MicroRNAome Profile of Euphorbia kansui in Response to Methyl Jasmonate

Abstract
:1. Introduction
2. Results
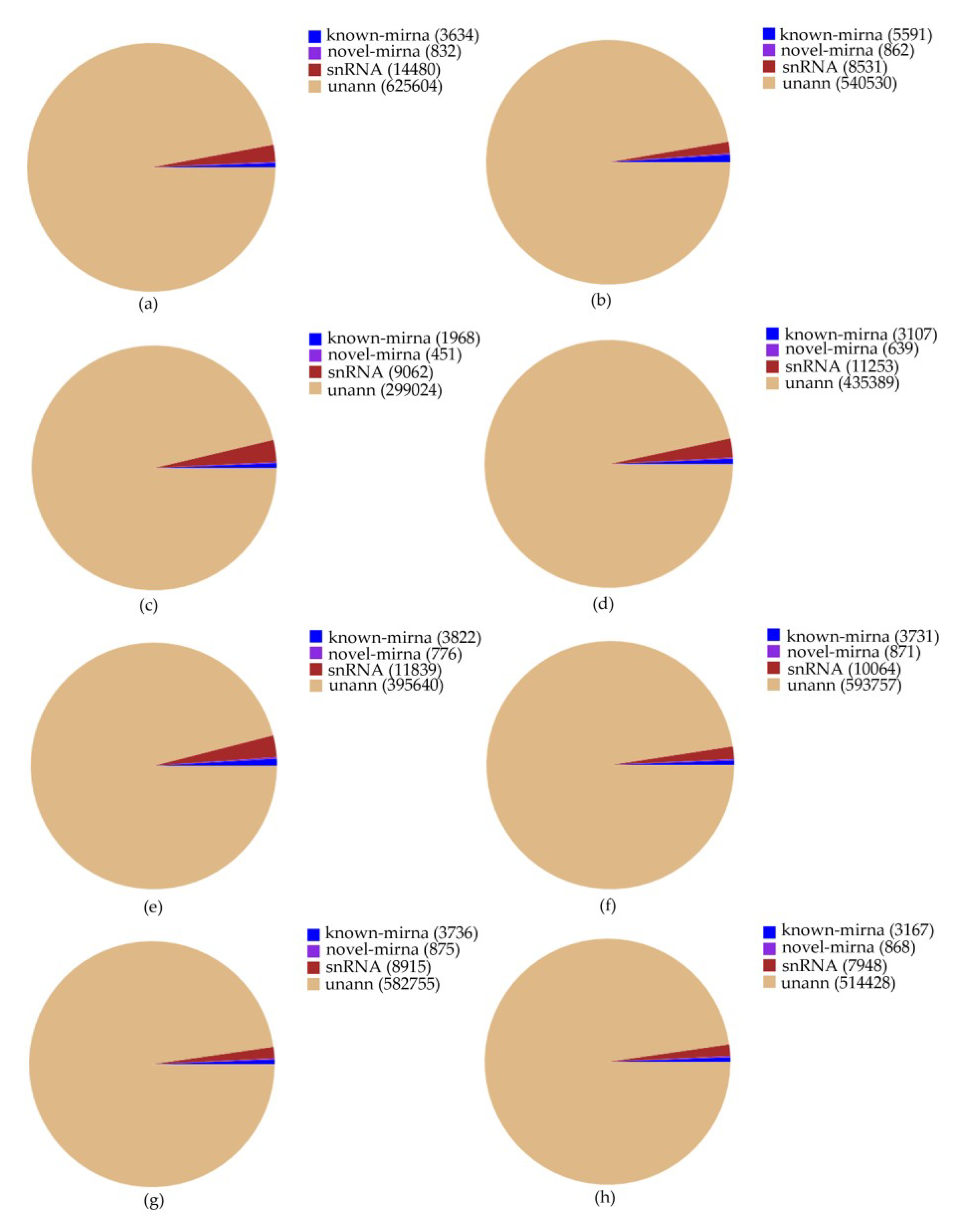
2.1. Sequencing of miRNA Library and Identification of miRNA Families
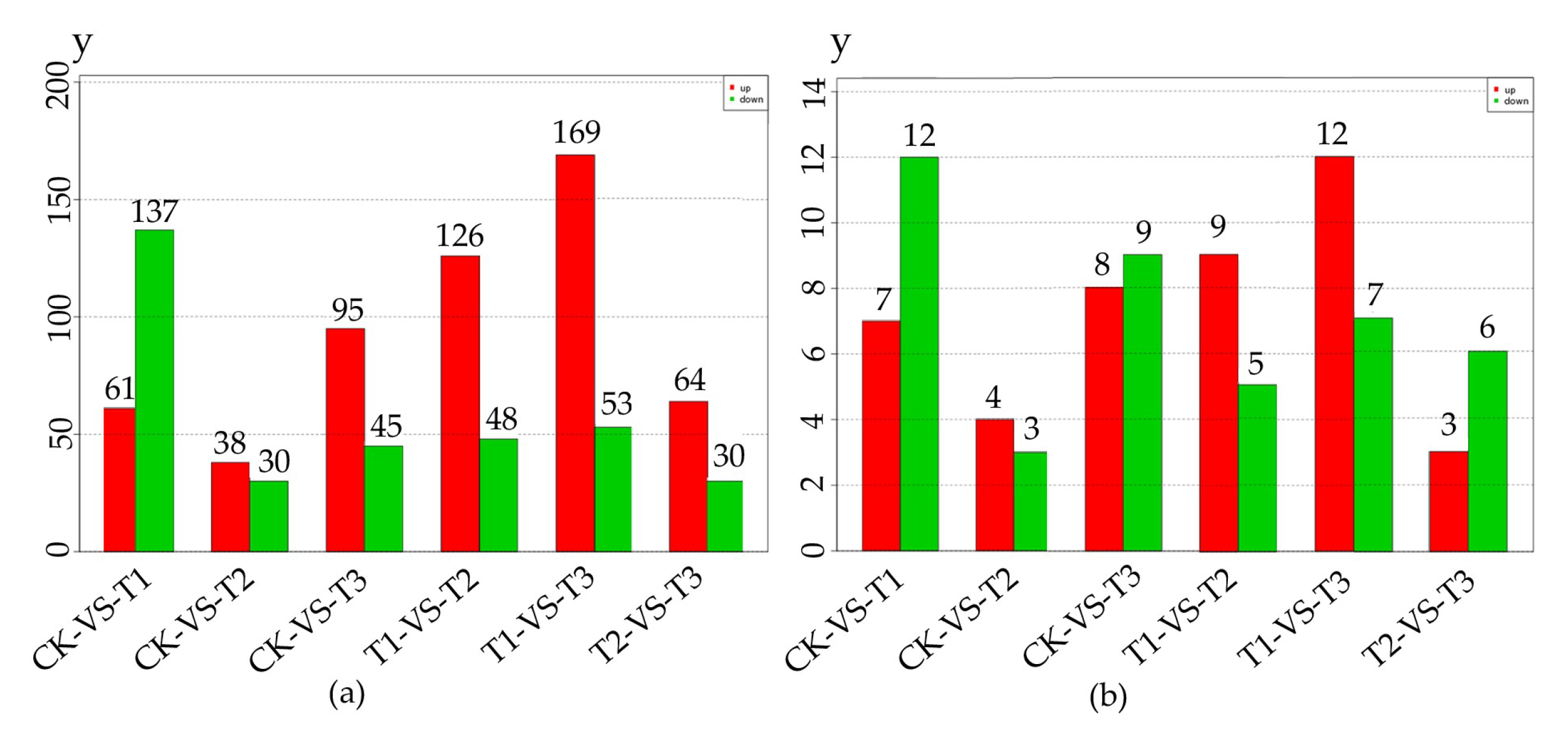
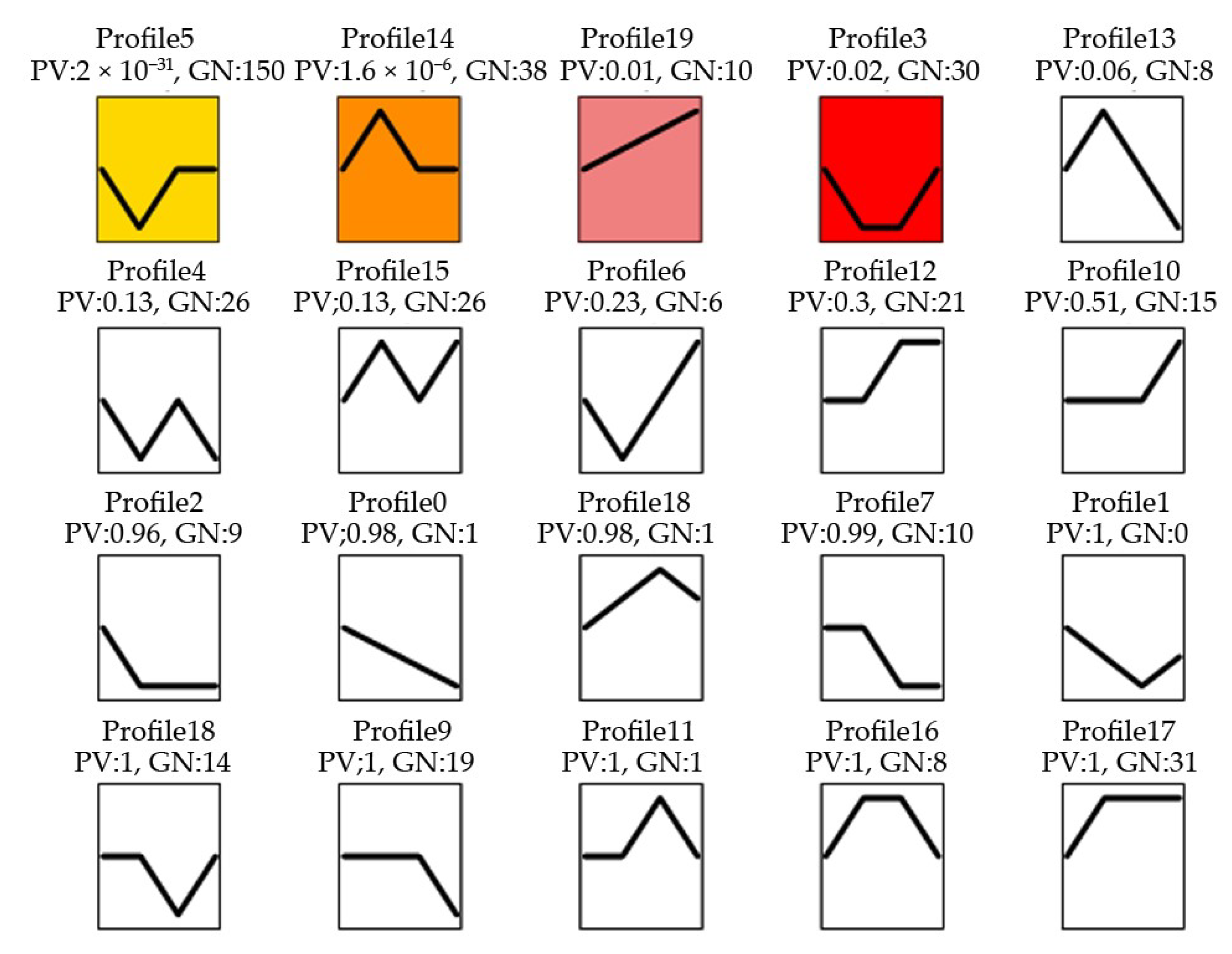
2.2. Differentially Expressed miRNAs (DEMs) Respond to Methyl Jasmonate (MeJA) Treatment
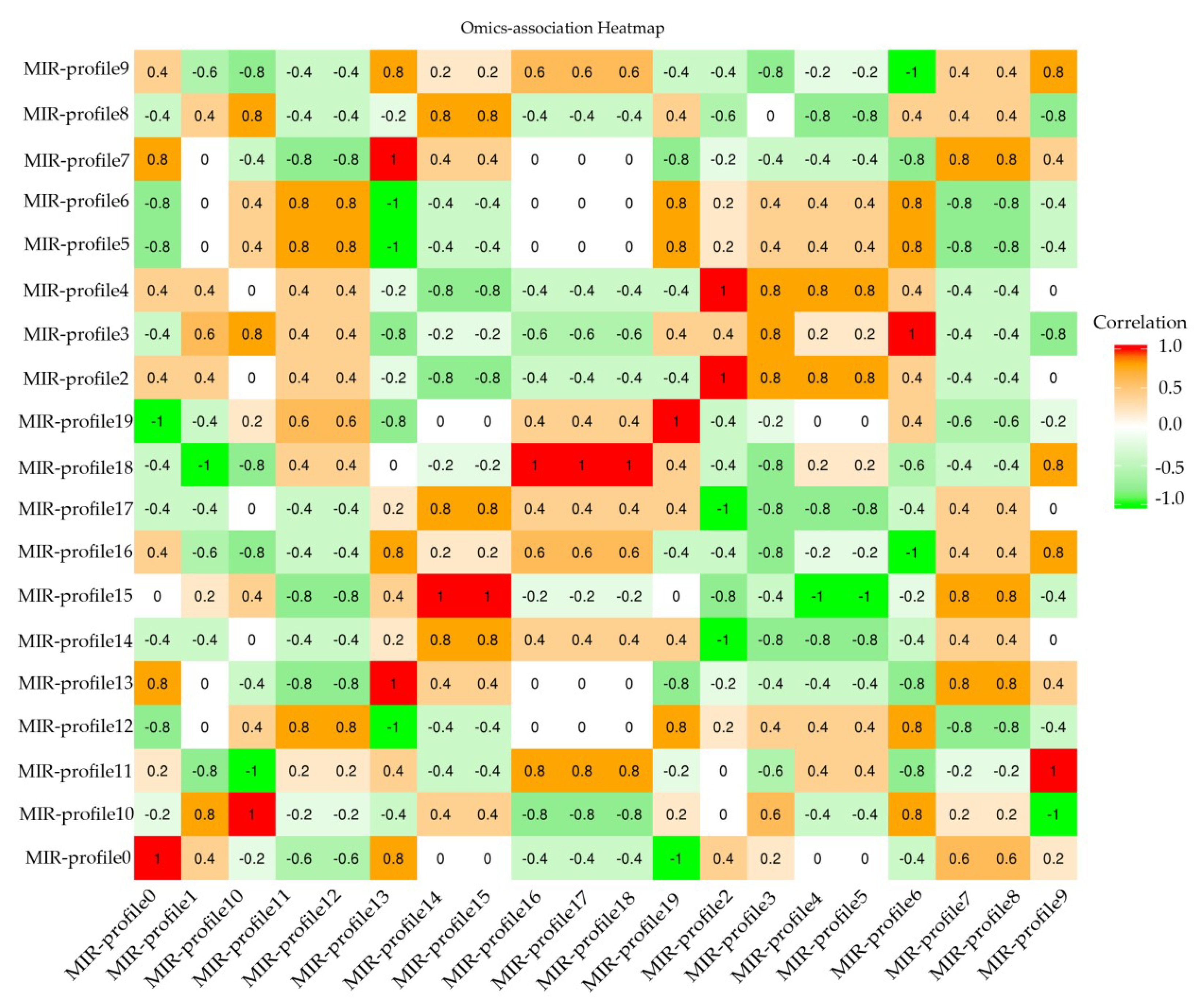
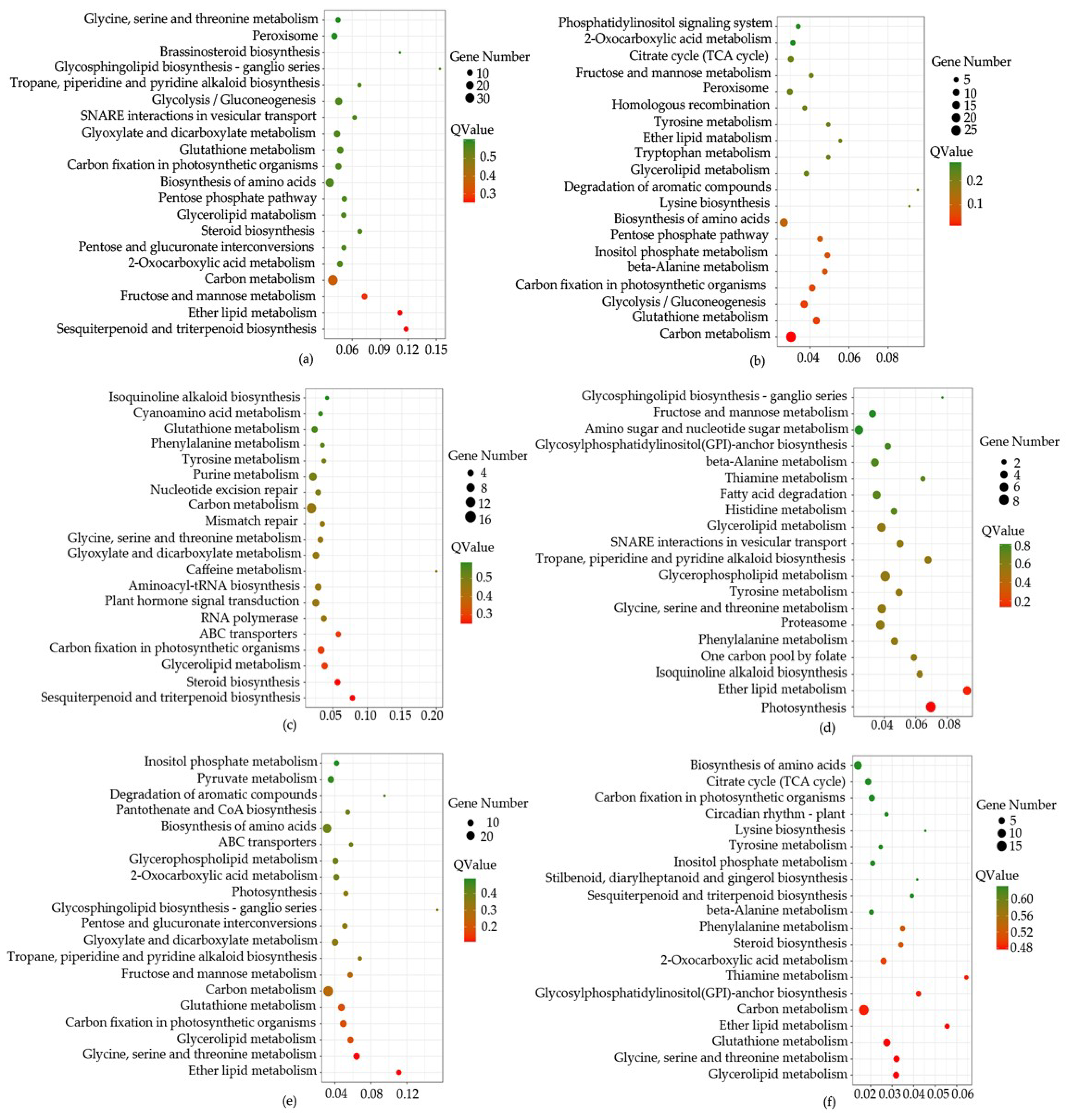
2.3. Target RNA Prediction and Enrichment Analysis
2.4. Differentially Expressed Genes (DEGs) and DEMs in Terpenoid Biosynthesis Associated with MeJA Treatment
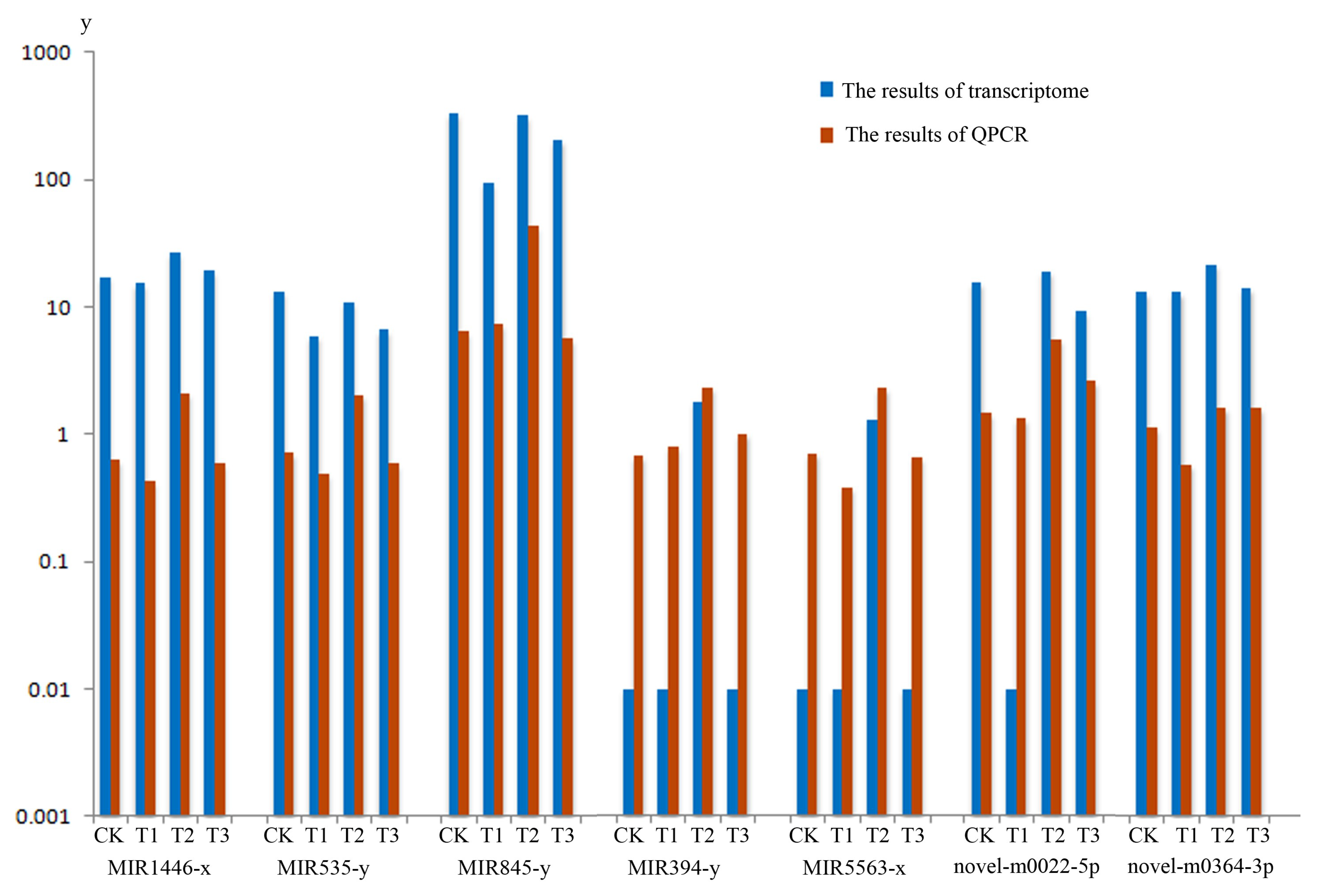
2.5. Verification of DEM Profiles Associated with MeJA Treatment with Real-time Quantitative PCR Detecting System (QPCR)
3. Discussion
3.1. miRNA Involved in the Response of Euphorbia kansui Plant to Exogenous MeJA
3.2. miRNA Involved in the Regulation of Terpenoid Biosynthesis after Exogenous MeJA Treatment in Euphorbia kansui
3.3. miRNA Involved in Other Pathways
4. Materials and Methods
4.1. Plant Material and MeJA Treatment
4.2. MicroRNA Sequencing
4.3. Sequencing Data Processing and Analysis
4.4. miRNA Expression Profiles
4.5. Differentially Expressed miRNAs Analysis and Target Prediction
4.6. Target Gene Prediction
4.7. Quantitative PCR (QPCR) Analysis and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Q.; Zhou, Q.R.; Lou, J.W.; Chen, P.D.; Yao, W.F.; Tao, W.W.; Tang, Y.P.; Dai, G.C.; Wang, K.; Zhang, L. Chemical Constituents from Euphorbia kansui. Molecular 2017, 22, 2176. [Google Scholar] [CrossRef]
- Zheng, W.F.; Cui, Z.; Zhu, Q. Cytotoxicity and antiviral activity of the compounds from Euphorbia kansui. Planta Med. 1998, 64, 754–756. [Google Scholar] [CrossRef]
- Li, X.R.; Zhang, Y.D.; Tang, H.H. Study of auxiliary therapeutic effect of kansui root on patients with severe acute pancreatitis. China J. Mod. Med. 2002, 12, 7–9. [Google Scholar]
- Yan, X.; Zhang, L.; Guo, J.; Cao, Y.; Shang, E.; Tang, Y.; Ding, A.; Duan, J.A. Processing of kansui roots stir-baked with vinegar reduces kansui-induced hepatocyte cytotoxicity by decreasing the contents of toxic terpenoids and regulating the cell apoptosis pathway. Molecular 2014, 19, 7237–7254. [Google Scholar] [CrossRef]
- Hsieh, W.T.; Lin, H.Y.; Chen, J.H.; Kuo, Y.H.; Fan, M.J.; Wu, R.S.; Wu, K.C.; Wood, W.G.; Chung, J.G. Latex of euphorbia antiquorum induces apoptosis in human cervical cancer cells via c-jun n-terminal kinase activation and reactive oxygen species production. Nutr. Cancer 2011, 63, 1339–1347. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Cai, X.; Hu, Z.H. Research progress on biology, chemical constituents in Euphorbia kansui, and their pharmacological effects. Chin. Tradit. Herb. Drugs 2014, 45, 3029–3033. [Google Scholar] [CrossRef]
- Kim, J.W.; Oh, S.R.; Ahn, K.S.; Kang, H.B.; Kwon, O.K.; Kim, S.H.; Son, J.M.; Song, H.H.; Lee, H.G.; Jeon, B.H.; et al. Pharmaceutical composition for treating wounds or revitalizing skin comprising Euphorbia kansui extracts, fractions thereof or diterpene compounds separated from the fractions as active ingredient. Uspto Appl. 2015, 20150093456. [Google Scholar]
- Fei, D.Q.; Dong, L.L.; Qi, F.M.; Fan, G.X.; Li, H.H.; Li, Z.Y.; Zhang, Z.X. Euphorikanin A, a diterpenoid lactone with a fused 5/6/7/3 ring system from Euphorbia kansui. Org. Lett. 2016, 18, 2844–2847. [Google Scholar] [CrossRef] [PubMed]
- Wasternack, C.; Hause, B. Jasmonates: Biosynthesis, perception, signal transduction and action in plant stress response, growth and development. An update to the 2007 review in Annals of Botany. Ann. Bot. 2013, 111, 1021–1058. [Google Scholar] [CrossRef]
- Wasternack, C. Jasmonates: An update on biosynthesis, signal transduction and action in plant stress response, growth and development. Ann. Bot. 2007, 100, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Jiang, Y.M.; Wang, N.; Xia, B.; Jiang, Y.L.; Li, X.D.; Zhang, Z.Z.; Li, Y.K.; Wang, R. Identification and differential regulation of microRNAs in response to methyl jasmonate treatment in Lycoris aurea by deep sequencing. BMC Genom. 2016, 17, 789–803. [Google Scholar] [CrossRef]
- De, G.N.; Gholami, A.; Goormachtig, S.; Goossens, A. Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends Plant Sci. 2012, 17, 349–359. [Google Scholar] [CrossRef]
- Vasconsuelo, A.; Boland, R. Molecular aspects of the early stages of elicitation of secondary metabolites in plants. Plant Sci. 2007, 172, 861–875. [Google Scholar] [CrossRef]
- Kuźma, L.; Kalemba, D.; Rózalski, M.; Rózalska, B.; Wieckowskaszakiel, M.; Krajewska, U.; Wysokińska, H. Chemical composition and biological activities of essential oil from Salvia sclarea plants regenerated in vitro. Molecules 2009, 14, 1438–1447. [Google Scholar] [CrossRef]
- Cheng, Q.; He, Y.; Li, G.; Liu, Y.; Gao, W.; Huang, L. Effects of combined elicitors on tanshinone metabolic profiling and SmCPS expression in Salvia miltiorrhiza hairy root cultures. Molecules 2013, 18, 7473–7485. [Google Scholar] [CrossRef]
- Zhang, S.; Yan, Y.; Wang, B.; Liang, Z.; Liu, Y.; Liu, F.; Qi, Z. Selective responses of enzymes in the two parallel pathways of rosmarinic acid biosynthetic pathway to elicitors in Salvia miltiorrhiza hairy root cultures. J. Biosci. Bioeng. 2014, 117, 645–651. [Google Scholar] [CrossRef]
- Wang, H.; Yang, J.F.; Deng, K.; He, X.Y.; Zhan, R.T.; Tang, L. Methyl Jasmonate affects metabolism and gene transcription of volatile terpenoids from Amomum villosum Lour. World Sci. Tech.-Mod. Tradit. Chin. Med. Mater Med. 2014, 16, 1528–1536. [Google Scholar]
- Zhang, W.J.; Cao, X.Y.; Jiang, J.H. Triterpene biosynthesis in Euphorbia pekinensis induced by methyl jasmonate. Guihaia 2015, 35, 590–596. [Google Scholar]
- Zhang, L.; Yang, B.; Lu, B.; Kai, G.; Wang, Z.; Xia, Y.; Ding, R.; Zhang, H.; Sun, X.; Chen, W.; et al. Tropane alkaloids production in transgenic Hyoscyamus niger hairy root cultures over-expressing putrescine n-methyltransferase is methyl jasmonate-dependent. Planta 2007, 225, 887–896. [Google Scholar] [CrossRef]
- Ferreira, T.H.; Gentile, A.; Vilela, R.D.; Costa, G.G.; Dias, L.; Endres, L.; Menossi, M. microRNAs associated with drought response in the bioenergy crop sugarcane (Saccharum spp.). PLoS ONE 2012, 7, e46703. [Google Scholar] [CrossRef]
- Sunkar, R.; Li, Y.F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef]
- Luo, Y.; Guo, Z.; Li, L. Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev. Biol. 2013, 380, 133–144. [Google Scholar] [CrossRef] [Green Version]
- He, X.H.; Sun, Q.; Jiang, H.Z.; Zhu, X.Y.; Mo, J.C.; Long, L.; Xiang, L.X.; Xie, Y.F.; Shi, Y.Z.; Yuan, Y.L.; et al. Identification of novel microRNAs in the Verticillium wilt-resistant upland cotton variety KV-1 by high-throughput sequencing. Springerplus 2014, 3, 564. [Google Scholar] [CrossRef]
- Budak, H.; Akpinar, P.J. Plant miRNAs: Biogenesis, organization and origins. Funct. Integr. Genom. 2015, 15, 523–531. [Google Scholar] [CrossRef]
- Pareek, M.; Yogindran, S.; Mukherjee, S.K.; Rajam, M.V. Plant MicroRNAs: Biogenesis, Functions and Applications. Plant Biotechnol. J. 2015, 32, 639–661. [Google Scholar] [CrossRef]
- Budak, H.; Kabtara, M.; Bulut, R.; Akpinar, B.A. Stress responsive miRNAs and isomiRs in cereals. Plant Sci. 2015, 235, 1–13. [Google Scholar] [CrossRef]
- Repka, V.; Čarná, M. Post-Transcriptional Gene Silencing Conferred by the Ectopic Expression of the Grapevine miRNA-g1 and Inhibition of the Response by Anti-miRNA-g1 Inhibitor. Agriculture 2011, 57, 137–143. [Google Scholar] [CrossRef]
- Biswas, C.; Dey, P.; Karmakar, P.G.; Satpathy, S. Next-generation sequencing and micro RNAs analysis reveal SA/MeJA1/ABA pathway genes mediated systemic acquired resistance (SAR) and its master regulation via production of phased, trans-acting siRNAs against stem rot pathogen Macrophomina phaseolina in a RIL population of jute (Corchorus capsularis). Physiol. Mol. Plant Pathol. 2014, 87, 76–85. [Google Scholar] [CrossRef]
- Shriram, V.; Kumar, V.; Devarumath, R.M.; Khare, T.S.; Wani, S.H. MicroRNAs as potential targets for abiotic stress tolerance in plants. Front. Plant Sci. 2016, 7, 817–835. [Google Scholar] [CrossRef]
- Bala, A.A.; Melda, K.; Hikmet, B. Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Funct. Integr. Genom. 2015, 15, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Gálvez, S.; Mérida-García, R.; Camino, C.; Borrill, P.; Abrouk, M.; Ramírez-González, R.H.; Biyiklioglu, S.; Amil-Ruiz, F.; Iwgsc, T.; Dorado, G.; et al. Hotspots in the genomic architecture of field drought responses in wheat as breeding targets. Funct. Integr. Genom. 2018, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Akpinar, A. Dehydration stress-responsive miRNA in Brachypodium distachyon: Evident by genome-wide screening of microRNAs expression. OMICS 2011, 15, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Biyiklioglu, S.; Alptekin, B.; Akpinar, B.A.; Varella, A.C.; Hofland, M.L.; Weaver, D.K.; Bothner, B.; Buthner, B.; Budak, H. A large-scale multiomics analysis of wheat stem solidness and the wheat stem sawfly feeding response, and syntenic associations in barley, Brachypodium, and rice. Funct. Integr. Genom. 2018, 18, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Xie, K.; Xiong, L. Global expression profiling of rice microRNAs by one-tube stem-loop reverse transcription quantitative PCR revealed important roles of microRNAs in abiotic stress responses. Mol. Genet. Genom. 2010, 284, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Kulcheski, F.R.; Oliveira, L.F.D.; Molina, L.G.; Almerão, M.P.; Rodrigues, F.A.; Marcolino, J.; Barbosa, J.F.; Stolf-Moreira, R.; Nepomuceno, A.L.; Marcelino-Guimarães, F.C. Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genom. 2011, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qin, Y.; Duan, H.; Yin, W.; Xia, X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J. Exp. Bot. 2011, 62, 3765–3779. [Google Scholar] [CrossRef]
- Qin, Y.; Duan, Z.; Xia, X.; Yin, W. Expression profiles of precursor and mature microRNAsunder dehydration and high salinity shock in Populus euphratica. Plant Cell Rep. 2011, 30, 1893–1907. [Google Scholar] [CrossRef]
- Song, H.; Zhang, X.; Shi, C.; Wang, S.S.; Wu, A.L.; Wei, C.L. Selection and verification of candidate reference genes for mature MicroRNA expression by quantitative RT-PCR in the tea plant (Camellia sinensis). Genes 2016, 7, 25. [Google Scholar] [CrossRef]
- Li, H.; Dong, Y.C.; Chang, J.J.; He, J.; Chen, H.J.; Liu, Q.Y.; Wei, C.H.; Ma, J.X.; Zhang, Y.; Yang, J.Q.; et al. High-throughput MicroRNA and mRNA sequencing reveals that MicroRNAs may be involved in melatonin-mediated cold tolerance in Citrullus lanatusl. Front. Plant Sci. 2016, 7, 1231. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, F.; Li, J.; Chen, J.P.; Zhang, H.M. Integrative Analysis of the microRNAome and Transcriptome Illuminates the Response of Susceptible Rice Plants to Rice Stripe Virus. PLoS ONE 2016, 11, e0146946. [Google Scholar] [CrossRef]
- Sun, Q.; Du, X.; Cai, C.; Long, L.; Zhang, S.; Qiao, P.; Wang, W.; Zhou, K.; Wang, G.; Liu, X.; et al. To be a flower or fruiting branch: Insights revealed by mRNA and small RNA transcriptomes from different cotton developmental stages. Sci. Rep. 2016, 17, 23212. [Google Scholar] [CrossRef]
- Chen, J.; Su, P.; Chen, P.Y.; Li, Q.; Yuan, X.L.; Liu, Z. Insights into the cotton anther development through association analysis of transcriptomic and small RNA sequencing. BMC Plant Biol. 2018, 18, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Watanabe, Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. USA 2004, 101, 12753–12758. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, P.; Sakvarelidze-Achard, L.; Bruun-Rasmussen, M.; Dunoyer, P.; Yamamoto, Y.Y.; Sieburth, L.; Voinnet, O. Widespread translational inhibition by plant miRNAs and siRNAs. Science 2008, 320, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.W.; Zhang, C.; Miller, M.; Palmer, G.; Whiteley, M.; Tholl, D.; Chen, Z.J. Cis- and trans-regulation of miR163 and target genes confers natural variation of secondary metabolites in two Arabidopsis species and their allopolyploids. Plant Cell 2011, 23, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.A.; MacLean, D.; Jikumaru, Y.; Hill, L.; Yamaguchi, S.; Kamiya, Y.; Jones, J.D. The microRNA miR393 re-directs secondary metabolite biosynthesis away from camalexin and towards glucosinolates. Plant J. 2011, 67, 218–231. [Google Scholar] [CrossRef]
- Boke, H.; Ozhuner, E.; Turktas, M.; Parmaksiz, I.; Ozcan, S.; Unver, T. Regulation of the alkaloid biosynthesis by miRNA in Opium poppy. Plant Biotechnol. J. 2015, 13, 409–420. [Google Scholar] [CrossRef]
- Jia, X.; Shen, J.; Liu, H.; Li, F.; Ding, N.; Gao, C.; Pattanaik, S.; Patra, B.; Li, R.; Yuan, L. Small tandem target mimic-mediated blockage of microRNA858 induces anthocyanin accumulation in tomato. Planta 2015, 242, 283–293. [Google Scholar] [CrossRef]
- Sharma, D.; Tiwari, M.; Pandey, A.; Bhatia, C.; Sharma, A.; Trivedi, P.K. MicroRNA858 is a potential regulator of phenylpropanoid pathway and plant development. Plant Physiol. 2016, 171, 944–959. [Google Scholar] [CrossRef]
- Shen, E.M.; Singh, S.K.; Ghosh, J.S.; Patra, B.; Paul, P.; Yuan, L.; Pattanaik, S. The mirnaome of Catharanthus roseus: Identification, expression analysis, and potential roles of micrornas in regulation of terpenoid indole alkaloid biosynthesis. Sci. Rep. 2017, 7, 43027. [Google Scholar] [CrossRef]
- Hazra, S.; Bhattacharyya, D.; Chattopadhyay, S. Methyl Jasmonate regulates podophyllotoxin accumulation in Podophyllum hexandrum by altering the ROS-responsive podophyllotoxin pathway gene expression additionally through the down regulation of few interfering miRNAs. Front. Plant Sci. 2017, 8, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Wang, W.; Zheng, Y.; Chen, X.; Bo, W.; Song, S.; Zhang, W.; Peng, M. Conservation and divergence of microRNAs and their functions in Euphorbiaceous plants. Nucleic Acids Res. 2009, 3, 981–995. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhu, J.K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.F.; Wang, G.D.; Zhang, W.X. UV-B responsive microRNA genes in Arabidopsis thaliana. Mol. Syst. Biol. 2007, 3, 103–112. [Google Scholar] [CrossRef]
- Dugas, D.V.; Bartel, B. Sucrose induction of Arabidopsis miR398 represses two Cu/Zn superoxide dismutases. Plant Mol. Biol. 2008, 67, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.F.; Sun, Y.H.; Chiang, V.L. Stress-responsive microRNAs in Populus. Plant J. 2008, 55, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, G.; Saini, A.; Sunkar, R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta 2009, 229, 1009–1014. [Google Scholar] [CrossRef]
- Gong, B.; Sun, S.S.; Yan, Y.Y.; Jing, X.; Shi, Q.H. Glutathione Metabolism and Its Function in Higher Plants Adapting to Stress. In Antioxidants and Antioxidant Enzymes in Higher Plants; Gupta, D., Palma, J., Corpas, F., Eds.; Springer: Cham, Switzerland, 2018; pp. 181–205. [Google Scholar]
- Harshavardhan, V.T.; Wu, T.M.; Hong, C.Y. Glutathione Reductase and Abiotic Stress Tolerance in Plants. In Glutathione in Plant Growth, Development, and Stress Tolerance; Hossain, M., Mostofa, M., Diaz-Vivancos, P., Burritt, D., Fujita, M., Tran, L.S., Eds.; Springer: Cham, Switzerland, 2017; pp. 265–286. [Google Scholar]
- Miao, L.H.; Pan, W.J.; Lin, Y.; Ge, X.P.; Liu, B.; Ren, M.C.; Zhou, Q.L. Bioinformatic prediction and analysis of glucolipid metabolic regulation by miR-34a in Megalobrama amblycephala. Genes Genom. 2017, 39, 1407–1417. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | DEGs Genes with Pathway Annotation | |||||
|---|---|---|---|---|---|---|
| CK-VS-T1 (318) | CK-VS-T2 (174) | CK-VS-T3 (156) | T1-VS-T2 (218) | T1-VS-T3 (262) | T2-VS-T3 (125) | |
| Terpenoid backbone biosynthesis | 2 (0.63%) | 1 (0.57%) | 1 (0.64%) | 4 (1.83%) | 4 (1.53%) | 1 (0.8%) |
| Ubiquinone and other terpenoid-quinone biosynthesis | 3 (0.94%) | 2 (1.15%) | 1 (0.64%) | 1 (0.46%) | 2 (0.76%) | 2 (1.6%) |
| Steroid biosynthesis | 6 (1.89%) | 2 (1.15%) | 5 (3.21%) | 1 (0.46%) | 3 (1.15%) | 3 (2.4%) |
| Sesquiterpenoid and triterpenoid biosynthesis | 6 (1.89%) | 1 (0.57%) | 4(2.56%) | 2 (0.92%) | 3 (1.15%) | 2 (1.6%) |
| Diterpenoid biosynthesis | 0 | 0 | 0 | 1 (0.46%) | 2 (0.76%) | 0 |
| RNA Name | Small RNA Primer |
|---|---|
| Kansui U6-11537-F | CATACAAATACCACGAAACCTC |
| Kansui U6-11537-R | TGATCTGCTTCACGATGCTAC |
| MIR1446-x | TTGAACTCTCTCCCTCAT |
| MIR535-y | TTGACAAAGAGAGAGAGCACA |
| MIR-845-y | TGCTCTGATACCACTTGA |
| MIR-394-y | CTCTGTTGGTCTCTCTTTG |
| MIR-5563-x | TATCAGGCAACTCTTTCC |
| novel-m0022-5p | TTGGAATACTGTTGAGAAGCAC |
| novel-m0364-3p | TTTGGGCATGTTGGATAAAA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Tian, Z.; Zhang, Q.; Zhang, Y.; Wang, M.; Fang, X.; Shi, W.; Cai, X. MicroRNAome Profile of Euphorbia kansui in Response to Methyl Jasmonate. Int. J. Mol. Sci. 2019, 20, 1267. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061267
Li P, Tian Z, Zhang Q, Zhang Y, Wang M, Fang X, Shi W, Cai X. MicroRNAome Profile of Euphorbia kansui in Response to Methyl Jasmonate. International Journal of Molecular Sciences. 2019; 20(6):1267. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061267
Chicago/Turabian StyleLi, Peng, Zheni Tian, Qing Zhang, Yue Zhang, Meng Wang, Xiaoai Fang, Wenjing Shi, and Xia Cai. 2019. "MicroRNAome Profile of Euphorbia kansui in Response to Methyl Jasmonate" International Journal of Molecular Sciences 20, no. 6: 1267. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061267