BCAS2 Enhances Carcinogenic Effects of Estrogen Receptor Alpha in Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
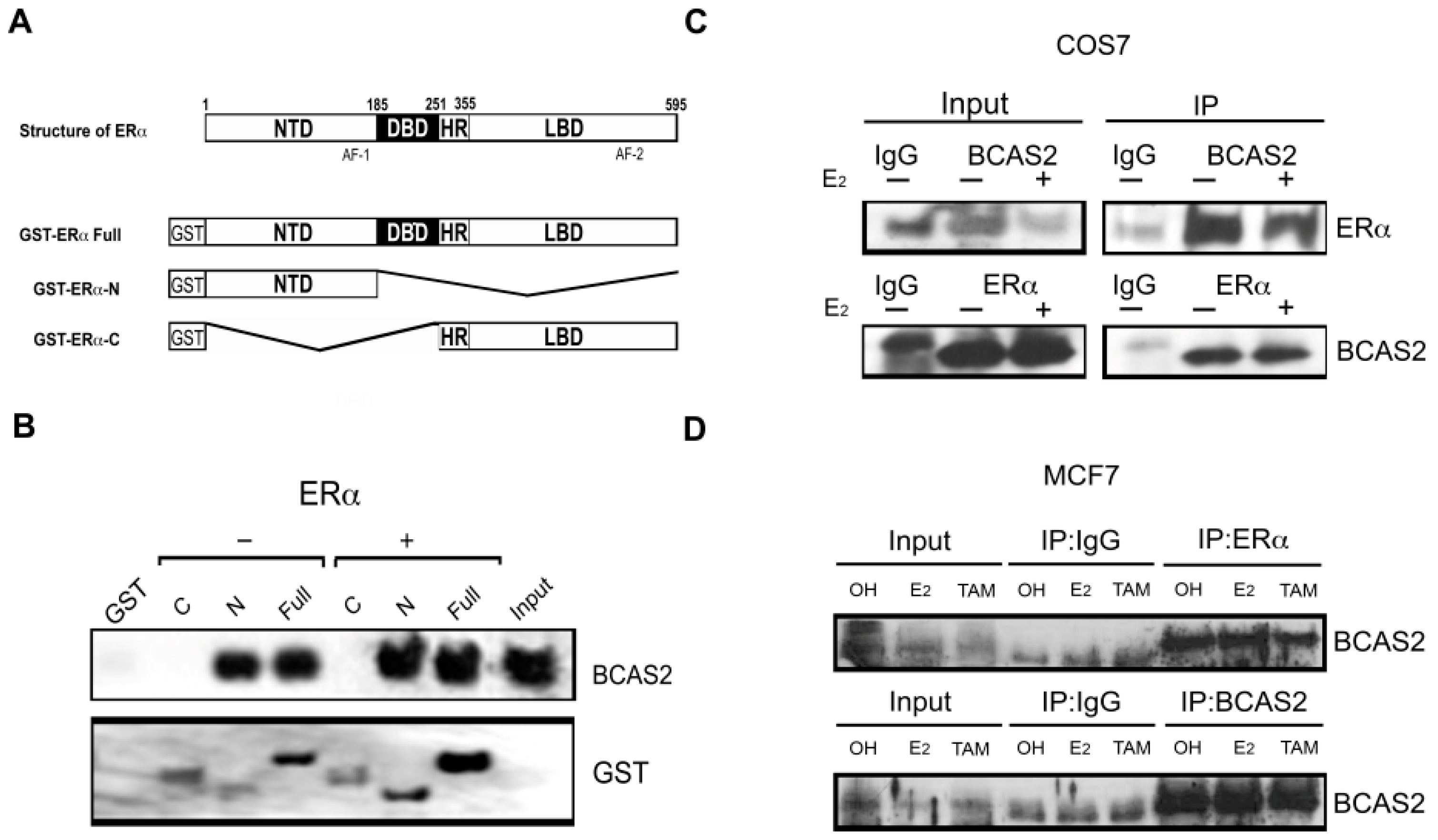
2.1. BCAS2 Interacts Directly with the N-Terminal Region of ERα
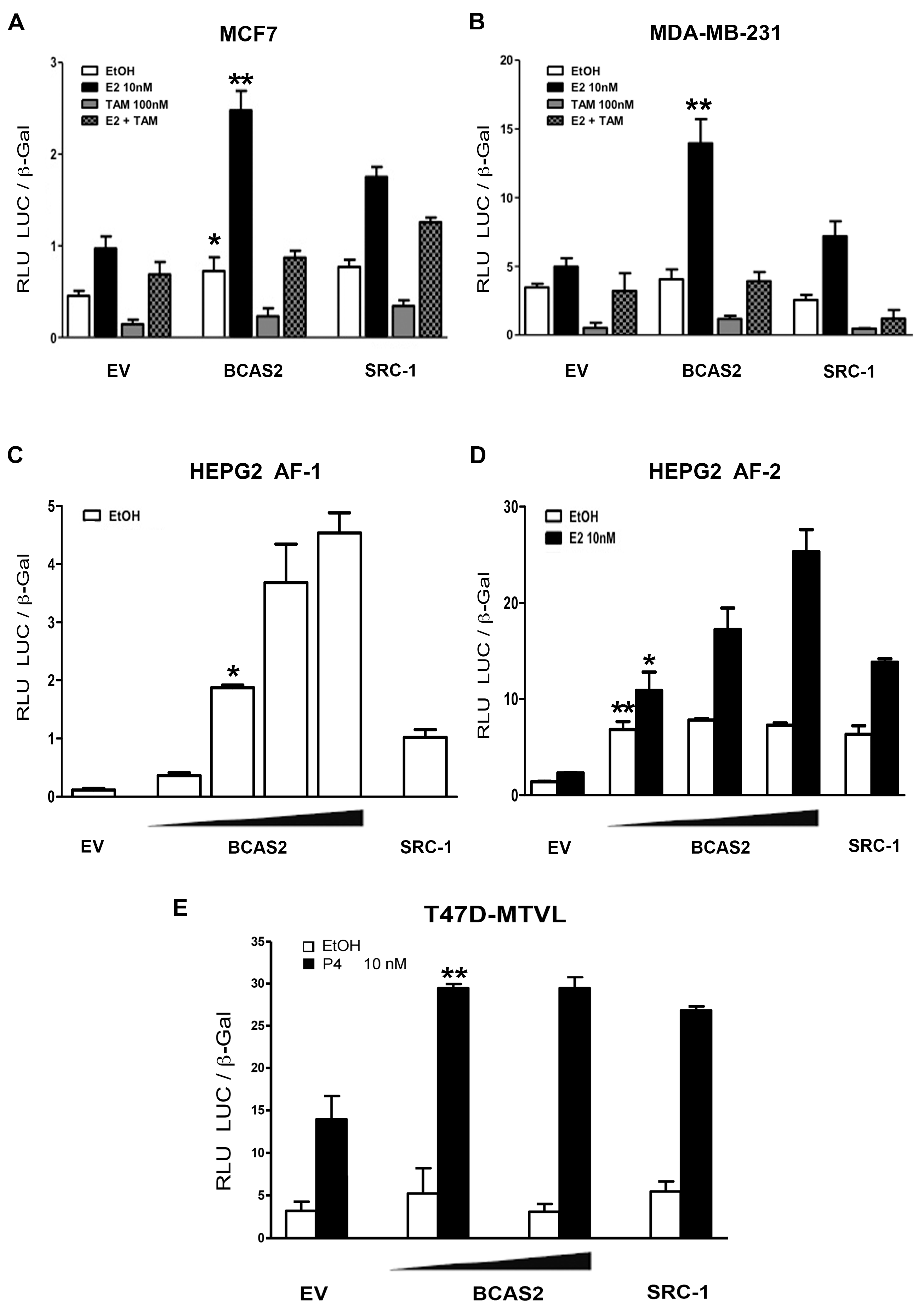
2.2. BCAS2 Enhances ERα Transcriptional Activity
2.3. BCAS2 Has a Positive Effect on the Transcriptional Activity of PR
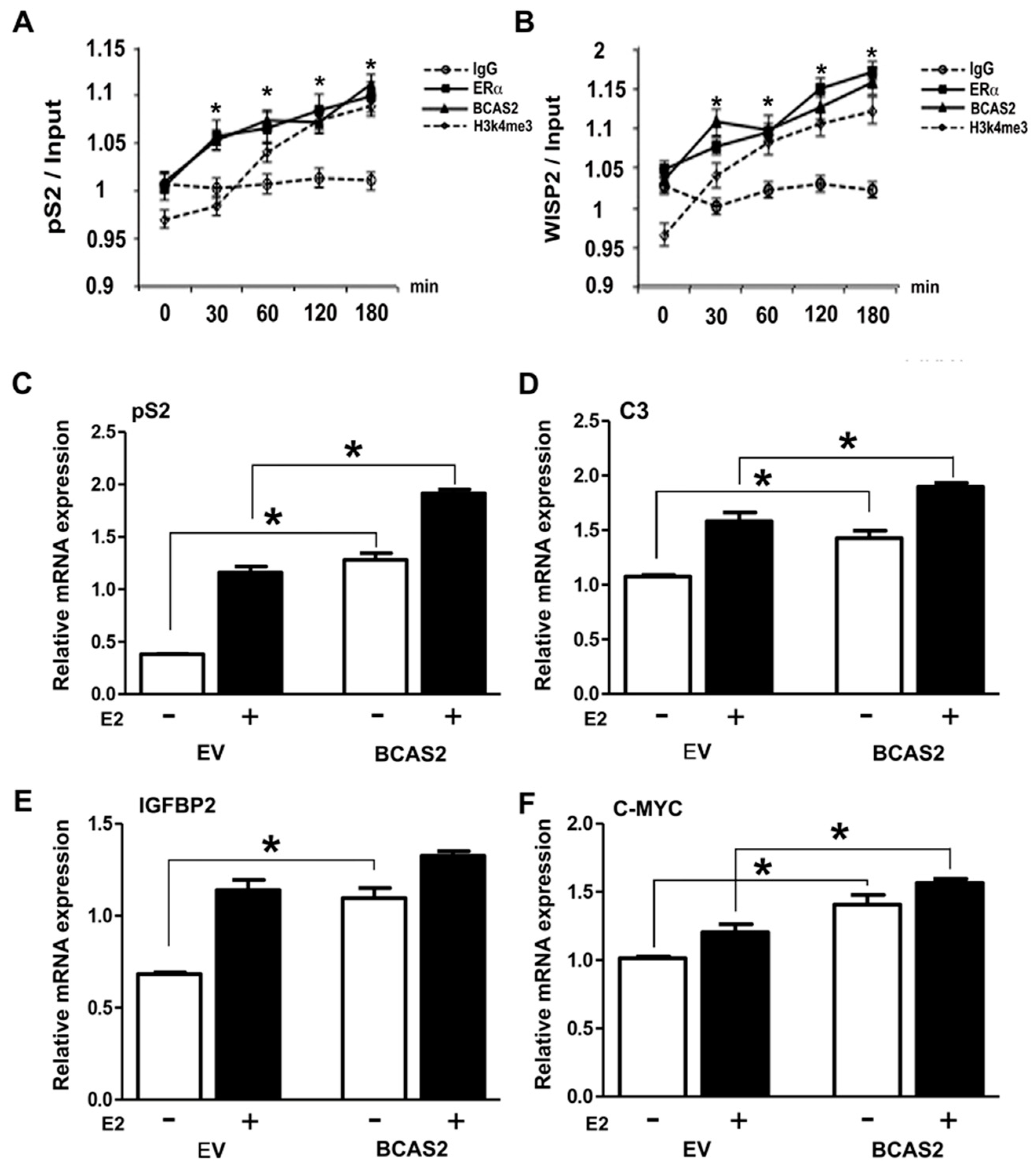
2.4. BCAS2 Is Recruited to ER Target Gene Promoters and Increases Target Gene Expression
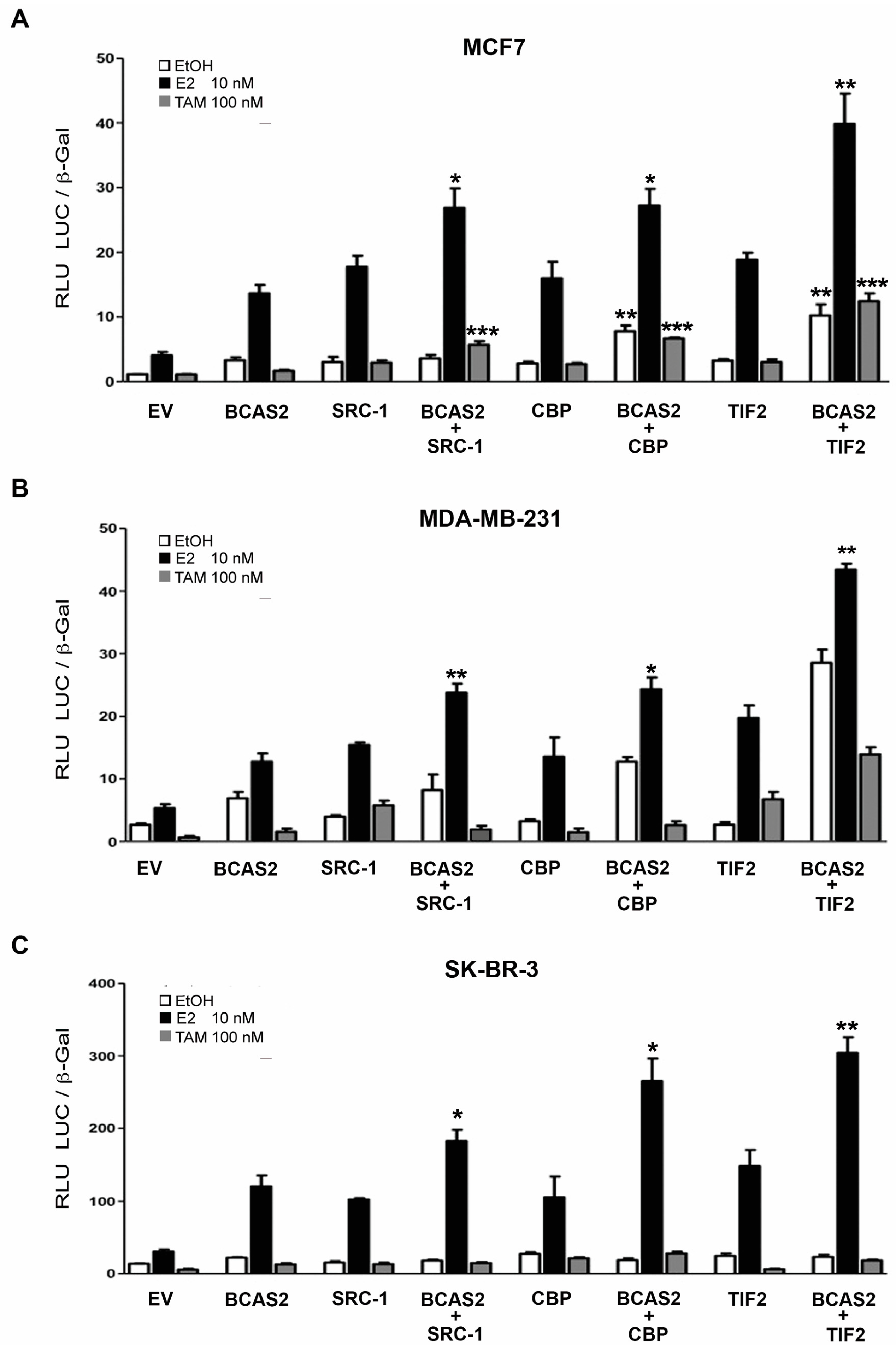
2.5. BCAS2 Acts Together with Other Coactivators to Enhance ERα Activity
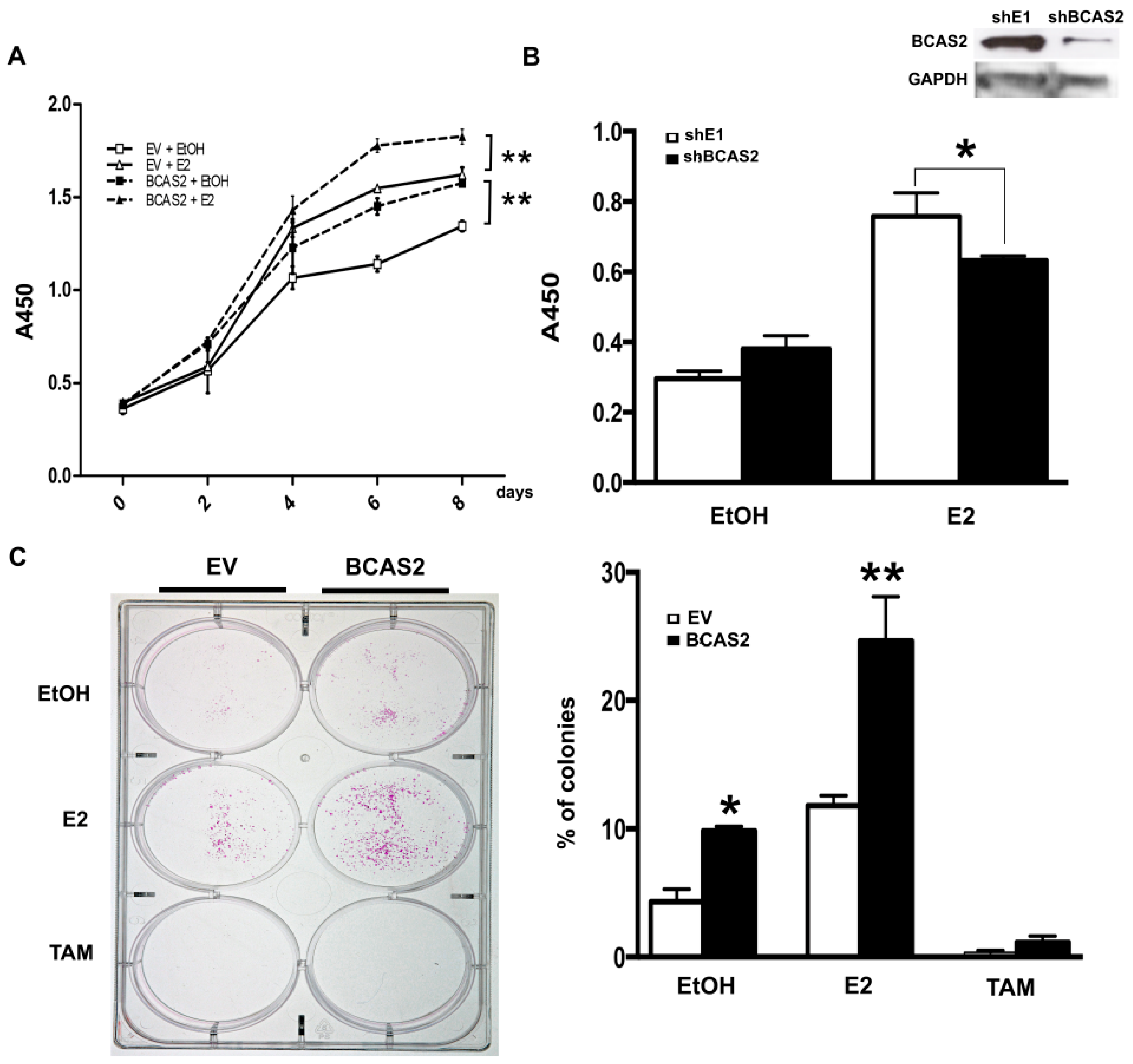
2.6. BCAS2 Promotes Cell Proliferation and Colony Formation in Breast Cancer Cells
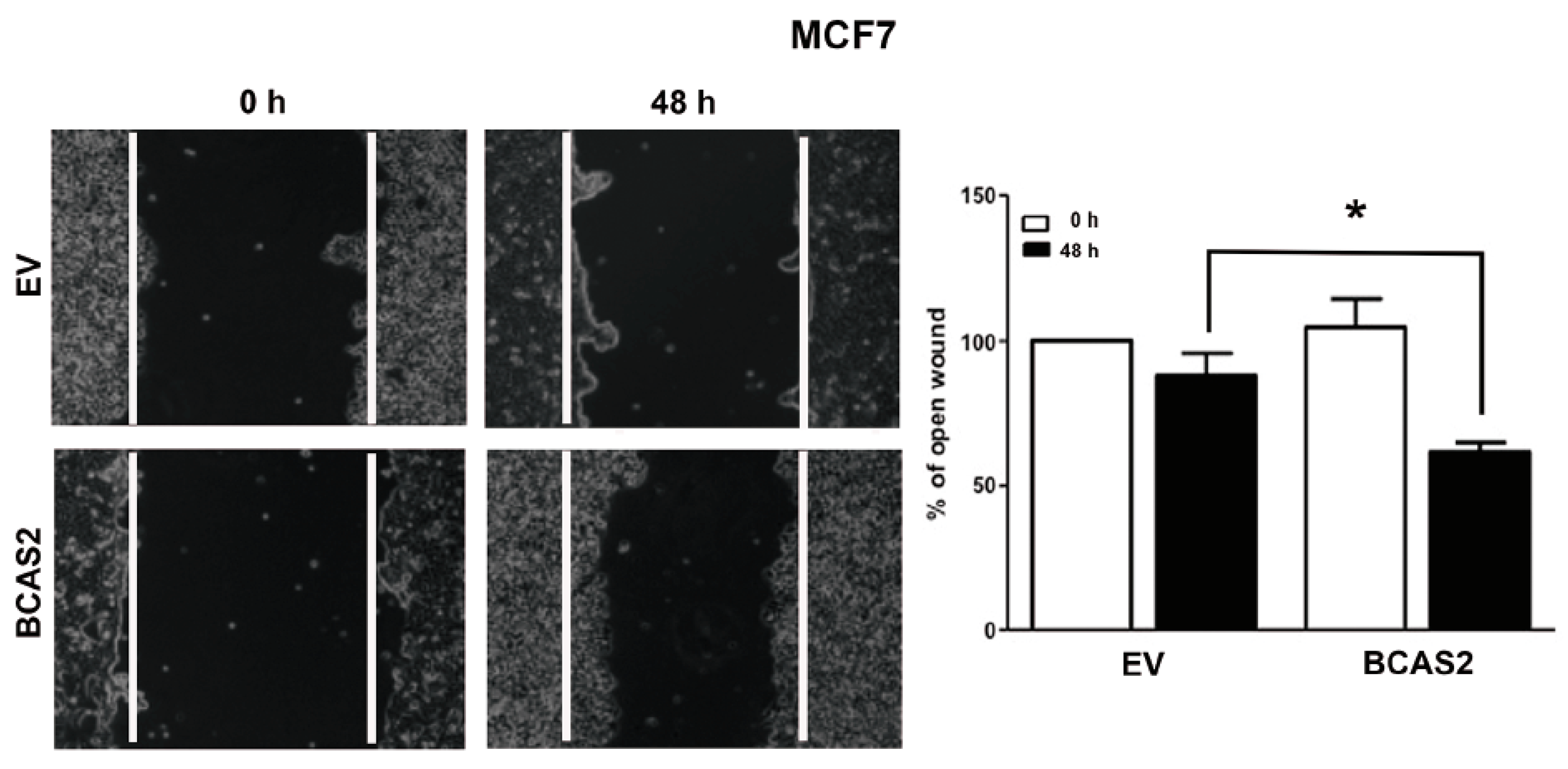
2.7. BCAS2 Promotes Estradiol Dependent Cell Migration
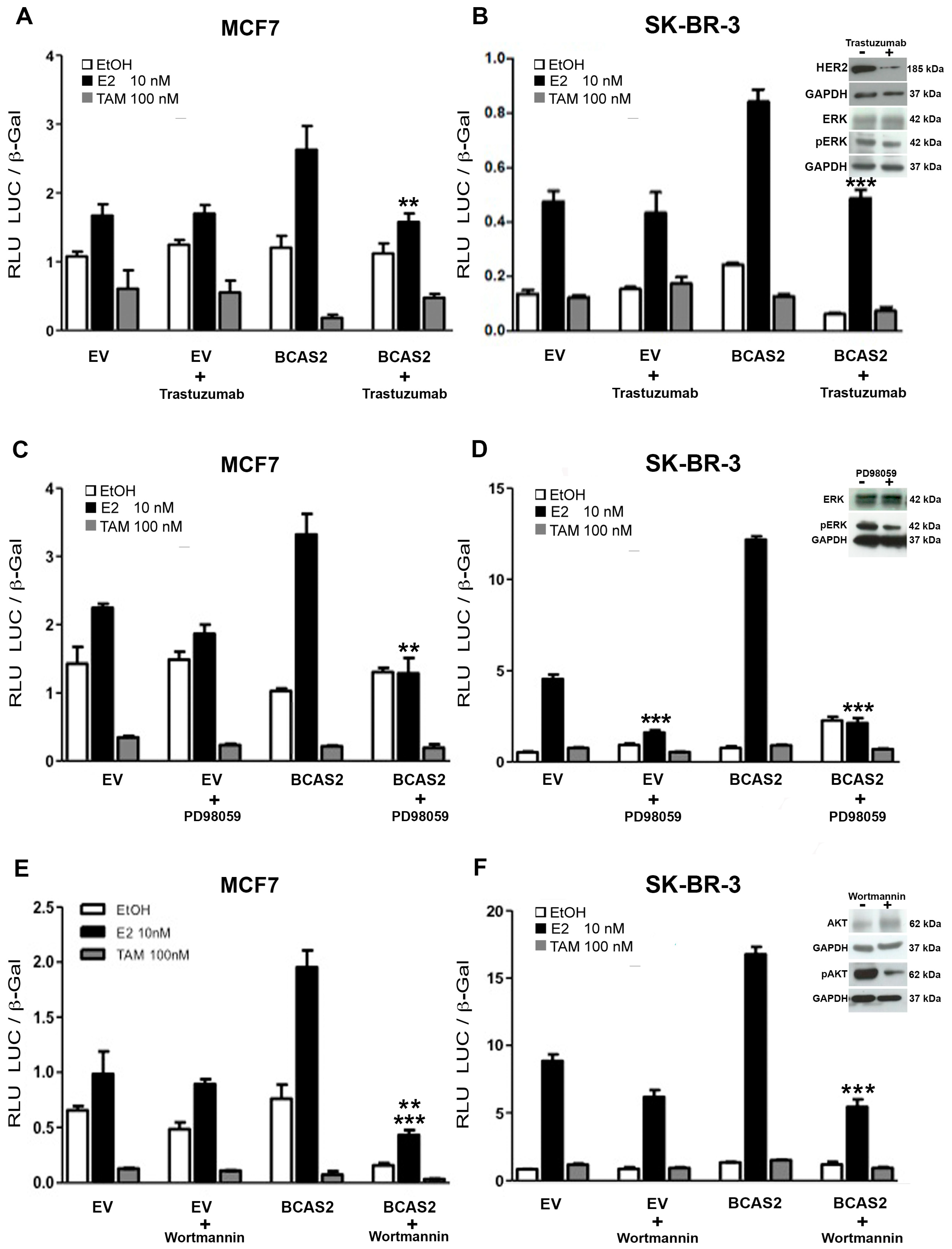
2.8. Various Signal Transduction Pathways Are Involved in BCAS2 Activation
3. Discussion
4. Materials and Methods
4.1. Yeast Two-Hybrid Screen
4.2. Cell Culture
4.3. Plasmids
4.4. Glutathione-S-Transferase (GST) Pull-Down Assays
4.5. Coimmunoprecipitation
4.6. Transient Transfection and Luciferase Assays
4.7. Chromatin Immunoprecipitation (ChIP)
4.8. Real-Time Quantitative PCR
4.9. Expression of Endogenous Genes
4.10. shRNA Transfection
4.11. Cell Viability Assays
4.12. Colony Formation and Wound Healing Assays
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ERα | Estrogen Receptor alpha |
| BCAS2 | Breast Cancer Amplified Sequence 2 |
| TAM | Tamoxifen |
| E2 | Estradiol |
| P4 | Progesterone |
| EtOH | Ethanol |
| aa | amino acids |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| SRC-1 | Steroid Receptor coactivator-1 |
| TIF2 | Transcriptional Intermediary Factor 2 |
| CBP | CREB Binding Protein |
References
- Sommer, S.; Fuqua, S.A. Estrogen receptor and breast cancer. Semin. Cancer Biol. 2001, 11, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Bender, L.M.; Nahta, R. Her2 cross talk and therapeutic resistance in breast cancer. Front. Biosci. 2008, 13, 3906–3912. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Pietras, R.J. Rational management of endocrine resistance in breast cancer: A comprehensive review of estrogen receptor biology, treatment options and future directions. Cancer 2008, 113, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.; Cavalieri, E.; Rogan, E.; Russo, J.; Guttenplan, J.; Ingle, J.; Yue, W. Estrogen mediation of breast tumor formation involves estrogen receptor-dependent, as well as independent, genotoxic effects. Ann. N. Y. Acad. Sci. 2009, 1155, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Gustafsson, J.Å. Estrogen receptor mutations and functional consequences for breast cancer. Trends Endocrinol. Metab. 2015, 26, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar] [PubMed]
- McKenna, N.J.; O’Malley, B.W. Combinatorial Control of Gene Expression by Nuclear Receptors and Coregulators. Cell 2002, 108, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Heery, D.M.; Kalkhoven, E.; Hoare, S.; Parker, M.G. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 1997, 387, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Torchia, J.; Glass, C.; Rosenfeld, M.G. Co-activators and co-repressors in the integration of transcriptional responses. Curr. Opin. Cell Biol. 1998, 10, 373–383. [Google Scholar] [CrossRef]
- Xu, J.; Wu, R.C.; O’Malley, B.W. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 2009, 9, 615–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perissi, V.; Jepsen, K.; Glass, C.K.; Rosenfeld, M.G. Deconstructing repression: Evolving models of co-repressor action. Nat. Rev. Genet. 2010, 11, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Glass, C.K.; Rosenfeld, M.G. Coactivator and corepressor complexes in nuclear receptor function. Curr. Opin. Genet. Dev. 1999, 9, 140–147. [Google Scholar] [CrossRef]
- Qi, C.; Zhu, Y.T.; Chang, J.; Yeldandi, A.V.; Rao, M.S.; Zhu, Y.J. Potentiation of estrogen receptor transcriptional activity by breast cancer amplified sequence 2. Biochem. Biophys. Res. Commun. 2005, 328, 393–398. [Google Scholar] [CrossRef]
- Kuo, P.C.; Tsao, Y.P.; Chang, H.W.; Chen, P.H.; Huang, C.W.; Lin, S.T.; Weng, Y.T.; Tsai, T.C.; Shieh, S.Y.; Chen, S.L. Breast cancer amplified sequence 2, a novel negative regulator of the p53 tumor suppressor. Cancer Res. 2009, 69, 8877–8885. [Google Scholar] [CrossRef]
- Tzukerman, M.T.; Esty, A.; Santiso-Mere, D.; Danielian, P.; Parker, M.G.; Stein, R.B.; Pike, J.W.; McDonnell, D.P. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol. Endocrinol. 1994, 8, 21–30. [Google Scholar] [CrossRef]
- Métivier, R.; Penot, G.; Flouriot, G.; Pakdel, F. Synergism between ERalpha transactivation function 1 (AF-1) and AF-2 mediated by steroid receptor coactivator protein-1: Requirement for the AF-1 alpha-helical core and for a direct interaction between the N- and C-terminal domains. Mol. Endocrinol. 2001, 15, 1953–1970. [Google Scholar] [CrossRef]
- Truss, M.; Bartsch, J.; Schelbert, A.; Hache, R.J.; Beato, M. Hormone induces binding of receptors and transcription factors to a rearranged nucleosome on the MMTV promoter in vivo. EMBO J. 1995, 14, 1737–1751. [Google Scholar] [CrossRef]
- Kuo, P.C.; Huang, C.W.; Lee, C.I.; Chang, H.W.; Hsieh, S.W.; Chung, Y.P.; Lee, M.S.; Huang, C.S.; Tsao, L.P.; Tsao, Y.P.; et al. BCAS2 promotes prostate cancer cells proliferation by enhancing AR mRNA transcription and protein stability. Br. J. Cancer 2015, 12, 391–402. [Google Scholar] [CrossRef]
- Merrell, K.W.; Crofts, J.D.; Smith, R.L.; Sin, J.H.; Kmetzsch, K.E.; Merrell, A.; Miguel, R.O.; Candelaria, N.R.; Lin, C.Y. Differential recruitment of nuclear receptor coregulators in ligand-dependent transcriptional repression by estrogen receptor-α. Oncogene 2011, 30, 1608–1614. [Google Scholar] [CrossRef]
- Fritah, A.; Redeuilh, G.; Sabbah, M. Molecular cloning and characterization of the human WISP-2/CCN5 gene promoter reveal its upregulation by oestrogens. J. Endocrinol. 2006, 191, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanaskova, N.; Keshamouni, V.G.; Krueger, J.S.; Schwartz, J.A.; Miller, F.; Reddy, K.B. MAP kinase/estrogen receptor cross-talk enhances estrogen-mediated signaling and tumor growth but does not confer tamoxifen resistance. Oncogene 2002, 21, 4000–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, V.S.; Kanaya, N.; Lo, C.; Mortimer, J.; Chen, S. From bench to bedside: What do we know about hormone receptor-positive and human epidermal growth factor receptor 2-positive breast cancer? J. Steroid Biochem. Mol. Biol. 2015, 153, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takimoto, G.S.; Graham, J.D.; Jackson, T.A.; Tung, L.; Powell, R.L.; Horwitz, L.D.; Horwitz, K.B. Tamoxifen resistant breast cancer: Coregulators determine the direction of transcription by antagonist-occupied steroid receptors. J. Steroid Biochem. Mol. Biol. 1999, 69, 45–50. [Google Scholar] [CrossRef]
- Osborne, C.K.; Shou, J.; Massarweh, S.; Schiff, R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin. Cancer Res. 2005, 11, 865s–870s. [Google Scholar] [PubMed]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.-M.; Corbo, L. Cracking the Estrogen Receptor’s Posttranslational Code in Breast Tumors. Endoc. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Hedman, A.C.; Ames, J.B.; Sacks, D.B. Calmodulin Lobes Facilitate Dimerization and Activation of Estrogen Receptor-α. J. Biol. Chem. 2017, 292, 4614–4622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.T.; Leung, Y.K.; Chung, I.; Tarapore, P.; Ho, S.M. Estrogen Receptor b (ERb1) Transactivation Is Differentially Modulated by the Transcriptional Coregulator Tip60 in a cis-Acting Element-dependent Manner. J. Biol. Chem. 2013, 288, 25038–25052. [Google Scholar] [CrossRef]
- Nagasaki, K.; Maass, N.; Manabe, T.; Hanzawa, H.; Tsukada, T.; Kikuchi, K.; Yamaguchi, K. Identification of a novel gene, DAM1, amplified at chromosome 1p13.3-21 region in human breast cancer cell lines. Cancer Lett. 1999, 140, 219–226. [Google Scholar] [CrossRef]
- Worsham, M.J.; Pals, G.; Schouten, J.P.; Miller, F.; Tiwari, N.; van Spaendonk, R.; Wolman, S.R. High-resolution mapping of molecular events associated with immortalization, transformation and progression to breast cancer in the MCF10 model. Breast Cancer Res. Treat. 2006, 96, 177–186. [Google Scholar] [CrossRef]
- Sengupta, D.; Bhargava, D.K.; Dixit, A.; Sahoo, B.S.; Biswas, S.; Biswas, G.; Mishra, S.K. ERRβ signaling through FST and BCAS2 inhibits cellular proliferation in breast cancer cells. Br. J. Cancer 2014, 110, 2144–2158. [Google Scholar] [CrossRef] [PubMed]
- Schiff, R.; Massarweh, S.; Shou, J.; Osborne, C.K. Breast cancer endocrine resistance: How growth factor signaling and estrogen receptor coregulators modulate response. Clin. Cancer Res. 2003, 9, 447S–454S. [Google Scholar] [PubMed]
- Zhao, W.; Zhang, Q.; Kang, X.; Jin, S.; Lou, C. AIB1 is required for the acquisition of epithelial growth factor receptor-mediated tamoxifen resistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2009, 380, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Nagalingam, A.; Tighiouart, M.; Ryden, L.; Joseph, L.; Landberg, G.; Saxena, N.K.; Sharma, D. Med1 plays a critical role in the development of tamoxifen resistance. Carcinogenesis 2012, 33, 918–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redmond, A.M.; Byrne, C.; Bane, F.T.; Brown, G.D.; Tibbitts, P.; O’Brien, K.; Hill, A.D.; Carroll, J.S.; Young, L.S. Genomic interaction between ER and HMGB2 identifies DDX18 as a novel driver of endocrine resistance in breast cancer cells. Oncogene 2015, 34, 3871–3880. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Chen, X.; Wu, Y.; Feng, Z.; He, T.; Wang, L.; Liao, L.; Xu, J. Steroid receptor coactivator-1 upregulates integrin α5 expression to promote breast cancer cell adhesion and migration. Cancer Res. 2011, 71, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.A.; Qin, L.; Tien, J.C.; Young, L.S.; Xu, J. The function of steroid receptor coactivator-1 in normal tissues and cancer. Int. J. Biol. Sci. 2012, 8, 470–485. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Z.; Chen, H.; Xu, J. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res. 2009, 69, 3819–3827. [Google Scholar] [CrossRef] [PubMed]
- Santer, F.R.; Höschele, P.S.; OH, S.J.; Erb, H.H.; Bouchal, J.; Cavarretta, I.T.; Parson, W.; Meyers, D.J.; Cole, P.A.; Culig, Z. Inhibition of the Acetyltransferases p300 and CBP Reveals a Targetable Function for p300 in the Survival and Invasion Pathways of Prostate Cancer Cell Lines. Mol. Cancer Ther. 2011, 10, 1644–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, G.; Gallo, D.; Cossy, J.; Laïos, I.; Larsimont, D.; Laurent, G.; Jacquot, Y. Peptides Targeting Estrogen Receptor Alpha-Potential Applications for Breast Cancer Treatment. Curr. Pharm. Des. 2011, 17, 2632–2653. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, H.; Lenferink, A.E.; Simpson, J.F.; Pisacane, P.I.; Sliwkowski, M.X.; Forbes, J.T.; Arteaga, C.L. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-over-expressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000, 60, 5887–5894. [Google Scholar] [PubMed]
- Arpino, G.; Green, S.J.; Allred, D.C.; Lew, D.; Martino, S.; Osborne, C.K.; Elledge, R.M. HER-2 amplification, HER-1 expression and tamoxifen response in estrogen receptor-positive metastatic breast cancer: A southwest oncology group study. Clin. Cancer Res. 2004, 10, 5670–5676. [Google Scholar] [CrossRef]
- Benz, C.C.; Scott, G.K.; Sarup, J.C.; Johnson, R.M.; Tripathy, D.; Coronado, E.; Shepard, H.M.; Osborne, C.K. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF7 cells transfected with HER2/neu. Breast Cancer Res. Treat. 1992, 24, 85–95. [Google Scholar] [CrossRef]
- Osborne, C.K.; Bardou, V.; Hopp, T.A.; Chamness, G.C.; Hilsenbeck, S.G.; Fuqua, S.A.; Wong, J.; Allred, D.C.; Clark, G.M.; Schiff, R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 2003, 95, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Noriega-Reyes, M.Y.; Rivas-Torres, M.A.; Oñate-Ocaña, L.F.; Vallés, A.J.; Baranda-Avila, N.; Langley, E. Novel role for PINX1 as a coregulator of nuclear hormone receptors. Mol. Cell. Endocrinol. 2015, 15, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Wiznerowicz, M.; Trono, D. Conditional suppression of cellular genes: Lentivirus vector-mediated drug-inducible RNA interference. J. Virol. 2003, 77, 8957–8961. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmerón-Hernández, Á.; Noriega-Reyes, M.Y.; Jordan, A.; Baranda-Avila, N.; Langley, E. BCAS2 Enhances Carcinogenic Effects of Estrogen Receptor Alpha in Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 966. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040966
Salmerón-Hernández Á, Noriega-Reyes MY, Jordan A, Baranda-Avila N, Langley E. BCAS2 Enhances Carcinogenic Effects of Estrogen Receptor Alpha in Breast Cancer Cells. International Journal of Molecular Sciences. 2019; 20(4):966. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040966
Chicago/Turabian StyleSalmerón-Hernández, Ángel, María Yamilet Noriega-Reyes, Albert Jordan, Noemi Baranda-Avila, and Elizabeth Langley. 2019. "BCAS2 Enhances Carcinogenic Effects of Estrogen Receptor Alpha in Breast Cancer Cells" International Journal of Molecular Sciences 20, no. 4: 966. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040966