Thinking Outside the Bug: Molecular Targets and Strategies to Overcome Antibiotic Resistance

Abstract
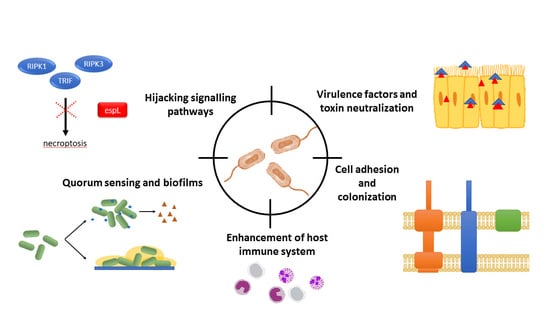
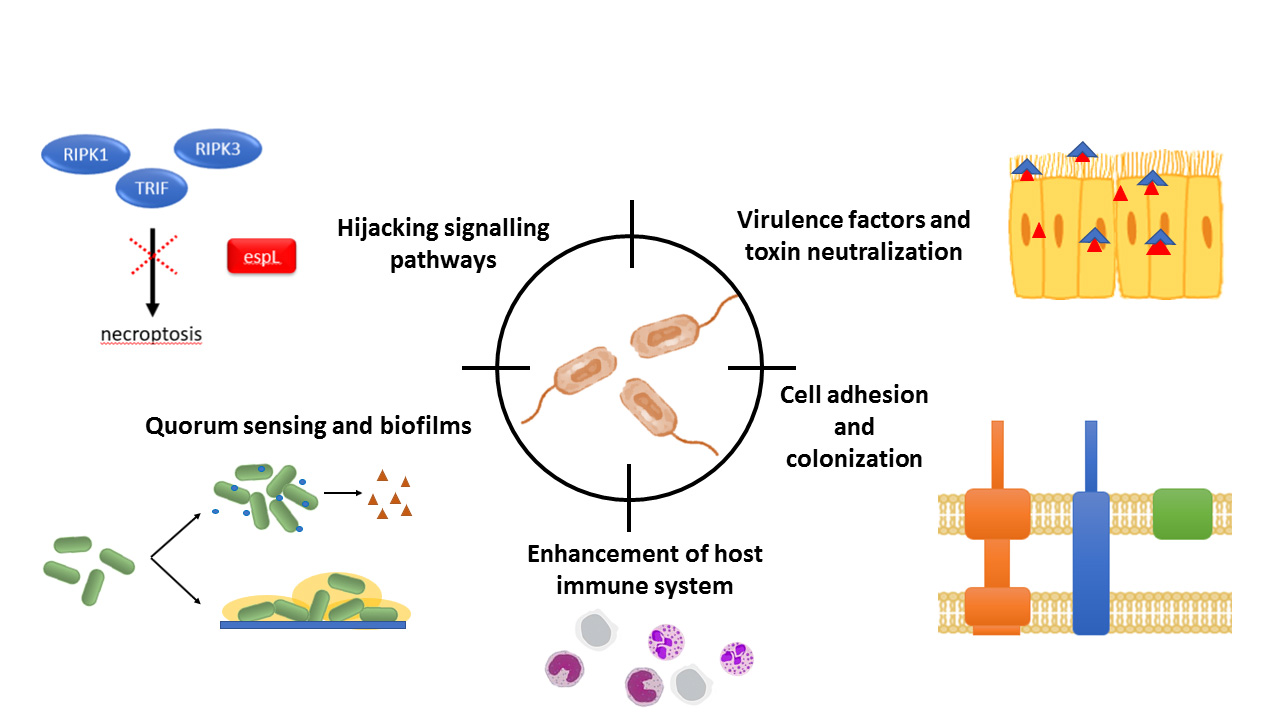
:
{kind=link}
{kind=link}
{kind=link}
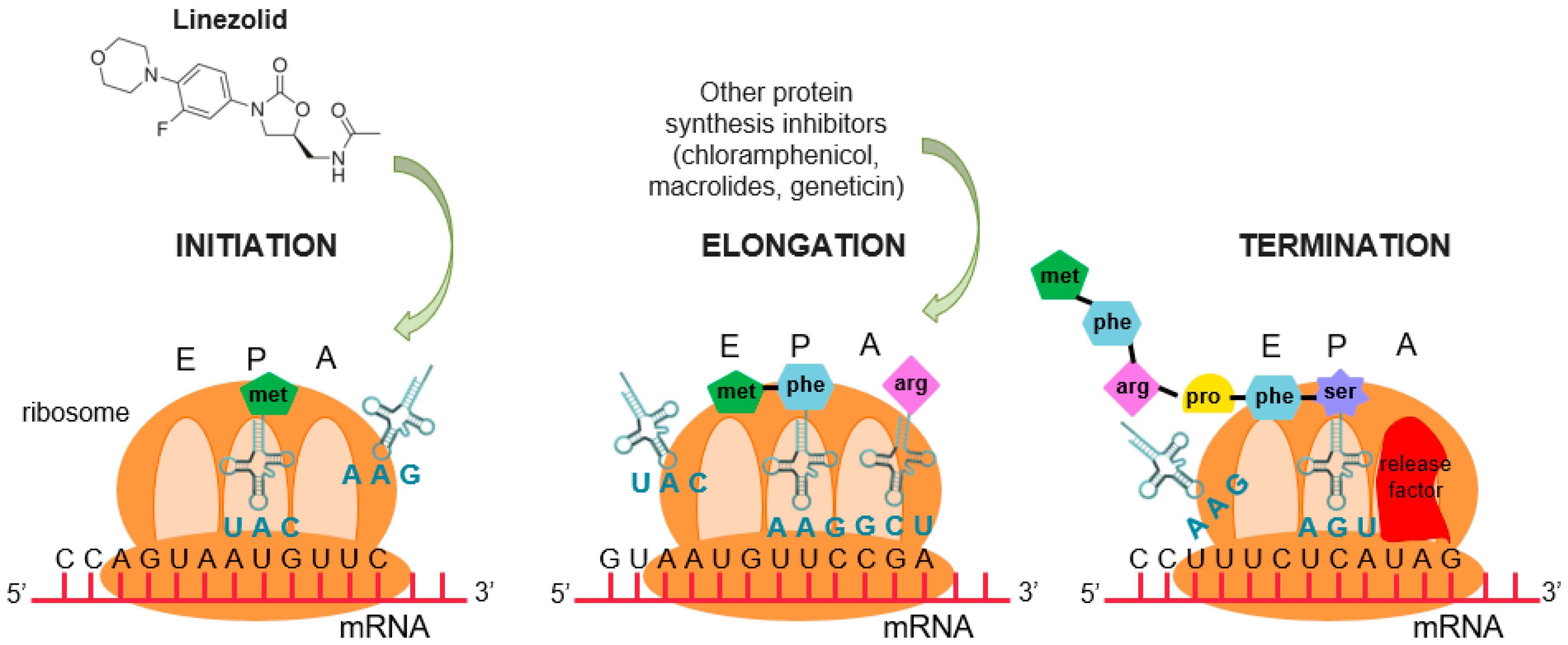{kind=link}
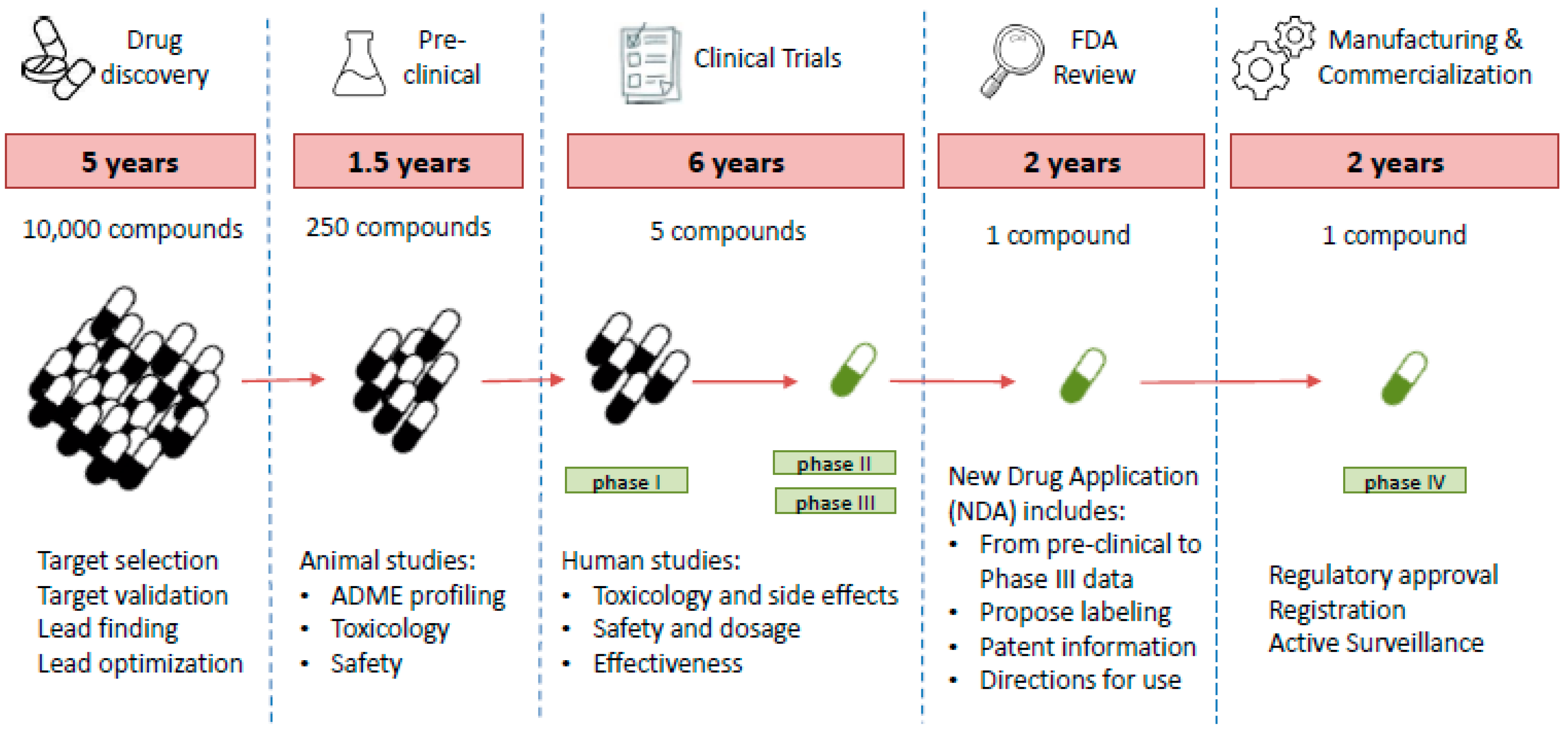{kind=link}
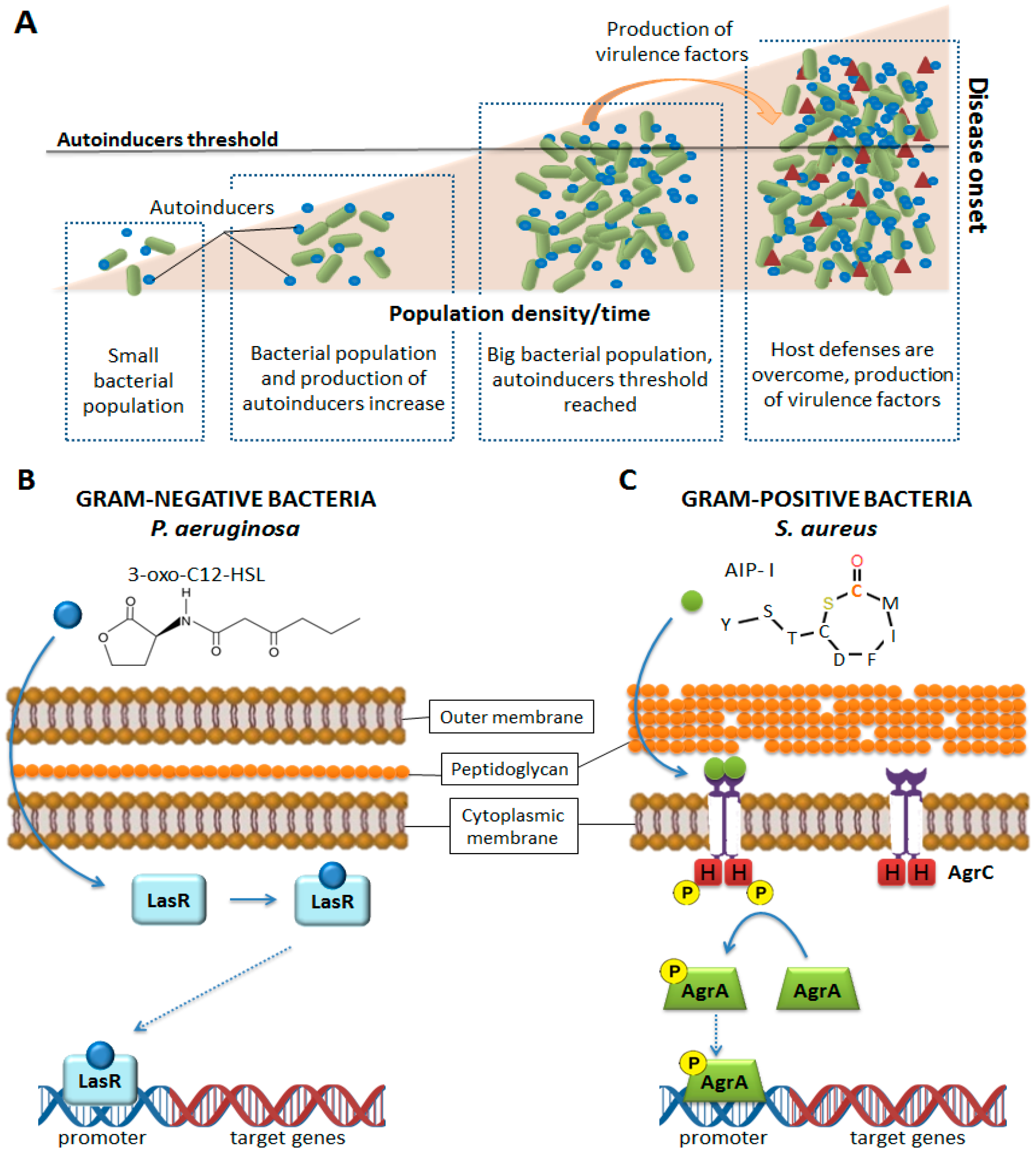{kind=link}
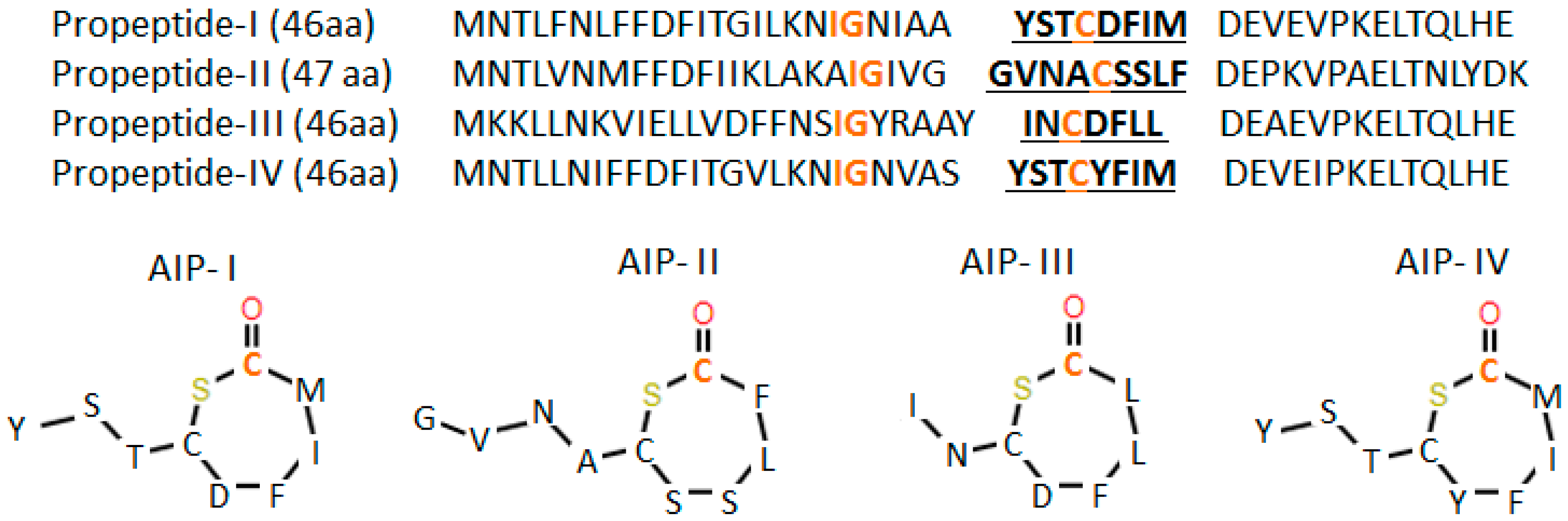{kind=link}
{kind=link}
1. Introduction
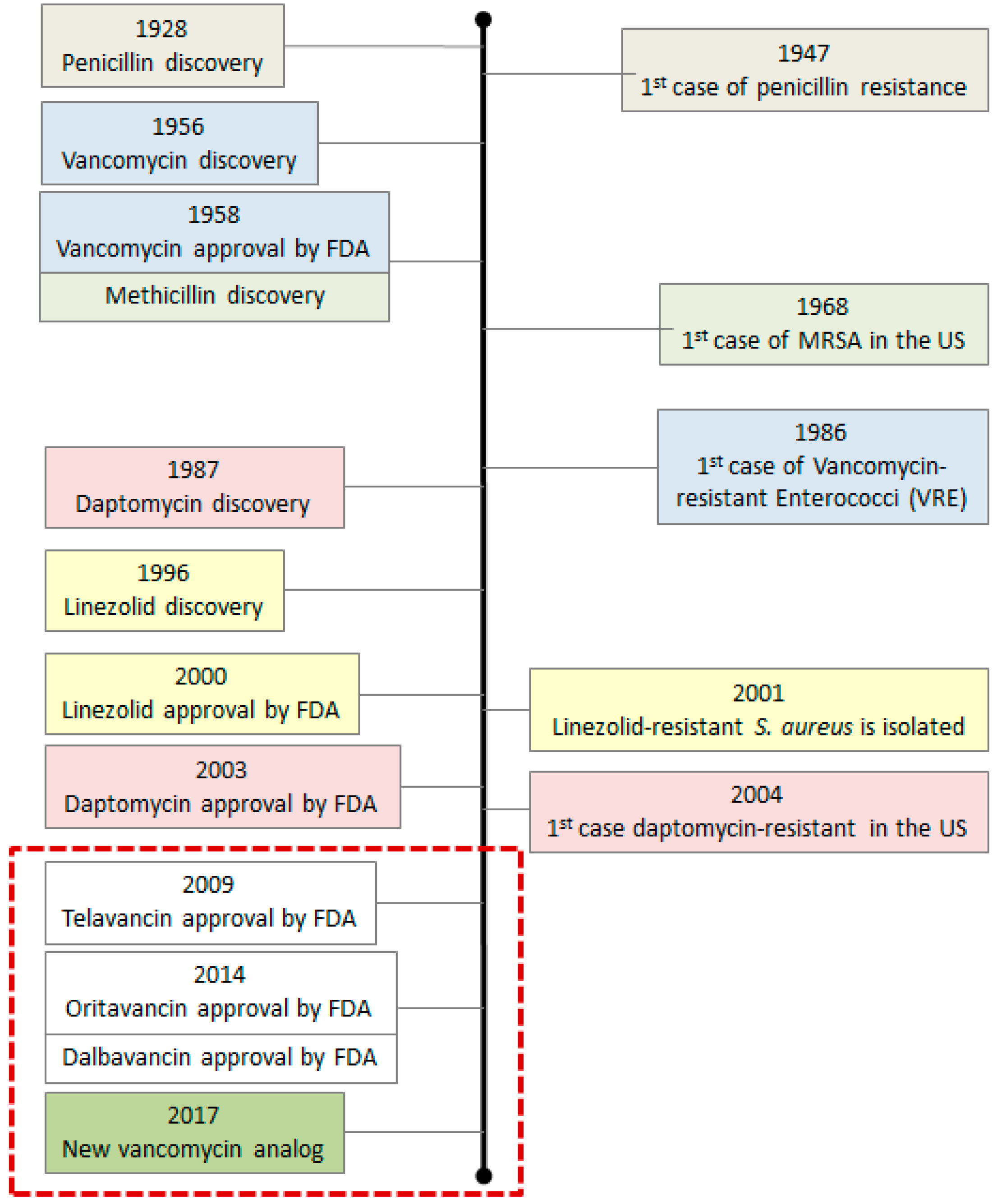
2. Bacterial Resistance and Evolution of Antibiotics
2.1. First Generation Antibiotics and Their Analogs
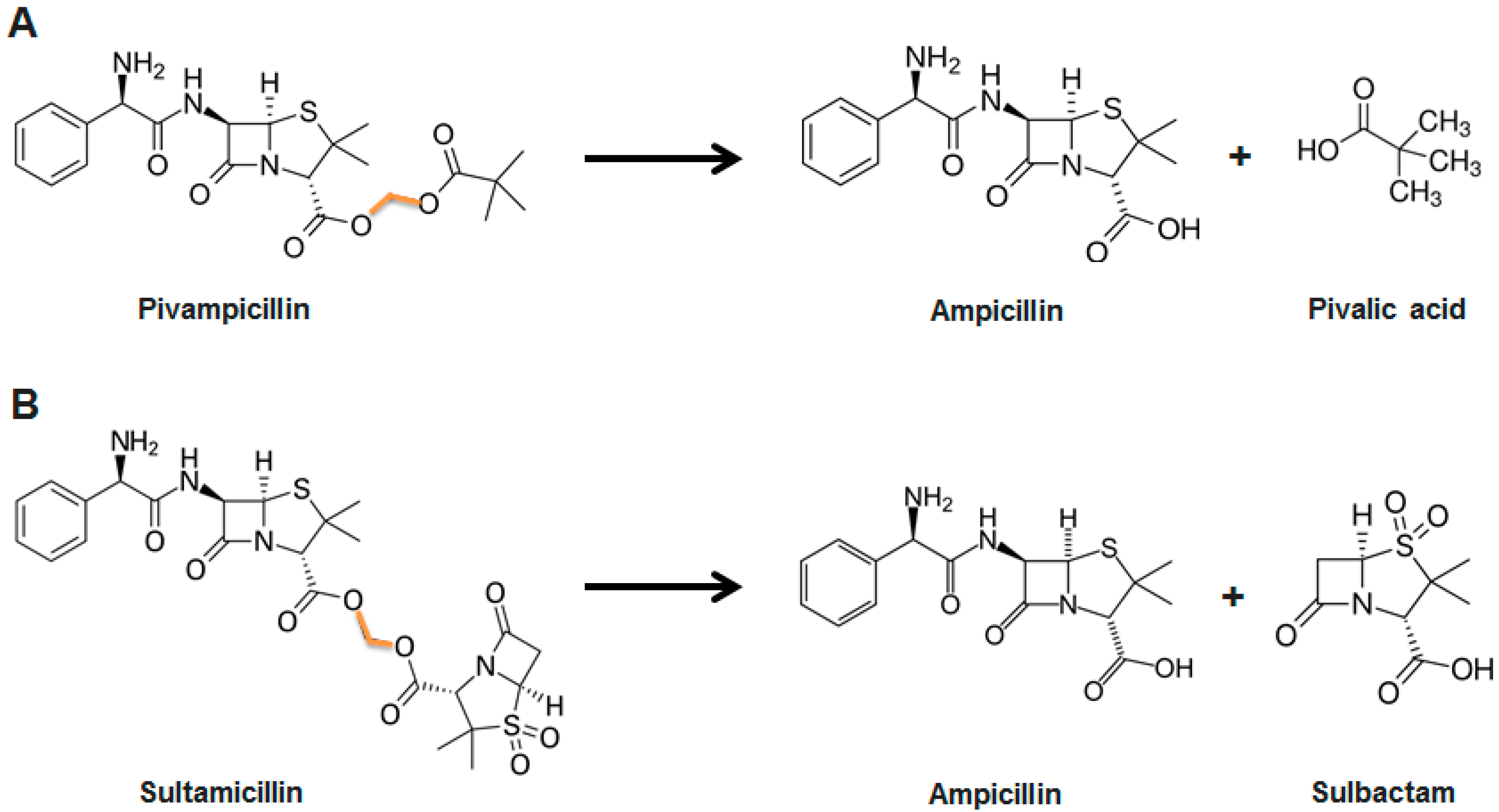
2.2. Prodrugs and Antibiotic Derivatives
2.3. Discovering New Molecules Using Bioinformatics and Rational Design
2.4. Combinatorial Chemistry and High Throughput Screening
2.5. Towards New Approaches for Antibacterial Drug Design
3. Targeting Non-Essential Processes: The Key to Overcome Resistance
3.1. Bacterial Communication: Quorum Sensing
3.2. Host–Pathogen Interactions
3.2.1. Cell Adhesion and Colonization
3.2.2. Virulence Factors and Toxins
3.2.3. Signaling
4. Another Alternative: Enhancing Host Cell Immune Response
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AHL | acyl-homoserine lactone |
| AIP | autoinducer peptide |
| AMPs | antimicrobial peptides |
| BID | BH3 interacting-domain death agonist |
| CBP | 4-chlorobiphenyl)methyl |
| d-Ala | d-Alanine |
| EPEC | Enteropathogenic E. coli |
| FDA | Food and Drug Administration |
| FPRL-1 | formyl peptide receptor like-1 |
| HIV | human immunodeficiency virus |
| MRSA | methicillin-resistant Staphylococcus aureus |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| QS | quorum sensing |
| RHIM | receptor-interacting protein homotypic interaction motif |
| RIP | receptor-interacting protein |
| RIPK1 | receptor-interacting serine/threonine-protein kinase 1 |
| RIPK3 | receptor-interacting serine/threonine-protein kinase 3 |
| SAM | S-adenosylmethionine |
| T3SS | type three secretion system |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| VRE | vancomycin-resistant enterococcus |
References
- Fernebro, J. Fighting bacterial infections—Future treatment options. Drug Resist. Updat. 2011, 14, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzæ. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- Rammelkamp, C.H.; Maxon, T. Resistance of Staphylococcus aureus to the action of penicillin. Proc. Soc. Exp. Biol. Med. 1942, 51, 386–389. [Google Scholar] [CrossRef]
- Enright, M.C.; Robinson, D.A.; Randle, G.; Feil, E.J.; Grundmann, H.; Spratt, B.G. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc. Natl. Acad. Sci. USA 2002, 99, 7687–7692. [Google Scholar] [CrossRef]
- Waness, A. Revisiting methicillin-resistant Staphylococcus aureus infections. J. Glob. Infect. Dis. 2010, 2, 49–56. [Google Scholar] [CrossRef]
- Fischer, J.; Ganellin, C.R. Analogue-Based Drug Discovery, 1st ed.; Wiley-VCH: Hoboken, NJ, USA, 2006; ISBN 9783527607495. [Google Scholar]
- Lemke, T. Foye’s Principles of Medicinal Chemistry, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008. [Google Scholar]
- Singh, J.; Arrieta, A.C. New cephalosporins. Semin. Pediatr. Infect. Dis. 1999, 10, 14–22. [Google Scholar] [CrossRef]
- North, D.S.; Pharm, D.; Fish, D.N.; Pharm, D.; Redington, J.J. Levofloxacin, a second-generation fluoroquinolone. Pharmacotherapy 1998, 5, 915–935. [Google Scholar]
- Levine, D.P. Vancomycin: A history. Clin. Infect. Dis. 2006, 42 (Suppl. 1), S5–S12. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Shlaes, D. Fix the antibiotics pipeline. Nature 2011, 472, 32. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef]
- Monciardini, P.; Iorio, M.; Maffioli, S.; Sosio, M.; Donadio, S. Discovering new bioactive molecules from microbial sources. Microb. Biotechnol. 2014, 7, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kardos, N.; Demain, A.L. Penicillin: The medicine with the greatest impact on therapeutic outcomes. Appl. Microbiol. Biotechnol. 2011, 92, 677. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Pages, J.M.; James, C.E.; Winterhalter, M. The porin and the permeating antibiotic: A selective diffusion barrier in gram-negative bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 2005, 56, 20–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, L.L. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 2007, 6, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Perkins, H.R. Vancomycin and related antibiotics. Pharmacol. Ther. 1982, 16, 181–197. [Google Scholar] [CrossRef]
- Romaniuk, J.A.H.; Cegelski, L. Bacterial cell wall composition and the influence of antibiotics by cell-wall and whole-cell NMR. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20150024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neu, H.; Gootz, T. Chapter 11: Antimicrobial Chemotherapy. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Murray, B.E. Vancomycin-resistant Enterococcal infections. N. Engl. J. Med. 2000, 342, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.C.; Virani, Z.; Cree, R.G.A. Co-transfer of vancomycin and other resistance genes from Enterococcus faecalis NCTC 12201 to Staphylococcus aureus. FEMS Microbiol. Lett. 1992, 93, 195–198. [Google Scholar] [CrossRef]
- McKessar, S.J.; Berry, A.M.; Bell, J.M.; Turnidge, J.D.; Paton, J.C. Genetic characterization of vanG, a novel vancomycin resistance locus of Enterococcus faecalis. Antimicrob. Agents Chemother. 2000, 44, 3224–3228. [Google Scholar] [CrossRef]
- Walsh, C.T.; Fisher, S.L.; Park, I.S.; Prahalad, M.; Wu, Z. Bacterial resistance to vancomycin: Five genes and one missing hydrogen bond tell the story. Chem. Biol. 1996, 3, 21–28. [Google Scholar] [CrossRef]
- Okano, A.; Isley, N.A.; Boger, D.L. Total syntheses of vancomycin-related glycopeptide antibiotics and key analogues. Chem. Rev. 2017, 117, 11952–11993. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Pierce, J.G.; James, R.C.; Okano, A.; Boger, D.L. A Redesigned vancomycin engineered for dual d-Ala-d-Ala and d-Ala-d-Lac binding exhibits potent antimicrobial activity against vancomycin-resistant bacteria. J. Am. Chem. Soc. 2011, 133, 13946–13949. [Google Scholar] [CrossRef]
- Baltz, R.H.; Miao, V.; Wrigley, S.K. Natural products to drugs: Daptomycin and related lipopeptide antibiotics. Nat. Prod. Rep. 2005, 22, 717–741. [Google Scholar] [CrossRef] [PubMed]
- Brickner, S.J.; Barbachyn, M.R.; Hutchinson, D.K.; Manninen, P.R. Linezolid (ZYVOX), the first member of a completely new class of antibacterial agents for treatment of serious gram-positive infections. J. Med. Chem. 2008, 51, 1981–1990. [Google Scholar] [CrossRef]
- Okano, A.; Isley, N.A.; Boger, D.L. Peripheral modifications of vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics. Proc. Natl. Acad. Sci. USA 2017, 26, 114. [Google Scholar]
- Rinehart, K.L.J.; Shield, L.S. Chemistry of the ansamycin antibiotics. Fortschr. Chem. Org. Naturst. 1976, 33, 231–307. [Google Scholar]
- Wehrli, W. Ansamycins. Chemistry, biosynthesis and biological activity. Top. Curr. Chem. 1977, 72, 21–49. [Google Scholar]
- Margalith, P.; Pagani, H. Rifomycin. XIV. Production of rifomycin B. Appl. Microbiol. 1961, 9, 325–334. [Google Scholar] [PubMed]
- Sensi, P.; Ballotta, R.; Greco, A.M.; Gallo, G.G. Rifamycin. Activation of rifamycin B and rifamycin O. Production and properties of rifamycin S and rifamycin SV. Il Farmaco, Ed. Sci. 1961, 16, 165–180. [Google Scholar]
- Sensi, P.; Thiemann, J.E. Production of rifamycins. Prog. Ind. Microbiol. 1967, 6, 21–59. [Google Scholar]
- Calvori, C.; Frontali, L.; Leoni, L.; Tecce, G. Effect of rifamycin on protein synthesis. Nature 1965, 207, 417–418. [Google Scholar] [CrossRef]
- Hartmann, G.; Honikel, K.O.; Knusel, F.; Nuesch, J. The specific inhibition of the DNA-directed RNA synthesis by rifamycin. Biochim. Biophys. Acta 1967, 145, 843–844. [Google Scholar] [CrossRef]
- Wehrli, W. Rifampin: Mechanisms of action and resistance. Rev. Infect. Dis. 1983, 5 (Suppl. 3), S407–S411. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.W. Rifamycin—Mode of Action, Resistance, and Biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef]
- David, H.L. Probability distribution of drug-resistant mutants in unselected populations of Mycobacterium tuberculosis. Appl. Microbiol. 1970, 20, 810–814. [Google Scholar]
- Gillespie, S.H. Evolution of drug resistance in Mycobacterium tuberculosis: Clinical and molecular perspective. Antimicrob. Agents Chemother. 2002, 46, 267–274. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Mizen, L.; Burton, G. The use of esters as prodrugs for oral delivery of beta-lactam antibiotics. Pharm. Biotechnol. 1998, 11, 345–365. [Google Scholar]
- Daehne, W.V.; Frederiksen, E.; Gundersen, E.; Lund, F.; Moerch, P.; Petersen, H.J.; Roholt, K.; Tybring, L.; Godtfredsen, W.O. Acyloxymethyl esters of ampicillin. J. Med. Chem. 1970, 13, 607–612. [Google Scholar] [CrossRef]
- Foltz, E.L.; West, J.W.; Breslow, I.H.; Wallick, H. Clinical pharmacology of pivampicillin. Antimicrob. Agents Chemother. 1970, 10, 442–454. [Google Scholar]
- Jordan, M.C.; de Maine, J.B.; Kirby, W.M. Clinical pharmacology of pivampicillin as compared with ampicillin. Antimicrob. Agents Chemother. 1970, 10, 438–441. [Google Scholar]
- Pines, A.; Greenfield, J.S.B.; Raafat, H.; Sreedharan, K.S. A comparison of pivampicillin and ampicillin in exacerbations of chronic bronchitis. Br. J. Dis. Chest 1973, 67, 221–226. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef]
- Baltzer, B.; Binderup, E.; von Daenhe, W.; Godtfredsen, W.O.; Hansen, K.; Nielsen, B.; Sorensen, H.; Vangedal, S. Mutual pro-drugs of β-lactam antibiotics and β-lactamase inhibitors. J. Antibiot. 1980, 33, 1183–1192. [Google Scholar] [CrossRef]
- Paladino, J.A.; Rainstein, M.A.; Serrianne, D.J.; Przylucki, J.E.; Welage, L.S.; Collura, M.L.; Schentag, J.J. Ampicillin-sulbactam versus cefoxitin for prophylaxis in high-risk patients undergoing abdominal surgery. Pharmacotherapy 1994, 14, 734–739. [Google Scholar]
- McKinnon, P.S.; Paladino, J.A.; Grayson, M.L.; Gibbons, G.W.; Karchmer, A.W. Cost-effectiveness of ampicillin/sulbactam versus imipenem/cilastatin in the treatment of limb-threatening foot infections in diabetic patients. Clin. Infect. Dis. 1997, 24, 57–63. [Google Scholar] [CrossRef]
- Messick, C.R.; Mamdani, M.; McNicholl, I.R.; Danziger, L.H.; Rodvold, K.A.; Condon, R.E.; Walker, A.P.; Edmiston, C.E.J. Pharmacoeconomic analysis of ampicillin-sulbactam versus cefoxitin in the treatment of intraabdominal infections. Pharmacotherapy 1998, 18, 175–183. [Google Scholar]
- Pfizer. Unasyn (Ampicillin Sodium/Sulbactam Sodium). 2010; Reviewed in 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/050608s041s042lbl.pdf (accessed on 11 March 2019).
- Clinical and Laboratory Standard Institute. Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Second Informational Supplement; CLSI Document M100-S22; Clinical and Laboratory Standard Institute: Suite, Wayne, NY, USA, 2012; Volume 32, ISBN 1562387855. [Google Scholar]
- Rafailidis, P.I.; Ioannidou, E.N.; Falagas, M.E. Ampicillin/sulbactam: Current status in severe bacterial infections. Drugs 2007, 67, 1829–1849. [Google Scholar] [CrossRef]
- Adam, M. Integrating research and development: The emergence of rational drug design in the pharmaceutical industry. Stud. Hist. Philos. Biol. Biomed. Sci. 2005, 36, 513–537. [Google Scholar] [CrossRef]
- Zahedi Bialvaei, A.; Rahbar, M.; Yousefi, M.; Asgharzadeh, M.; Samadi Kafil, H. Linezolid: A promising option in the treatment of gram-positives. J. Antimicrob. Chemother. 2017, 72, 354–364. [Google Scholar] [CrossRef]
- European Medicines Agency. CHP Assessment Report for Xarelto (EMA/CHMP/301607/2011). 2011. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/xarelto (accessed on 15 March 2012).
- Wilcox, M.H.; Tack, K.J.; Bouza, E.; Herr, D.L.; Ruf, B.R.; Ijzerman, M.M.; Croos-Dabrera, R.V.; Kunkel, M.J.; Knirsch, C. Complicated skin and skin-structure infections and catheter-related bloodstream infections: Noninferiority of linezolid in a phase 3 study. Clin. Infect. Dis. 2009, 48, 203–212. [Google Scholar] [CrossRef]
- Linezolid Side Effects in Detail. Available online: www.drugs.com (accessed on 1 December 2018).
- Okafor, N. Chapter 28.2 Newer methods of drug discovery. In Modern Industrial Microbiology and Biotechnology; Science Publishers: Enfield, NH, USA, 2016; ISBN 978-1-57808-434-0 (HC). [Google Scholar]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef]
- Geysen, H.M.; Meloen, R.H.; Barteling, S.J. Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc. Natl. Acad. Sci. USA 1984, 81, 3998–4002. [Google Scholar] [CrossRef]
- Houghten, R.A. General method for the rapid solid-phase synthesis of large numbers of peptides: Specificity of antigen-antibody interaction at the level of individual amino acids. Proc. Natl. Acad. Sci. USA 1985, 82, 5131–5135. [Google Scholar] [CrossRef]
- Pandeya, S.N.; Thakkar, D. Combinatorial chemistry: A novel method in drug discovery and its application. Indian J. Chem. Sect. B Org. Med. Chem. 2005, 44, 335–348. [Google Scholar] [CrossRef]
- Carroll, J. Will combinatorial chemistry keep its promise? Biotechnol. Healthc. 2005, 2, 26–32. [Google Scholar]
- Kennedy, J.P.; Williams, L.; Bridges, T.M.; Daniels, R.N.; Weaver, D.; Lindsley, C.W. Application of combinatorial chemistry science on modern drug discovery. J. Comb. Chem. 2008, 10, 345–354. [Google Scholar] [CrossRef]
- Gordeeva, E.V.; Lushniknov, D.E.; Zefirov, N.S. COMPASS Program—An original semi-empirical approach to computer-assisted synthesis. Tetrahedron 1992, 48, 3789–3804. [Google Scholar] [CrossRef]
- Ho, C. Access to Medicine in the Global Economy; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Gwynne, P.; Heebner, G. LABORATORY TECHNOLOGY TRENDS: Drug Discovery: 3. Available online: https://www.sciencemag.org/site/products/dd3text.xhtml (accessed on 11 March 2019).
- Lange, R.P.; Locher, H.H.; Wyss, P.C.; Then, R.L. The targets of currently used antibacterial agents: Lessons for drug discovery. Curr. Pharm. Des. 2007, 13, 3140–3154. [Google Scholar] [CrossRef]
- Fernandes, P. Antibacterial discovery and development-the failure of success? Nat. Biotechnol. 2006, 24, 1497–1503. [Google Scholar] [CrossRef]
- Galán, J.C.; Baquero, M.R.; Morosini, M.I.; Baquero, F. Bacterias con alta tasa de mutación:los riesgos de una vida acelerada. Asociación Colombianade Infectología 2006, 10, 22–29. [Google Scholar]
- Bromham, L. Why do species vary in their rate of molecular evolution? Biol. Lett. 2009, 5, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Dunning Hotopp, J.C. Horizontal gene transfer between bacteria and animals. Trends Genet. 2011, 27, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Gagneux, S.; Long, C.D.; Small, P.M.; Van, T.; Schoolnik, G.K.; Bohannan, B.J.M. The Competitive Cost of Antibiotic Resistance in Mycobacterium tuberculosis. Science 2006, 312, 1944–1946. [Google Scholar] [CrossRef]
- Barnard, F.; Maxwell, A. Interaction between DNA gyrase and quinolones: Effects of alanine mutations at GyrA subunit residues Ser83 and Asp87. Antimicrob. Agents Chemother. 2001, 45, 1994–2000. [Google Scholar] [CrossRef]
- Heddle, J.; Maxwell, A. Quinolone-binding pocket of DNA gyrase: Role of GyrB. Antimicrob. Agents Chemother. 2002, 46, 1805–1815. [Google Scholar] [CrossRef]
- Parry, C.M.; Threlfall, E.J. Antimicrobial resistance in typhoidal and nontyphoidal salmonellae. Curr. Opin. Infect. Dis. 2008, 21, 531–538. [Google Scholar] [CrossRef]
- Ruiz, J. Mechanisms of resistance to quinolones: Target alterations, decreased accumulation and DNA gyrase protection. J. Antimicrob. Chemother. 2003, 51, 1109–1117. [Google Scholar] [CrossRef]
- Njoroge, J.; Sperandio, V. Jamming bacterial communication: New approaches for the treatment of infectious diseases. EMBO Mol. Med. 2009, 1, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Khor, C.-C.; Hibberd, M.L. Revealing the molecular signatures of host-pathogen interactions. Genome Biol. 2011, 12, 229. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.K.; Daniell, S.; Ebel, F.; Frankel, G.; Knutton, S. EspA filament-mediated protein translocation into red blood cells. Cell. Microbiol. 2001, 3, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Ide, T.; Laarmann, S.; Greune, L.; Schillers, H.; Oberleithner, H.; Schmidt, M.A. Characterization of translocation pores inserted into plasma membranes by type III-secreted Esp proteins of enteropathogenic Escherichia coli. Cell. Microbiol. 2001, 3, 669–679. [Google Scholar] [CrossRef]
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal enterotoxins. Toxins 2010, 2, 2177–2197. [Google Scholar] [CrossRef]
- Perrett, C.A.; Lin, D.Y.-W.; Zhou, D. Interactions of bacterial proteins with host eukaryotic ubiquitin pathways. Front. Microbiol. 2011, 2, 143. [Google Scholar] [CrossRef]
- Jimenez, A.; Chen, D.; Alto, N.M. How bacteria subvert animal cell structure and function. Annu. Rev. Cell Dev. Biol. 2016, 32, 373–397. [Google Scholar] [CrossRef]
- Gilbert, R.J.C.; Bayley, H.; Anderluh, G. Membrane pores: From structure and assembly, to medicine and technology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160208. [Google Scholar] [CrossRef]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-Cell Communication in Bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef]
- Vasil, M. DNA microarrays in analysis of quorum sensing: Strengths and limitations. J. Bacteriol. 2003, 185, 2061–2065. [Google Scholar] [CrossRef]
- Deep, A.; Chaudhary, U.; Gupta, V. Quorum sensing and Bacterial Pathogenicity: From Molecules to Disease. J. Lab. Physicians 2011, 3, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.; Gray, K.; Passadort, L.; Tuckert, K.; Eberhard, A.; Iglewskit, B.; Greenberg, E. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proc. Natl. Acad. Sci. USA 1994, 91, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, N.A.; Barnard, A.M.; Slater, H.; Simpson, N.J.; Salmond, G.P. Quorum sensing in Gram-negative bacteria. FEMS Microbiol. Rev. 2001, 25, 365–404. [Google Scholar] [CrossRef] [PubMed]
- Papenfort, K.; Bassler, B. Quorum sensing signal-response systems in gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Case, R.J.; Labbate, M.; Kjelleberg, S. AHL-driven quorum sensing circuits: Their frequency and function among the Proteobacteria. ISME J. 2008, 2, 345–349. [Google Scholar] [CrossRef]
- Hao, Y.; Winans, S.C.; Glick, B.R.; Charles, T.C. Identification and characterization of new LuxR/LuxI-type quorum sensing systems from metagenomic libraries. Environ. Microbiol. 2010, 12, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.; Wang, J.-H.; Swatton, J.E.; Davenport, P.; Price, B.; Mikkelsen, H.; Stickland, H.; Nishikawa, K.; Gardiol, N.; Spring, D.R.; et al. Variations on a theme: Diverse N-acyl homoserine lactone-mediated quorum sensing mechanisms in gram-negative bacteria. Sci. Prog. 2006, 89, 167–211. [Google Scholar] [CrossRef]
- Stevens, A.M.; Dolan, K.M.; Greenberg, E.P. Synergistic binding of the Vibrio fischeri LuxR transcriptional activator domain and RNA polymerase to the lux promoter region. Proc. Natl. Acad. Sci. USA 1994, 91, 12619–12623. [Google Scholar] [CrossRef]
- Zhang, R.; Pappas, K.M.; Brace, J.L.; Miller, P.C.; Oulmassov, T.; Molyneaux, J.M.; Anderson, J.C.; Bashkin, J.K.; Winans, S.C.; Joachimiak, A. Structure of a bacterial quorum sensing transcription factor complexed with pheromone and DNA. Nature 2002, 417, 971–974. [Google Scholar] [CrossRef]
- Rutherford, S.T.; Bassler, B.L. Bacterial quorum sensing: Its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med. 2012, 2, 1–25. [Google Scholar] [CrossRef]
- LaSarre, B.; Federle, M.J. Exploiting Quorum Sensing To Confuse Bacterial Pathogens. Microbiol. Mol. Biol. Rev. 2013, 77, 73–111. [Google Scholar] [CrossRef] [Green Version]
- Ng, W.-L.; Bassler, B.L. Bacterial quorum sensing network architectures. Annu. Rev. Genet. 2009, 43, 197–222. [Google Scholar] [CrossRef]
- Wang, B.; Muir, T.W. Regulation of Virulence in Staphylococcus aureus: Molecular Mechanisms and Remaining Puzzles. Cell Chem. Biol. 2016, 23, 214–224. [Google Scholar] [CrossRef]
- Dufour, P.; Jarraud, S.; Vandenesch, F.; Greenland, T.; Novick, R.P.; Bes, M.; Etienne, J.; Lina, G. High genetic variability of the agr locus in Staphylococcus species. J. Bacteriol. 2002, 184, 1180–1186. [Google Scholar] [CrossRef]
- Ji, G.; Beavis, R.; Novick, R.P. Bacterial interference caused by autoinducing peptide variants. Science 1997, 276, 2027–2030. [Google Scholar] [CrossRef]
- Otto, M.; Sussmuth, R.; Jung, G.; Gotz, F. Structure of the pheromone peptide of the Staphylococcus epidermidis agr system. FEBS Lett. 1998, 424, 89–94. [Google Scholar] [CrossRef]
- Jarraud, S.; Lyon, G.J.; Figueiredo, A.M.; Lina, G.; Vandenesch, F.; Etienne, J.; Muir, T.W.; Novick, R.P. Exfoliatin-producing strains define a fourth agr specificity group in Staphylococcus aureus. J. Bacteriol. 2000, 182, 6517–6522. [Google Scholar] [CrossRef]
- Pestova, E.V.; Havarstein, L.S.; Morrison, D.A. Regulation of competence for genetic transformation in Streptococcus pneumoniae by an auto-induced peptide pheromone and a two-component regulatory system. Mol. Microbiol. 1996, 21, 853–862. [Google Scholar] [CrossRef]
- Novick, R.P.; Geisinger, E. Quorum sensing in staphylococci. Annu. Rev. Genet. 2008, 42, 541–564. [Google Scholar] [CrossRef]
- Lyon, G.J.; Wright, J.S.; Christopoulos, A.; Novick, R.P.; Muir, T.W. Reversible and specific extracellular antagonism of receptor-histidine kinase signaling. J. Biol. Chem. 2002, 277, 6247–6253. [Google Scholar] [CrossRef]
- Leadbetter, J.R.; Greenberg, E.P. Metabolism of Acyl-Homoserine Lactone Quorum Sensing Signals by Variovorax paradoxus. J. Bacteriol. 2000, 182, 6921–6926. [Google Scholar] [CrossRef]
- Dong, Y.H.; Xu, J.L.; Li, X.Z.; Zhang, L.H. AiiA, an enzyme that inactivates the acylhomoserine lactone quorum sensing signal and attenuates the virulence of Erwinia carotovora. Proc. Natl. Acad. Sci. USA 2000, 97, 3526–3531. [Google Scholar] [CrossRef]
- Dong, Y.H.; Wang, L.H.; Xu, J.L.; Zhang, H.B.; Zhang, X.F.; Zhang, L.H. Quenching quorum sensing-dependent bacterial infection by an N-acyl homoserine lactonase. Nature 2001, 411, 813–817. [Google Scholar] [CrossRef]
- Uroz, S.; Chhabra, S.R.; Camara, M.; Williams, P.; Oger, P.; Dessaux, Y. N-Acylhomoserine lactone quorum-sensing molecules are modified and degraded by Rhodococcus erythropolis W2 by both amidolytic and novel oxidoreductase activities. Microbiology 2005, 151, 3313–3322. [Google Scholar] [CrossRef]
- Zhang, H.-B.; Wang, L.-H.; Zhang, L.-H. Genetic control of quorum sensing signal turnover in Agrobacterium tumefaciens. Proc. Natl. Acad. Sci. USA 2002, 99, 4638–4643. [Google Scholar] [CrossRef]
- MDowell, P.; Affas, Z.; Reynolds, C.; Holden, M.T.G.; Wood, S.J.; Saint, S.; Cockayne, A.; Hill, P.J.; Dodd, C.E.R.; Bycroft, B.W.; et al. Structure, activity and evolution of the group I thiolactone peptide quorum sensing system of Staphylococcus aureus. Mol. Microbiol. 2001, 41, 503–512. [Google Scholar] [CrossRef]
- Lyon, G.J.; Mayville, P.; Muir, T.W.; Novick, R.P. Rational design of a global inhibitor of the virulence response in Staphylococcus aureus, based in part on localization of the site of inhibition to the receptor-histidine kinase, AgrC. Proc. Natl. Acad. Sci. USA 2000, 97, 13330–13335. [Google Scholar] [CrossRef]
- Park, J.; Jagasia, R.; Kaufmann, G.F.; Mathison, J.C.; Ruiz, D.I.; Moss, J.A.; Meijler, M.M.; Ulevitch, R.J.; Janda, K.D. Infection control by antibody disruption of bacterial quorum sensing signaling. Chem. Biol. 2007, 14, 1119–1127. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L. Host-pathogen interactions: Redefining the basic concepts of virulence and pathogenicity. Infect. Immun. 1999, 67, 3703–3713. [Google Scholar]
- Green, E.R.; Mecsas, J. Bacterial secretion systems—An overview. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Bacon, G.A.; Burrows, T.W. The basis of virulence in Pasteurella pestis: An antigen determining virulence. Br. J. Exp. Pathol. 1956, 37, 481–493. [Google Scholar]
- Guan, K.L.; Dixon, J.E. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science 1990, 249, 553–556. [Google Scholar] [CrossRef]
- Andersson, K.; Carballeira, N.; Magnusson, K.E.; Persson, C.; Stendahl, O.; Wolf-Watz, H.; Fallman, M. YopH of Yersinia pseudotuberculosis interrupts early phosphotyrosine signalling associated with phagocytosis. Mol. Microbiol. 1996, 20, 1057–1069. [Google Scholar] [CrossRef]
- Rosqvist, R.; Bolin, I.; Wolf-Watz, H. Inhibition of phagocytosis in Yersinia pseudotuberculosis: A virulence plasmid-encoded ability involving the Yop2b protein. Infect. Immun. 1988, 56, 2139–2143. [Google Scholar]
- High, N.; Mounier, J.; Prevost, M.C.; Sansonetti, P.J. IpaB of Shigella flexneri causes entry into epithelial cells and escape from the phagocytic vacuole. EMBO J. 1992, 11, 1991–1999. [Google Scholar] [CrossRef]
- Menard, R.; Sansonetti, P.J.; Parsot, C. Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J. Bacteriol. 1993, 175, 5899–5906. [Google Scholar] [CrossRef]
- Tran Van Nhieu, G.; Ben-Ze’ev, A.; Sansonetti, P.J. Modulation of bacterial entry into epithelial cells by association between vinculin and the Shigella IpaA invasin. EMBO J. 1997, 16, 2717–2729. [Google Scholar] [CrossRef]
- Clark, E.A.; Brugge, J.S. Integrins and signal transduction pathways: The road taken. Science 1995, 268, 233–239. [Google Scholar] [CrossRef]
- Watarai, M.; Funato, S.; Sasakawa, C. Interaction of Ipa proteins of Shigella flexneri with alpha5beta1 integrin promotes entry of the bacteria into mammalian cells. J. Exp. Med. 1996, 183, 991–999. [Google Scholar] [CrossRef]
- Wilson, R.K.; Shaw, R.K.; Daniell, S.; Knutton, S.; Frankel, G. Role of EscF, a putative needle complex protein, in the type III protein translocation system of enteropathogenic Escherichia coli. Cell. Microbiol. 2001, 3, 753–762. [Google Scholar] [CrossRef]
- Sawa, T.; Yahr, T.L.; Ohara, M.; Kurahashi, K.; Gropper, M.A.; Wiener-Kronish, J.P.; Frank, D.W. Active and passive immunization with the Pseudomonas V antigen protects against type III intoxication and lung injury. Nat. Med. 1999, 5, 392–398. [Google Scholar] [CrossRef]
- Goure, J.; Broz, P.; Attree, O.; Cornelis, G.R.; Attree, I. Protective anti-V antibodies inhibit Pseudomonas and Yersinia translocon assembly within host membranes. J. Infect. Dis. 2005, 192, 218–225. [Google Scholar] [CrossRef]
- Baer, M.; Sawa, T.; Flynn, P.; Luehrsen, K.; Martinez, D.; Wiener-Kronish, J.P.; Yarranton, G.; Bebbington, C. An engineered human antibody fab fragment specific for Pseudomonas aeruginosa PcrV antigen has potent antibacterial activity. Infect. Immun. 2009, 77, 1083–1090. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Ton-that, H. MicroReview Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 2001, 40, 1049–1057. [Google Scholar] [CrossRef]
- Ton-That, H.; Schneewind, O. Assembly of pili in gram-positive bacteria. Trends Microbiol. 2004, 12, 228–234. [Google Scholar] [CrossRef]
- Schneewind, O.; Model, P.; Fischetti, V.A. Sorting of protein A to the staphylococcal cell wall. Cell 1992, 70, 267–281. [Google Scholar] [CrossRef]
- Ton-That, H.; Schneewind, O. Assembly of pili on the surface of Corynebacterium diphtheriae. Mol. Microbiol. 2003, 50, 1429–1438. [Google Scholar] [CrossRef]
- Reichert, J.M.; Rosensweig, C.J.; Faden, L.B.; Dewitz, M.C. Monoclonal antibody successes in the clinic. Nat. Biotechnol. 2005, 23, 1073–1078. [Google Scholar] [CrossRef]
- Sparrow, E.; Friede, M.; Torvaldsen, S. Therapeutic antibodies for infectious diseases. Bull. World Health Organ. 2017, 95, 235–237. [Google Scholar] [CrossRef]
- López, E.L.; Contrini, M.M.; Glatstein, E.; González Ayala, S.; Santoro, R.; Allende, D.; Ezcurra, G.; Teplitz, E.; Koyama, T.; Matsumoto, Y.; et al. Safety and pharmacokinetics of urtoxazumab, a humanized monoclonal antibody, against Shiga-like toxin 2 in healthy adults and in pediatric patients infected with Shiga-like toxin-producing Escherichia coli. Antimicrob. Agents Chemother. 2010, 54, 239–243. [Google Scholar] [CrossRef]
- Bitzan, M.; Poole, R.; Mehran, M.; Sicard, E.; Brockus, C.; Thuning-Roberson, C.; Rivière, M. Safety and pharmacokinetics of chimeric anti-Shiga toxin 1 and anti-Shiga toxin 2 monoclonal antibodies in healthy volunteers. Antimicrob. Agents Chemother. 2009, 53, 3081–3087. [Google Scholar] [CrossRef]
- Greig, S.L. Obiltoxaximab: First Global Approval. Drugs 2016, 76, 823–830. [Google Scholar] [CrossRef]
- Mazumdar, S. Raxibacumab. MAbs 2009, 1, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Rudel, T.; Kepp, O.; Kozjak-Pavlovic, V. Interactions between bacterial pathogens and mitochondrial cell death pathways. Nat. Rev. Microbiol. 2010, 8, 693–705. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P.-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in inflammation and immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell death and infection: A double-edged sword for host and pathogen survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef] [Green Version]
- Haslett, C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am. J. Respir. Crit. Care Med. 1999, 160, S5–S11. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010, 8, 44–54. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311. [Google Scholar] [CrossRef]
- Sun, X.; Yin, J.; Starovasnik, M.A.; Fairbrother, W.J.; Dixit, V.M. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 2002, 277, 9505–9511. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Offermann, M.K. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 2005, 174, 4942–4952. [Google Scholar] [CrossRef]
- Rebsamen, M.; Heinz, L.X.; Meylan, E.; Michallet, M.-C.; Schroder, K.; Hofmann, K.; Vazquez, J.; Benedict, C.A.; Tschopp, J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009, 10, 916–922. [Google Scholar] [CrossRef]
- Pearson, J.S.; Giogha, C.; Mühlen, S.; Nachbur, U.; Pham, C.L.L.; Zhang, Y.; Hildebrand, J.M.; Oates, C.V.; Lung, T.W.F.; Ingle, D.; et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat. Microbiol. 2017, 2, 16258. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef]
- Zhang, L.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Reddy, K.V.R.; Yedery, R.D.; Aranha, C. Antimicrobial peptides: Premises and promises. Int. J. Antimicrob. Agents 2004, 24, 536–547. [Google Scholar] [CrossRef]
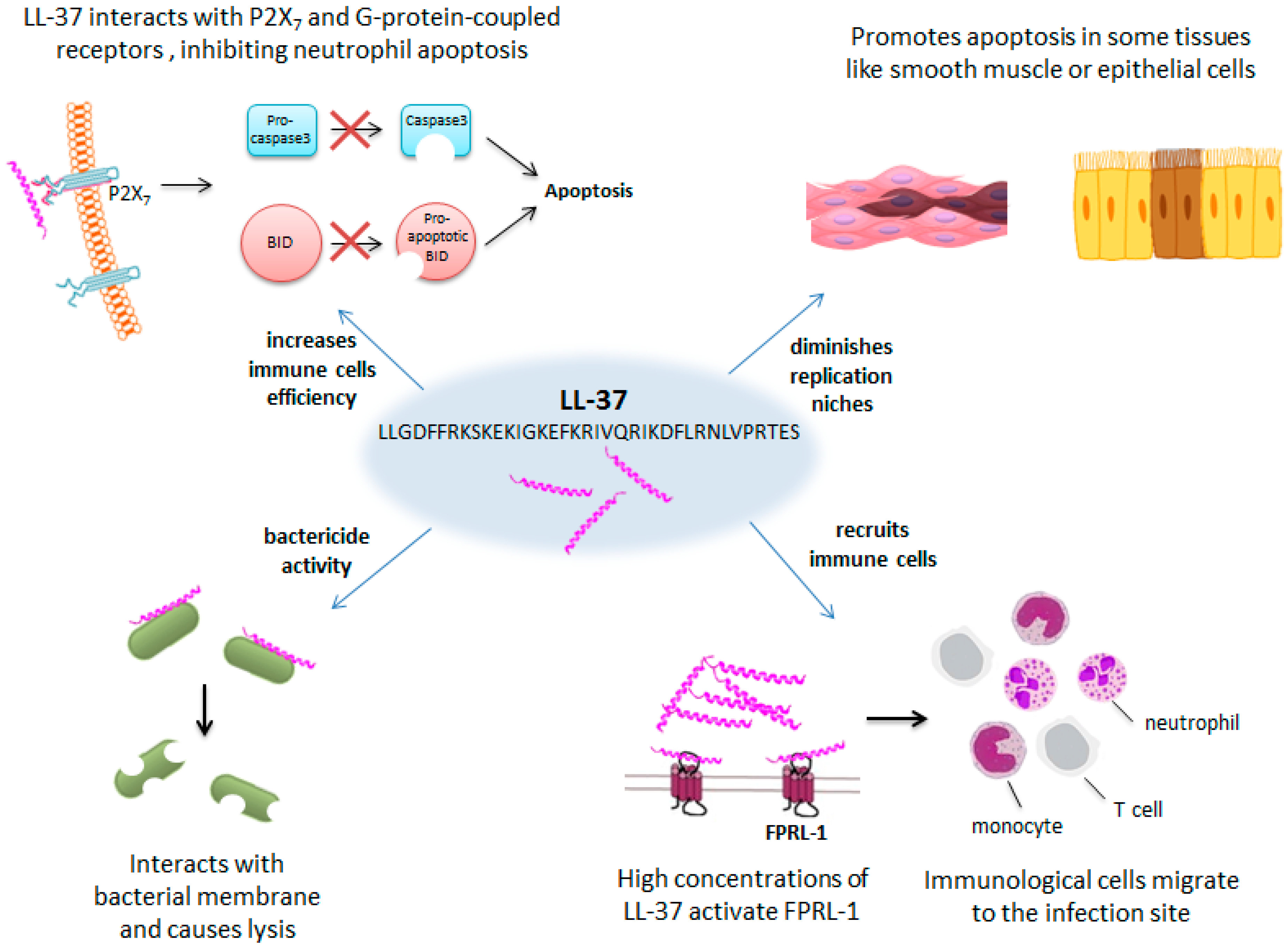
- Kurosaka, K.; Chen, Q.; Yarovinsky, F.; Oppenheim, J.J.; Yang, D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J. Immunol. 2005, 174, 6257–6265. [Google Scholar] [CrossRef]
- Yang, D.; Chen, Q.; Schmidt, A.P.; Anderson, G.M.; Wang, J.M.; Wooters, J.; Oppenheim, J.J.; Chertov, O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med. 2000, 192, 1069–1074. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Iwabuchi, K.; Matsuda, H.; Ogawa, H.; Nagaoka, I. Epithelial cell-derived human beta-defensin-2 acts as a chemotaxin for mast cells through a pertussis toxin-sensitive and phospholipase C-dependent pathway. Int. Immunol. 2002, 14, 421–426. [Google Scholar] [CrossRef]
- Yang, D.; Biragyn, A.; Kwak, L.W.; Oppenheim, J.J. Mammalian defensins in immunity: More than just microbicidal. Trends Immunol. 2002, 23, 291–296. [Google Scholar] [CrossRef]
- Territo, M.C.; Ganz, T.; Selsted, M.E.; Lehrer, R. Monocyte-chemotactic activity of defensins from human neutrophils. J. Clin. Investig. 1989, 84, 2017–2020. [Google Scholar] [CrossRef]
- Heilborn, J.D.; Nilsson, M.F.; Kratz, G.; Weber, G.; Sorensen, O.; Borregaard, N.; Stahle-Backdahl, M. The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J. Investig. Dermatol. 2003, 120, 379–389. [Google Scholar] [CrossRef]
- Baroni, A.; Donnarumma, G.; Paoletti, I.; Longanesi-Cattani, I.; Bifulco, K.; Tufano, M.A.; Carriero, M.V. Antimicrobial human beta-defensin-2 stimulates migration, proliferation and tube formation of human umbilical vein endothelial cells. Peptides 2009, 30, 267–272. [Google Scholar] [CrossRef]
- Tokumaru, S.; Sayama, K.; Shirakata, Y.; Komatsuzawa, H.; Ouhara, K.; Hanakawa, Y.; Yahata, Y.; Dai, X.; Tohyama, M.; Nagai, H.; et al. Induction of keratinocyte migration via transactivation of the epidermal growth factor receptor by the antimicrobial peptide LL-37. J. Immunol. 2005, 175, 4662–4668. [Google Scholar] [CrossRef]
- Gallo, Y.L.; Gallo, R.L. AMPed Up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141. [Google Scholar]
- Scott, M.G.; Davidson, D.J.; Gold, M.R.; Bowdish, D.; Hancock, R.E.W. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J. Immunol. 2002, 169, 3883–3891. [Google Scholar] [CrossRef]
- Tjabringa, G.S.; Aarbiou, J.; Ninaber, D.K.; Drijfhout, J.W.; Sorensen, O.E.; Borregaard, N.; Rabe, K.F.; Hiemstra, P.S. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J. Immunol. 2003, 171, 6690–6696. [Google Scholar] [CrossRef]
- Kai-larsen, Y.; Agerberth, B. The role of the multifunctional peptide LL-37 in host defense. Front. Biosci. 2008, 13, 3760–3767. [Google Scholar] [CrossRef]
- Johansson, J.; Gudmundsson, G.H.; Rottenberg, M.E.; Berndt, K.D.; Agerberth, B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 1998, 273, 3718–3724. [Google Scholar] [CrossRef]
- Tjabringa, G.S.; Ninaber, D.K.; Drijfhout, J.W.; Rabe, K.F.; Hiemstra, P.S. Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. Int. Arch. Allergy Immunol. 2006, 140, 103–112. [Google Scholar] [CrossRef]
- Nagaoka, I.; Tamura, H.; Hirata, M. An antimicrobial cathelicidin peptide, human CAP18/LL-37, suppresses neutrophil apoptosis via the activation of formyl-peptide receptor-like 1 and P2X7. J. Immunol. 2006, 176, 3044–3052. [Google Scholar] [CrossRef]
- Barlow, P.G.; Li, Y.; Wilkinson, T.S.; Bowdish, D.M.E.; Lau, Y.E.; Cosseau, C.; Haslett, C.; Simpson, A.J.; Hancock, R.E.W.; Davidson, D.J. The human cationic host defense peptide LL-37 mediates contrasting effects on apoptotic pathways in different primary cells of the innate immune system. J. Leukoc. Biol. 2006, 80, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Aarbiou, J.; Tjabringa, G.S.; Verhoosel, R.M.; Ninaber, D.K.; White, S.R.; Peltenburg, L.T.C.; Rabe, K.F.; Hiemstra, P.S. Mechanisms of cell death induced by the neutrophil antimicrobial peptides alpha-defensins and LL-37. Inflamm. Res. 2006, 55, 119–127. [Google Scholar] [CrossRef]
- Ciornei, C.D.; Tapper, H.; Bjartell, A.; Sternby, N.H.; Bodelsson, M. Human antimicrobial peptide LL-37 is present in atherosclerotic plaques and induces death of vascular smooth muscle cells: A laboratory study. BMC Cardiovasc. Disord. 2006, 6, 49. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Nijnik, A.; Philpott, D.J. Modulating immunity as a therapy for bacterial infections. Nat. Rev. Microbiol. 2012, 10, 243–254. [Google Scholar] [CrossRef]
- Subbalakshmi, C.; Sitaram, N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol. Lett. 1998, 160, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Agerberth, B.; Boman, A.; Andersson, M.; JornvalL, H.; Mutt, V.; Boman, H.G. Isolation of three antibacterial peptides from pig intestine: Gastric inhibitory polypeptide(7–42), diazepam-binding inhibitor(32–86) and a novel factor, peptide 3910. Eur. J. Biochem. 1993, 216, 623–629. [Google Scholar] [CrossRef]
- Le, C.F.; Fang, C.M.; Sekaran, S.D. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef]
- Park, C.B.; Yi, K.S.; Matsuzaki, K.; Kim, M.S.; Kim, S.C. Structure-activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: The proline hinge is responsible for the cell-penetrating ability of buforin II. Proc. Natl. Acad. Sci. USA 2000, 97, 8245–8250. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, D.A.; Hurst, M.A.; Fujii, C.A.; Kung, A.H.; Ho, J.F.; Cheng, F.C.; Loury, D.J.; Fiddes, J.C. Protegrin-1: A broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob. Agents Chemother. 1997, 41, 1738–1742. [Google Scholar] [CrossRef]
- Aoki, W.; Ueda, M. Characterization of antimicrobial peptides toward the development of novel antibiotics. Pharmaceuticals 2013, 6, 1055–1081. [Google Scholar] [CrossRef]
- Liu, D.; DeGrado, W.F. De Novo Design, Synthesis, and Characterization of Antimicrobial β-Peptides. J. Am. Chem. Soc. 2001, 123, 7553–7559. [Google Scholar] [CrossRef]
- Loose, C.; Jensen, K.; Rigoutsos, I.; Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867–869. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monserrat-Martinez, A.; Gambin, Y.; Sierecki, E. Thinking Outside the Bug: Molecular Targets and Strategies to Overcome Antibiotic Resistance. Int. J. Mol. Sci. 2019, 20, 1255. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061255
Monserrat-Martinez A, Gambin Y, Sierecki E. Thinking Outside the Bug: Molecular Targets and Strategies to Overcome Antibiotic Resistance. International Journal of Molecular Sciences. 2019; 20(6):1255. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061255
Chicago/Turabian StyleMonserrat-Martinez, Ana, Yann Gambin, and Emma Sierecki. 2019. "Thinking Outside the Bug: Molecular Targets and Strategies to Overcome Antibiotic Resistance" International Journal of Molecular Sciences 20, no. 6: 1255. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061255