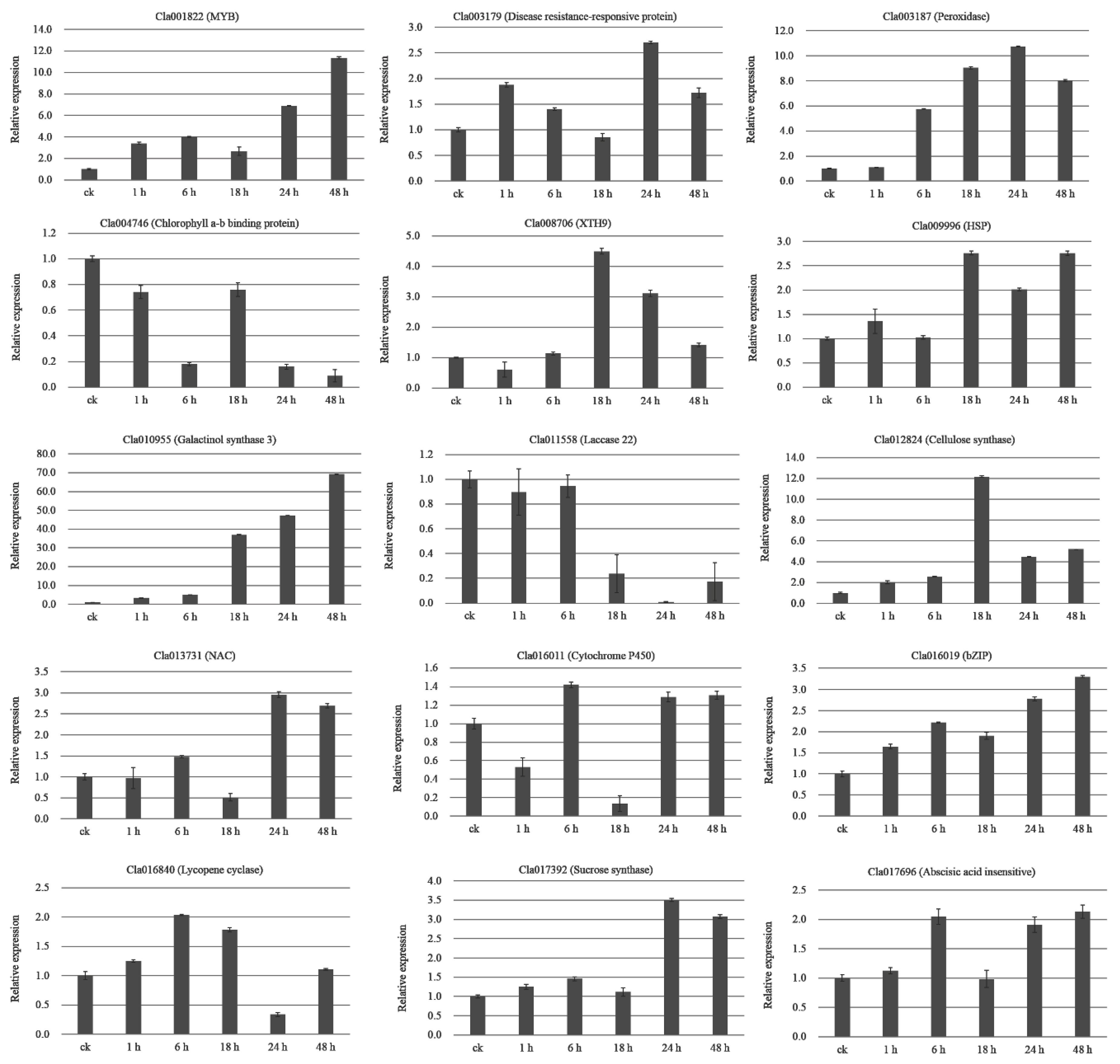
2.10. qRT-PCR Validation of DEGs
3. Discussion
3.1. DEGs in Watermelon Leaves and Fruit
3.2. Changes in Photosynthesis After CGMMV Infection
3.3. Changes in Plant–Pathogen Interactions After CGMMV Infection
3.4. The Main TF Families Responding to CGMMV-Induced Stress
4. Materials and Methods
4.1. Plant Materials and the CGMMV Treatment
4.2. mRNA Library Construction and Sequencing
4.3. RNA-Seq Read Mapping
4.4. Transcript Abundance Estimation and Differential Expression Analysis
4.5. GO and KEGG Enrichment Analyses of DEGs
4.6. Validation of RNA-Seq Gene Expression Using qRT-PCR
5. Conclusions
Supplementary Materials
Availability of Sequencing Data
Author Contributions
Acknowledgments
Conflicts of Interest
Compliance with Ethical Standards
References
- Tian, T.; Posis, K.; Maroon-Lango, C.J.; Mavrodieva, V.; Haymes, S.; Pitman, T.L.; Falk, W. First report of cucumber green mottle mosaic virus on melon in the United States. Plant Dis. 2014, 98, 1163. [Google Scholar] [CrossRef]
- Liu, H.W.; Luo, L.X.; Li, J.Q.; Liu, P.F.; Chen, X.Y.; Hao, J.J. Pollen and seed transmission of Cucumber green mottle mosaic virus in cucumber. Plant Pathol. 2014, 63, 72–77. [Google Scholar] [CrossRef]
- Mink, G.I. Pollen- and seed-transmitted viruses and viroids. Ann. Rev. Phytopathol. 1993, 31, 375–402. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, G.C. Mosaic disease of the cucumber. Ann. Appl. Biol. 1935, 22, 55–67. [Google Scholar] [CrossRef]
- Pop, I.; Jilaveanu, A. Identification of cucumber green mottle virus in Romania. Analele Institutului de Cercetari Pentru Protectia Plantelor 1985, 18, 43–47. [Google Scholar]
- Varveri, C.; Vassilakos, N.; Bem, F. Characterization and detection of Cucumber green mottle mosaic virus in Greece. Phytoparasitica 2002, 30, 493–501. [Google Scholar] [CrossRef]
- Li, R.; Zheng, Y.; Fei, Z.; Ling, K.S. First complete genome sequence of an emerging cucumber green mottle mosaic virus isolate in North America. Genome Announc. 2015, 3, e00452-15. [Google Scholar] [CrossRef]
- Tesoriero, L.A.; Chambers, G.; Srivastava, M.; Smith, S.; Conde, B.; Tran-Nguyen, L.T.T. First report of cucumber green mottle mosaic virus in Australia. Australas. Plant Dis. Notes 2016, 11, 1. [Google Scholar] [CrossRef]
- Dombrovsky, A.; Tran-Nguyen, L.T.T.; Jones, R.A.C. Cucumber green mottle mosaic virus: Rapidly increasing global distribution, etiology, epidemiology and management. Ann. Rev. Phytopathol. 2017, 55, 231–256. [Google Scholar] [CrossRef]
- Liu, H.W.; Luo, L.X.; Liang, C.Q.; Jiang, N.; Liu, P.F.; Li, J.Q. High-throughput sequencing identifies novel and conserved cucumber (Cucumis sativus L.) microRNAs in response to Cucumber green mottle mosaic virus infection. PLoS ONE 2015, 10, e0129002. [Google Scholar] [CrossRef]
- Li, Y.; Deng, C.; Shang, Q.; Zhao, X.; Liu, X.; Zhou, Q. Characterization of siRNAs derived from cucumber green mottle mosaic virus in infected cucumber plants. Arch. Virol. 2016, 161, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, H.; Zhang, C.; Han, K.; Wang, S.; Peng, J.; Lu, Y.; Zhao, J.; Xu, P.; Wu, X.; et al. Different virus-derived siRNAs profiles between leaves and fruits in Cucumber green mottle mosaic virus-infected Lagenaria siceraria plants. Front. Microbiol. 2016, 7, 1797. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; An, M.; Xia, Z.; Bai, X.; Wu, Y. Transcriptome analysis of watermelon (Citrullus lanatus) fruits in response to Cucumber green mottle mosaic virus (CGMMV) infection. Sci. Rep. 2017, 7, 16747. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Niu, X.; Fan, M. Genome-wide identification of cucumber green mottle mosaic virus-responsive microRNAs in watermelon. Arch. Virol. 2017, 162, 2591–2602. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Niu, X.; Cui, D.; Fan, M. High-throughput sequencing reveals vsiRNAs derived from Cucumber green mottle mosaic virus-infected watermelon. Hortic. Plant J. 2017, 3, 17–22. [Google Scholar] [CrossRef]
- Rahoutei, J.; García-Luque, I.; Barón, M. Inhibition of photosynthesis by viral infection: Effect on PSII structure and function. Physiol. Plant 2000, 110, 286–292. [Google Scholar] [CrossRef]
- Scharte, J.; Schon, H.; Weis, E. Photosynthesis and carbohydrate metabolism in tobacco leaves during an incompatible interaction with Phytophthora nicotianae. Plant Cell Environ. 2005, 28, 1421–1435. [Google Scholar] [CrossRef]
- Parker, D.; Beckmann, M.; Zubair, H.; Enot, D.P.; Caracuel-Rios, Z.; Overy, D.P.; Snowdon, S.; Talbot, N.J.; Draper, J. Metabolomic analysis reveals a common pattern of metabolic re-programming during invasion of three host plant species by Magnaporthe grisea. Plant J. 2009, 59, 723–737. [Google Scholar] [CrossRef]
- Fanciullino, A.L.; Bidel, L.P.R.; Urban, L. Carotenoid responses to environmental stimuli: Integrating redox and carbon controls into a fruit model. Plant Cell Environ. 2014, 37, 273–289. [Google Scholar] [CrossRef]
- Fujiwara, A.; Togawa, S.; Hikawa, T.; Matsuura, H.; Masuta, C.; Inukai, T. Ascorbic acid accumulates as a defense response to Turnip mosaic virus in resistant Brassica rapa cultivars. J. Exp. Bot. 2016, 67, 4391–4402. [Google Scholar] [CrossRef]
- Collum, T.D.; Culver, J.N. The impact of phytohormones on virus infection and disease. Curr. Opin. Virol. 2016, 17, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, H.; Urao, T.; Ito, T.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 2003, 15, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Ito, Y.; Yamaguchi-Shinozaki, K. Transcriptional regulatory networks in response to abiotic stresses in Arabidopsis and grasses. Plant Physiol. 2009, 149, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Reingold, V.; Lachman, O.; Koren, A.; Dombrovsky, A. First report of Cucumber green mottle mosaic virus (CGMMV) symptoms in watermelon used for the discrimination of non-marketable fruits in Israeli commercial fields. Plant Pathol. 2013, 28, 11. [Google Scholar]
- Fan, H.; Zhang, Y.; Sun, H.; Liu, J.; Wang, Y.; Wang, X.; Li, D.; Yu, J.; Han, C. Transcriptome analysis of beta macrocarpa and identification of differentially expressed transcripts in response to Beet necrotic yellow vein virus infection. PLoS ONE 2015, 10, e0132277. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Singh, J.; Hill, J.H.; Whitham, S.A.; Cannon, S.B. Dynamic transcriptome profiling of Bean Common Mosaic Virus (BCMV) infection in Common Bean (Phaseolus vulgaris L.). BMC Genom. 2016, 17, 613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zheng, H.; Wu, X.; Xu, H.; Han, K.; Peng, J.; Lu, Y.; Lin, L.; Xu, P.; Wu, X.; et al. Genome-wide identification of new reference genes for RT-qPCR normalization in CGMMV-infected Lagenaria siceraria. Peer J. 2018, 6, e5642. [Google Scholar] [CrossRef] [PubMed]
- Nelson, N.; Yocum, C.F. Structure and function of Photosystems I and II. Ann. Rev. Plant Biol. 2006, 57, 521–565. [Google Scholar] [CrossRef]
- Bricker, T.M.; Frankel, L.K. Auxiliary functions of the PsbO, PsbP and PsbQ proteins of higher plant photosystem II: A critical analysis. J. Photochem. Photobiol. B Biol. 2011, 104, 165–178. [Google Scholar] [CrossRef]
- Lu, Y.K.; Stemler, A.J. Extrinsic Photosystem II carbonic anhydrase in maize mesophyll chloroplasts. Plant Physiol. 2002, 128, 643–649. [Google Scholar] [CrossRef]
- Spetea, C.; Hundal, T.; Lundin, B.; Heddad, M.; Adamska, I.; Andersson, B. Multiple evidence for nucleotide metabolism in the chloroplast thylakoid lumen. Proc. Natl. Acad. Sci. USA 2004, 101, 1409–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondarava, N.; Krieger-Liszkay, A. Manganese binding to the 23 kDa extrinsic protein of Photosystem II. Biochim. Biophys. Acta 2007, 1767, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Las Rivas, J.; Heredia, P.; Roman, A. Oxygen-evolving extrinsic proteins (PsbO, P, Q, R): Bioinformatic and functional analysis. Biochim. Biophys. Acta 2007, 1767, 575–582. [Google Scholar] [CrossRef] [PubMed]
- García-Cerdán, J.G.; Kovács, L.; Tóth, T.; Kereïche, S.; Aseeva, E.; Boekema, E.J.; Mamedov, F.; Funk, C.; Schröder, W.P. The PsbW protein stabilizes the supramolecular organization of photosystem II in higher plants. Plant J. 2011, 65, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Fu, A.; Garcia, V.J.; Buchanan, B.B.; Luan, S. PSB27: A thylakoid protein enabling Arabidopsis to adapt to changing light intensity. Proc. Natl. Acad. Sci. USA 2015, 112, 1613–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrzykowska, M.; Suorsa, M.; Semchonok, D.A.; Tikkanen, M.; Boekema, E.J.; Aro, E.M.; Jansson, S. The light-harvesting chlorophyll a/b binding proteins Lhcb1 and Lhcb2 play complementary roles during state transitions in Arabidopsis. Plant Cell 2014, 26, 3646–3660. [Google Scholar] [CrossRef] [PubMed]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef]
- Yuenyong, W.; Chinpongpanich, A.; Comai, L.; Chadchawan, S.; Buaboocha, T. Downstream components of the calmodulin signaling pathway in the rice salt stress response revealed by transcriptome profiling and target identification. BMC Plant Biol. 2018, 18, 335. [Google Scholar] [CrossRef]
- Heo, W.D.; Lee, S.H.; Kim, M.C.; Kim, J.C.; Chung, W.S.; Chun, H.J.; Lee, K.J.; Park, C.Y.; Park, H.C.; Choi, J.Y.; et al. Involvement of specific calmodulin isoforms in salicylic acid-independent activation of plant disease resistance responses. Proc. Natl. Acad. Sci. USA 1999, 96, 766–771. [Google Scholar] [CrossRef] [Green Version]
- Yamakawa, H.; Mitsuhara, I.; Ito, N.; Seo, S.; Kamada, H.; Ohashi, Y. Transcriptionally and post-transcriptionally regulated response of 13 calmodulin genes to tobacco mosaic virus-induced cell death and wounding in tobacco plant. Eur. J. Biochem. 2001, 268, 3916–3929. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Deng, F.; Ramonell, K.M. Receptor-like kinases and receptor-like proteins: Keys to pathogen recognition and defense signaling in plant innate immunity. Frontiers Biol. 2012, 7, 155–166. [Google Scholar] [CrossRef]
- Kanzaki, H.; Saitoh, H.; Takahashi, Y.; Berberich, T.; Ito, A.; Kamoun, S.; Terauchi, R. NbLRK1, a lectin-like receptor kinase protein of Nicotiana benthamiana, interacts with Phytophthora infestans INF1 elicitin and mediates INF1-induced cell death. Planta 2008, 228, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.T.; Oh, J.; Kim, K.H.; Uhm, J.Y.; Lee, B.M. Isolation and characterization of NgRLK1, a receptor-like kinase of Nicotiana glutinosa that interacts with the elicitin of Phytophthora capsici. Mol. Biol. Rep. 2010, 37, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.H.; Shih, M.C.; Li, W.H. Transcription factor families have much higher expansion rates in plants than in animals. Plant Physiol. 2005, 139, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [Green Version]
- Dang, F.; Wang, Y.; She, J.; Lei, Y.; Liu, Z.; Eulgem, T.; Lai, Y.; Lin, J.; Yu, L.; Lei, D.; et al. Overexpression of CaWRKY27, a subgroup IIe WRKY transcription factor of Capsicum annuum, positively regulates tobacco resistance to Ralstonia solanacearum infection. Physiol. Plant 2014, 150, 397–411. [Google Scholar] [CrossRef]
- Huang, Y.; Li, M.Y.; Wu, P.; Xu, Z.S.; Que, F.; Wang, F.; Xiong, A.S. Members of WRKY Group III transcription factors are important in TYLCV defense signaling pathway in tomato (Solanum lycopersicum). BMC Genom. 2016, 17, 788. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.N.; Ernst, H.A.; Lo Leggio, L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 1360–1385. [Google Scholar] [CrossRef]
- Hu, R.; Qi, G.; Kong, Y.; Kong, D.; Gao, Q.; Zhou, G. Comprehensive analysis of NAC domain transcription factor gene family in Populus trichocarpa. BMC Plant Biol. 2010, 10, 145. [Google Scholar] [CrossRef]
- Wang, X.; Basnayake, B.M.; Zhang, H.; Li, G.; Li, W.; Virk, N.; Mengiste, T.; Song, F. The Arabidopsis ATAF1, a NAC transcription factor, is a negative regulator of defense responses against necrotrophic fungal and bacterial pathogens. Mol. Plant-Microbe Interact. 2009, 22, 1227–1238. [Google Scholar] [CrossRef]
- Suyal, G.; Rana, V.S.; Mukherjee, S.K.; Wajid, S.; Choudhury, N.R. Arabidopsis thaliana NAC083 protein interacts with Mungbean yellow mosaic India virus (MYMIV) Rep protein. Virus Genes 2014, 48, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Zhang, G.; Liu, X.Y.; Deng, L.; Cai, G.L.; Zhang, Y.; Wang, X.J.; Zhao, J.; Huang, L.L.; Kang, Z.S. Characterization of a novel wheat NAC transcription factor gene involved in defense response against stripe rust pathogen infection and abiotic stresses. Mol. Biol. Rep. 2010, 37, e3703–e3712. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Zhang, G.; Sun, Y.F.; Zhu, L.; Xu, L.S.; Chen, X.M.; Liu, B.; Yu, Y.T.; Wang, X.J.; Huang, L.L.; et al. TaNAC8, a novel NAC transcription factor gene in wheat, responds to stripe rust pathogen infection and abiotic stresses. Physiol. Mol. Plant 2010, 74, e394–e402. [Google Scholar] [CrossRef]
- Huang, Y.; Li, T.; Xu, Z.; Wang, F.; Xiong, A. Six NAC transcription factors involved in response to TYLCV infection in resistant and susceptible tomato cultivars. Plant Physiol. Biochem. 2017, 120, e61–e74. [Google Scholar] [CrossRef] [PubMed]
- Al-Attala, M.N.; Wang, X.; Abou-Attia, M.A.; Duan, X.; Kang, Z. A novel TaMYB4 transcription factor involved in the defence response against Puccinia striiformis f. sp. tritici and abiotic stresses. Plant Mol. Biol. 2014, 84, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, J.; Ghosal, D.; Wienand, U.; Peterson, P.A.; Saedler, H. The regulatory c1 locus of Zea mays encodes a protein with homology to myb proto-oncogene products and with structural similarities to transcriptional activators. EMBO. J. 1987, 6, 3553–3558. [Google Scholar] [CrossRef]
- Mengiste, T.; Chen, X.; Salmeron, J.; Dietrich, R. The BOTRYTIS SUSCEPTIBLE1 gene encodes an R2R3MYB transcription factor protein that is required for biotic and abiotic stress responses in Arabidopsis. Plant Cell 2003, 15, 2551–2565. [Google Scholar] [CrossRef]
- Chang, C.; Yu, D.; Jiao, J.; Jing, S.; Schulze-Lefert, P.; Shen, Q.H.; Barley, M.L. A immune receptors directly interfere with antagonistically acting transcription factors to initiate disease resistance signaling. Plant Cell 2013, 25, 1158–1173. [Google Scholar] [CrossRef]
- Kuhlmann, M.; Horvay, K.; Stathmann, A.; Heinekamp, T.; Fischer, U.; Bottner, S.; Droge-Laser, W. The alpha-helical D1 domain of the bZIP transcription factor BZI-1 interacts with the ankyrin-repeat protein ANK1, and is essential for BZI-1 function, both in auxin signaling and pathogen response. J. Biol. Chem. 2003, 278, 8786–8794. [Google Scholar] [CrossRef]
- Murmu, J.; Bush, M.J.; DeLong, C.; Li, S.; Xu, M.; Khan, M.; Malcolmson, C.; Fobert, P.R.; Zachgo, S.; Hepworth, S.R. Arabidopsis basic leucine-zipper transcription factors TGA9 and TGA10 interact with floral glutaredoxins ROXY1 and ROXY2 and are redundantly required for anther development. Plant Physiol. 2010, 154, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.; Fahad, S.; Aqeel, M.; Ali, U.; Amanullah; Anwar, S.; Baloch, S.K.; Zainab, M. miRNAs: Major modulators for crop growth and development under abiotic stresses. Biotechnol. Lett. 2017, 39, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhang, J.; Sun, H.; Salse, J.; Lucas, W.J.; Zhang, H.; Zheng, Y.; Mao, L.; Ren, Y.; Wang, Z.; et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 2013, 45, 51–58. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Frazee, A.C.; Pertea, G.; Jaffe, A.E.; Langmead, B.; Salzberg, S.L.; Leek, J.T. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015, 33, 243–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Raw Base | Clean Reads | Mapped Reads | Unique Mapped Reads | Q20% | Q30% |
|---|---|---|---|---|---|---|---|
| ck_1 | 52,659,470 | 7.90 G | 52,037,998 (98.82%) | 50,088,200 (96.25%) | 38,860,793 (74.68%) | 99.75 | 96.64 |
| ck_2 | 49,566,772 | 7.44 G | 49,101,272 (99.06%) | 46,572,421 (94.85%) | 32,009,543 (65.19%) | 99.67 | 96.41 |
| ck_3 | 43,812,198 | 6.57 G | 43,327,768 (98.89%) | 41,915,065 (96.74%) | 32,382,435 (74.74%) | 99.71 | 96.34 |
| 24h_1 | 41,190,724 | 6.18 G | 36,924,504 (89.64%) | 35,371,699 (95.79%) | 27,425,778 (74.28%) | 99.60 | 96.56 |
| 24h_2 | 46,758,132 | 7.01 G | 46,328,460 (99.08%) | 44,965,010 (97.06%) | 33,083,469 (71.41%) | 99.83 | 97.30 |
| 24h_3 | 48,964,964 | 7.34 G | 48,456,442 (98.96%) | 47,145,884 (97.30%) | 36,449,461 (75.22%) | 99.71 | 96.06 |
| Gene Name | Description | ck_1 | ck_2 | ck_3 | 24h_1 | 24h_2 | 24h_3 | log2(fc) | Regulation |
|---|---|---|---|---|---|---|---|---|---|
| Cla000152 | Sex-linked protein 9 (Fragment) | 275.6 | 343.6 | 292.0 | 165.5 | 117.5 | 124.1 | −1.16 | down |
| Cla001715 | PsbQ | 23.4 | 41.5 | 27.0 | 17.7 | 13.2 | 10.5 | −1.15 | down |
| Cla001790 | Oxygen-evolving enhancer protein 1 of photosystem II | 223.6 | 66.0 | 231.7 | 25.5 | 20.8 | 144.1 | −1.45 | down |
| Cla004698 | PsbP | 56.5 | 98.3 | 56.1 | 26.9 | 25.5 | 22.6 | −1.49 | down |
| Cla004704 | PsbP | 47.6 | 60.1 | 44.4 | 26.3 | 17.6 | 21.4 | −1.22 | down |
| Cla004703 | PsbP | 47.6 | 60.1 | 44.4 | 26.3 | 17.6 | 21.4 | −1.22 | down |
| Cla007741 | Photosystem II protein Psb27 | 33.5 | 40.9 | 37.7 | 20.2 | 12.0 | 13.7 | −1.29 | down |
| Cla007940 | Photosystem I reaction center subunit XI | 13.9 | 17.2 | 15.0 | 6.4 | 6.5 | 7.3 | −1.19 | down |
| Cla008429 | Photosystem I reaction center subunit N | 230.9 | 231.3 | 221.6 | 128.3 | 92.9 | 105.8 | −1.06 | down |
| Cla008898 | Ferredoxin--NADP reductase | 607.9 | 870.4 | 595.8 | 381.3 | 298.7 | 291.4 | −1.09 | down |
| Cla019799 | Photosystem II reaction center W protein | 25.1 | 11.2 | 12.5 | 4.7 | 7.2 | 8.2 | −1.27 | down |
| Cla019798 | Photosystem II reaction center W protein | 25.1 | 11.2 | 12.5 | 4.7 | 7.2 | 8.2 | −1.27 | down |
| Cla004746 | Chlorophyll a-b binding protein 6A | 308.0 | 547.5 | 409.9 | 201.0 | 146.4 | 115.9 | −1.45 | down |
| Cla011145 | Chlorophyll a-b binding protein | 92.2 | 154.3 | 87.8 | 47.6 | 34.8 | 51.2 | −1.32 | down |
| Cla011748 | Chlorophyll a-b binding protein 13 | 192.7 | 320.0 | 218.0 | 152.7 | 105.8 | 74.4 | −1.13 | down |
| Cla012368 | Chlorophyll a-b binding protein 8 | 733.7 | 1294.7 | 799.6 | 545.9 | 441.2 | 373.9 | −1.06 | down |
| Cla013826 | Chlorophyll a-b binding protein | 208.0 | 246.4 | 260.1 | 90.2 | 85.7 | 95.0 | −1.40 | down |
| Cla018117 | Chlorophyll a-b binding protein 6 | 34.1 | 31.5 | 36.9 | 13.9 | 11.8 | 15.4 | −1.32 | down |
| Cla019105 | Chlorophyll a-b binding protein P4 | 903.5 | 1298.7 | 1142.1 | 647.1 | 447.7 | 408.2 | −1.15 | down |
| Cla019595 | Chlorophyll a-b binding protein 21 | 4.7 | 20.2 | 6.3 | 4.2 | 1.6 | 2.5 | −1.89 | down |
| Cla022573 | Chlorophyll a-b binding protein 4 | 21.1 | 41.4 | 26.2 | 10.8 | 8.9 | 10.0 | −1.58 | down |
| Cla022963 | Chlorophyll a-b binding protein 7 | 1106.7 | 1615.0 | 1306.7 | 786.1 | 563.1 | 628.6 | −1.03 | down |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Fan, M.; He, Y. Transcriptome Analysis of Watermelon Leaves Reveals Candidate Genes Responsive to Cucumber green mottle mosaic virus Infection. Int. J. Mol. Sci. 2019, 20, 610. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030610
Sun Y, Fan M, He Y. Transcriptome Analysis of Watermelon Leaves Reveals Candidate Genes Responsive to Cucumber green mottle mosaic virus Infection. International Journal of Molecular Sciences. 2019; 20(3):610. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030610
Chicago/Turabian StyleSun, Yuyan, Min Fan, and Yanjun He. 2019. "Transcriptome Analysis of Watermelon Leaves Reveals Candidate Genes Responsive to Cucumber green mottle mosaic virus Infection" International Journal of Molecular Sciences 20, no. 3: 610. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030610