Population Substructure Has Implications in Validating Next-Generation Cancer Genomics Studies with TCGA

 ,
, 
Abstract
:1. Introduction
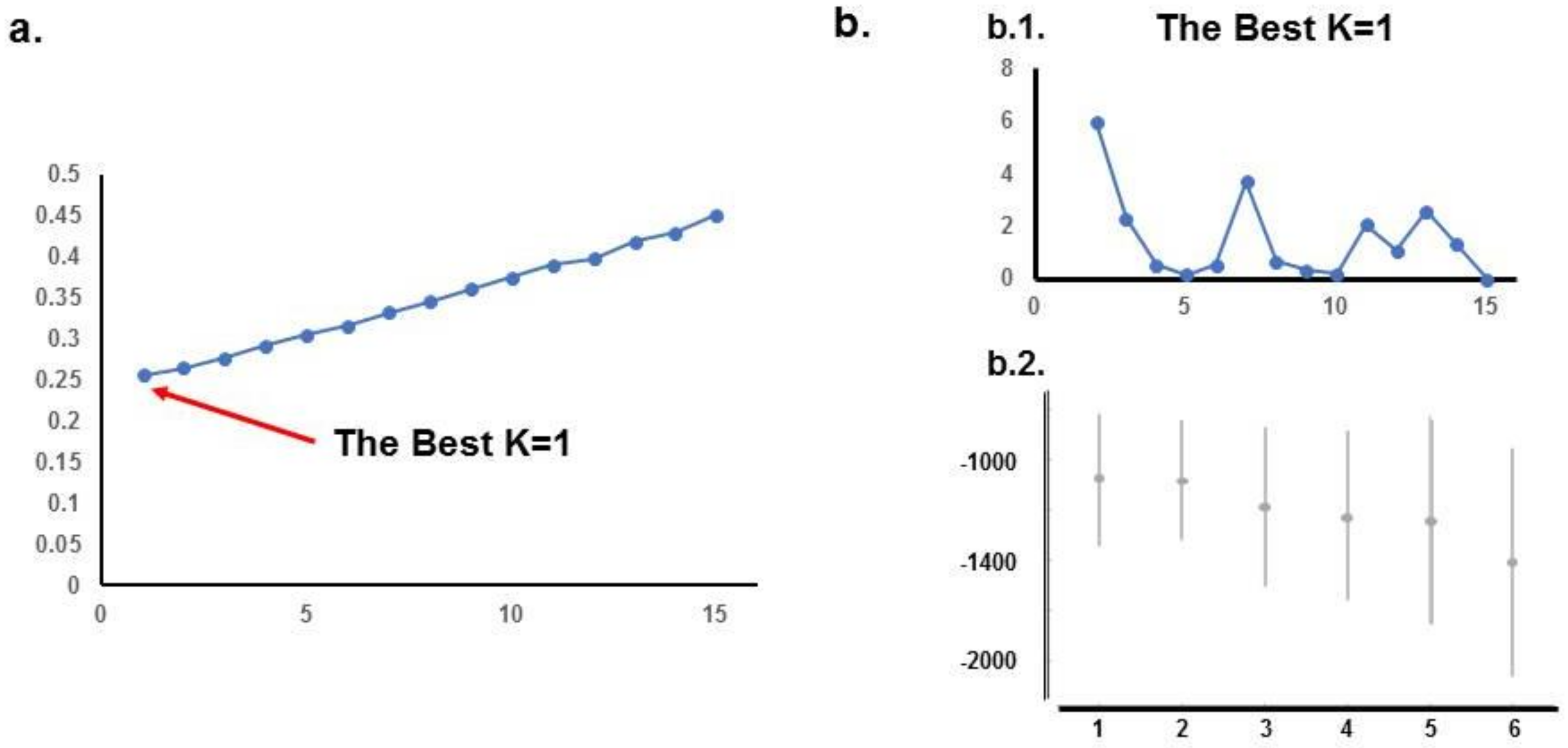
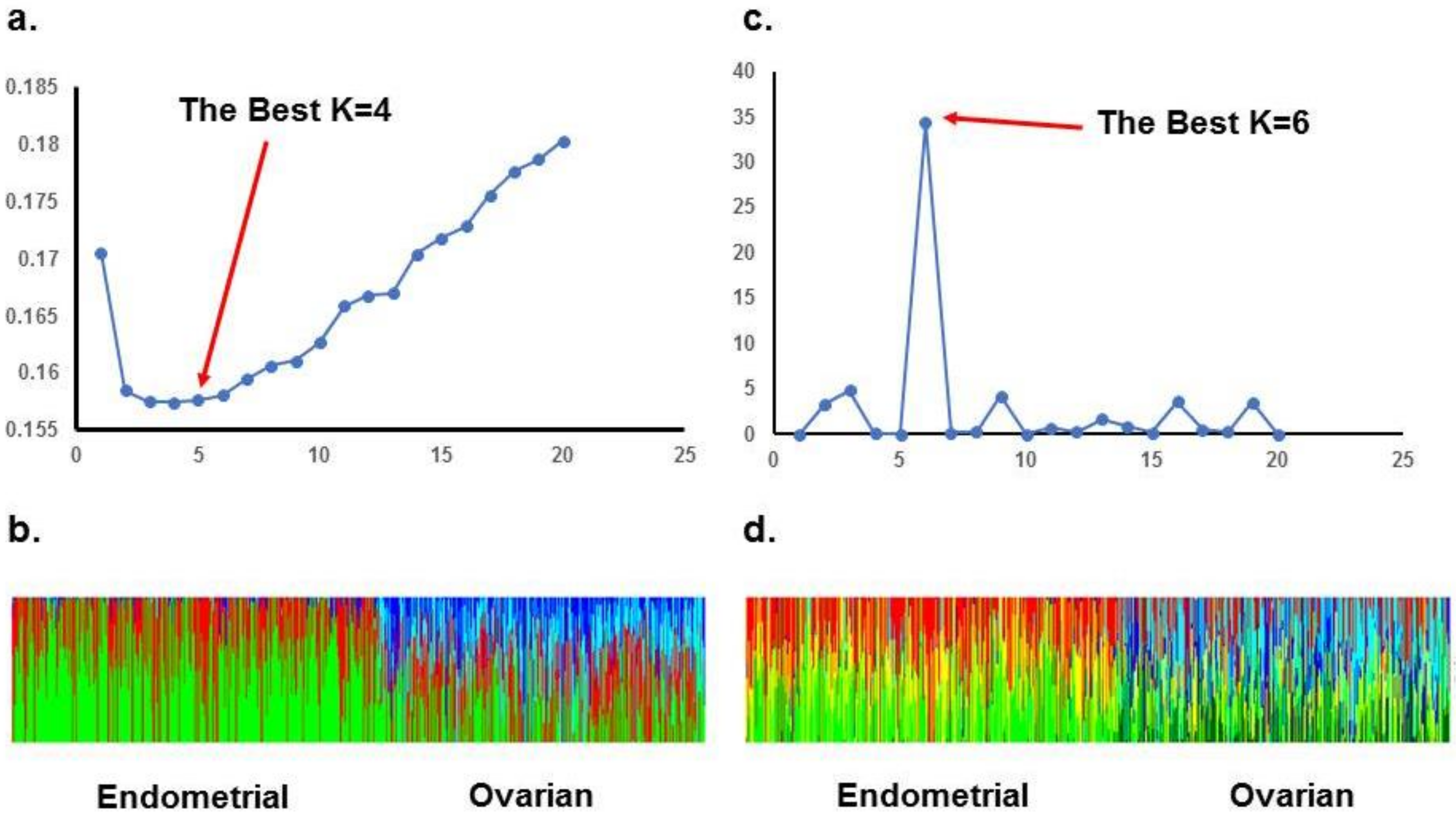
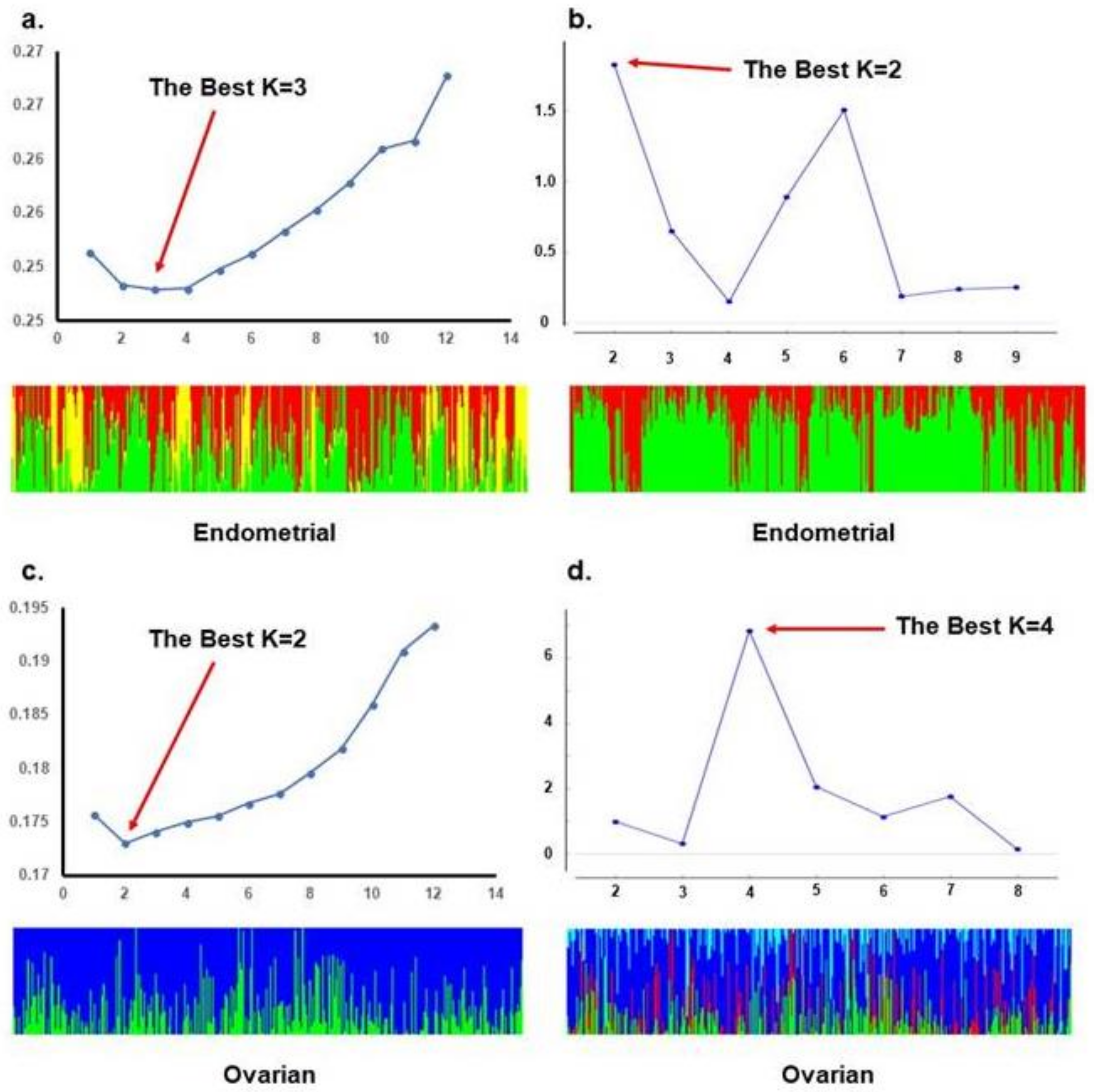
2. Results
3. Discussion
4. Materials and Methods
4.1. Tissue Procurement
4.2. RNA Purification and Sequencing
4.3. TCGA Cohort
4.4. File Pre-Processing
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TCGA | The Cancer Genome Atlas |
| UIHC | University of Iowa Hospitals and Clinics |
| RNA | ribonucleic acid |
| NHW | non-Hispanic white |
| NHB | non-Hispanic black |
References
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. (Pozn) 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Salinas, E.A.; Miller, M.D.; Newtson, A.M.; Sharma, D.; McDonald, M.E.; Me, K.; Smith, B.J.; Bender, B.J.; Goodheart, M.J.; Thiel, K.W.; et al. A prediction model for preoperative risk assessment in endometrial cancer utilizing clinical and molecular variables. Int. J. Mol. Sci. 2019. under review. [Google Scholar]
- Miller, M.D.; Salinas, E.A.; Newtson, A.M.; Sharma, D.; Me, K.; Warrier, A.; Smith, B.J.; Bender, B.J.; Goodheart, M.J.; Thiel, K.W.; et al. An Integrated Prediction Model of Recurrence in Endometrial Endometrioid Cancers. Cancer Manag. Res. 2019. under review. [Google Scholar]
- Grunda, J.M.; Steg, A.D.; He, Q.; Steciuk, M.R.; Byan-Parker, S.; Johnson, M.R.; Grizzle, W.E. Differential expression of breast cancer-associated genes between stage- and age-matched tumor specimens from African- and Caucasian-American Women diagnosed with breast cancer. BMC Res. Notes 2012, 5, 248. [Google Scholar] [CrossRef] [PubMed]
- Dubil, E.A.; Tian, C.; Wang, G.; Tarney, C.M.; Bateman, N.W.; Levine, D.A.; Conrads, T.P.; Hamilton, C.A.; Maxwell, G.L.; Darcy, K.M. Racial disparities in molecular subtypes of endometrial cancer. Gynecol. Oncol. 2018, 149, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Ruterbusch, J.J.; Cote, M.L. Recent Trends in Ovarian Cancer Incidence and Relative Survival in the United States by Race/Ethnicity and Histologic Subtypes. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1511–1518. [Google Scholar] [CrossRef]
- Facts, Q. United States Census Bureau Quick Facts: Iowa; U.S. Department of Commerce: Washington, DC, USA, 2019. Available online: https://www.census.gov/quickfacts/IA (accessed on 30 January 2019).
- Spratt, D.E.; Chan, T.; Waldron, L.; Speers, C.; Feng, F.Y.; Ogunwobi, O.O.; Osborne, J.R. Racial/Ethnic Disparities in Genomic Sequencing. JAMA Oncol. 2016, 2, 1070–1074. [Google Scholar] [CrossRef] [Green Version]
- Cote, M.L.; Ruterbusch, J.J.; Olson, S.H.; Lu, K.; Ali-Fehmi, R. The Growing Burden of Endometrial Cancer: A Major Racial Disparity Affecting Black Women. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1407–1415. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, C.E.; Siegel, R.L.; Sauer, A.G.; Miller, K.D.; Fedewa, S.A.; Alcaraz, K.I.; Jemal, A. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities. CA Cancer J. Clin. 2016, 66, 290–308. [Google Scholar] [CrossRef]
- Bryc, K.; Velez, C.; Karafet, T.; Moreno-Estrada, A.; Reynolds, A.; Auton, A.; Hammer, M.; Bustamante, C.D.; Ostrer, H. Colloquium paper: Genome-wide patterns of population structure and admixture among Hispanic/Latino populations. Proc. Natl. Acad. Sci. USA 2010, 107, 8954–8961. [Google Scholar] [CrossRef]
- Sillanpaa, M.J. Overview of techniques to account for confounding due to population stratification and cryptic relatedness in genomic data association analyses. Heredity (Edinb) 2011, 106, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Janes, J.K.; Miller, J.M.; Dupuis, J.R.; Malenfant, R.M.; Gorrell, J.C.; Cullingham, C.I.; Andrew, R.L. The K = 2 conundrum. Mol. Ecol. 2017, 26, 3594–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Browning, S.R. Genotype Imputation with Millions of Reference Samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Francis, R.M. Pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Evolution in Mendelian Populations. Genetics 1931, 16, 97–159. [Google Scholar] [PubMed]
- Meirmans, P.G.; Hedrick, P.W. Assessing population structure: F(ST) and related measures. Mol. Ecol. Resour. 2011, 11, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| UIHC | TCGA | |||
|---|---|---|---|---|
| Cancer | Ovarian | Endometrial | Ovarian | Endometrial |
| Histological Type | High grade serous | Endometrioid | High grade serous | Endometrioid |
| Samples | 50 | 62 | 351 | 395 |
| Age (mean) | 59 | 61 | 59 | 65 |
| * Race: | ||||
| White | 48 | 57 | 302 | 288 |
| Black | 1 | 0 | 25 | 61 |
| Asian | 0 | 0 | 10 | 17 |
| Pacific Islander | 0 | 1 | 1 | 7 |
| American Indian | 0 | 0 | 2 | 3 |
| Unknown | 1 | 4 | 12 | 20 |
| * Ethnicity | ||||
| Hispanic | 0 | 0 | 8 | 9 |
| Non-Hispanic | 49 | 58 | 201 | 275 |
| Unknown | 1 | 4 | 142 | 111 |
| Stage: | ||||
| I | 0 | 44 | 1 | 281 |
| II | 0 | 4 | 20 | 34 |
| III | 34 | 11 | 274 | 66 |
| IV | 13 | 3 | 53 | 14 |
| Unknown | 3 | 0 | 1 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, M.D.; Devor, E.J.; Salinas, E.A.; Newtson, A.M.; Goodheart, M.J.; Leslie, K.K.; Gonzalez-Bosquet, J. Population Substructure Has Implications in Validating Next-Generation Cancer Genomics Studies with TCGA. Int. J. Mol. Sci. 2019, 20, 1192. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051192
Miller MD, Devor EJ, Salinas EA, Newtson AM, Goodheart MJ, Leslie KK, Gonzalez-Bosquet J. Population Substructure Has Implications in Validating Next-Generation Cancer Genomics Studies with TCGA. International Journal of Molecular Sciences. 2019; 20(5):1192. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051192
Chicago/Turabian StyleMiller, Marina D., Eric J. Devor, Erin A. Salinas, Andreea M. Newtson, Michael J. Goodheart, Kimberly K. Leslie, and Jesus Gonzalez-Bosquet. 2019. "Population Substructure Has Implications in Validating Next-Generation Cancer Genomics Studies with TCGA" International Journal of Molecular Sciences 20, no. 5: 1192. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051192