The Selection of NFκB Inhibitors to Block Inflammation and Induce Sensitisation to FasL-Induced Apoptosis in HNSCC Cell Lines Is Critical for Their Use as a Prospective Cancer Therapy

 ,
,
Abstract
:1. Introduction
2. Results
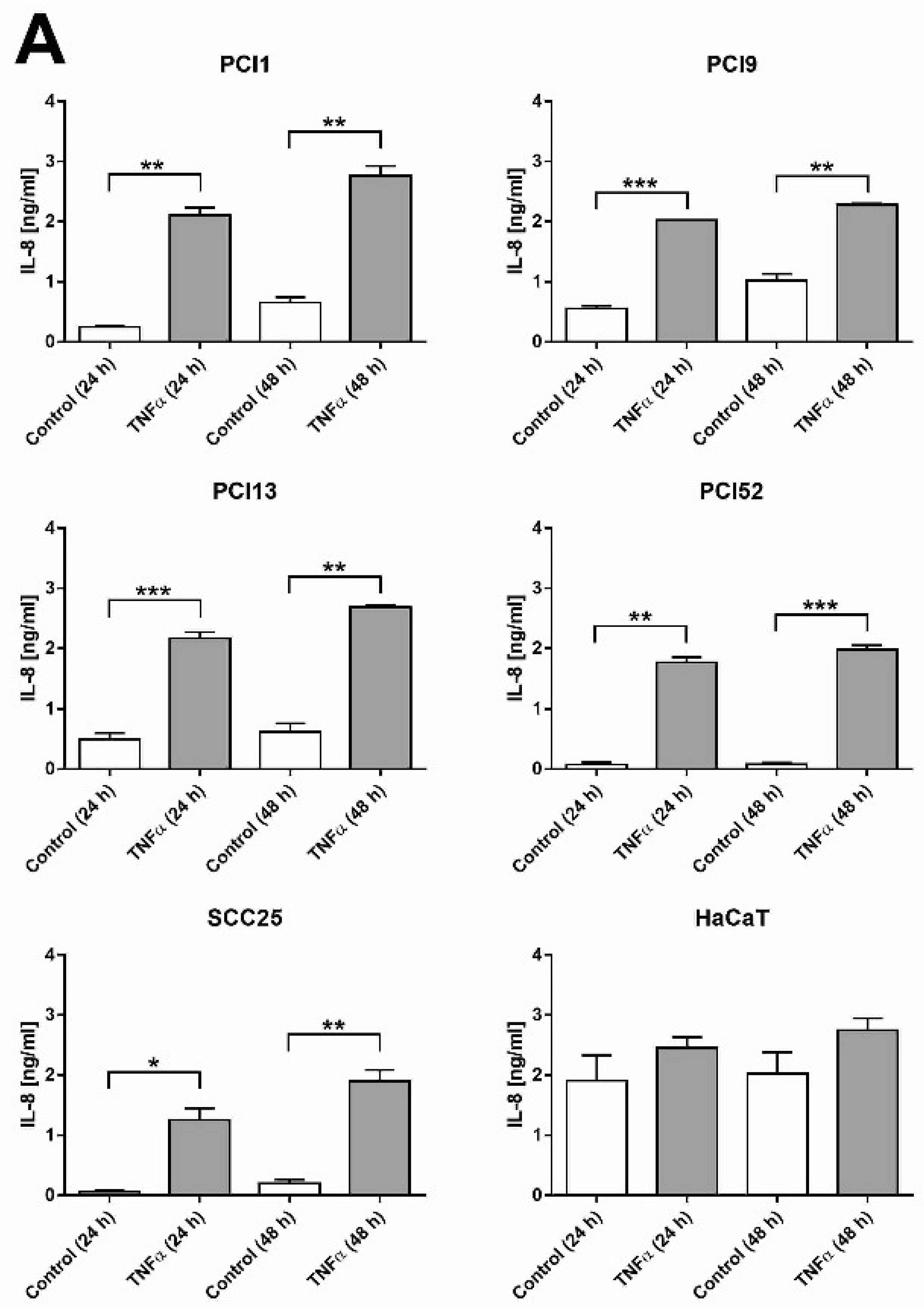
2.1. HNSCC Cell Lines Secrete the Pro-Inflammatory Marker IL-8 and Are Responsive to TNFα
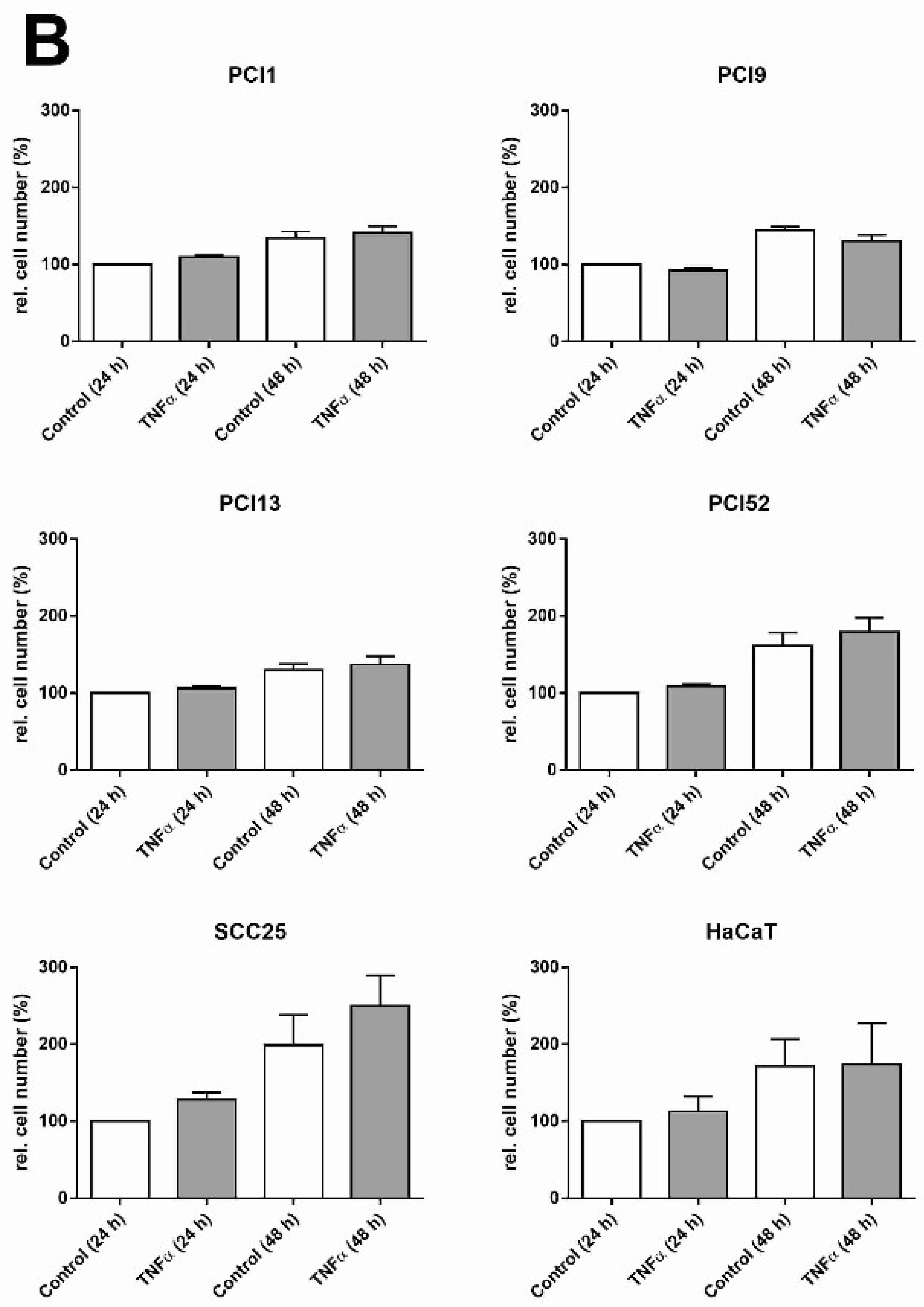
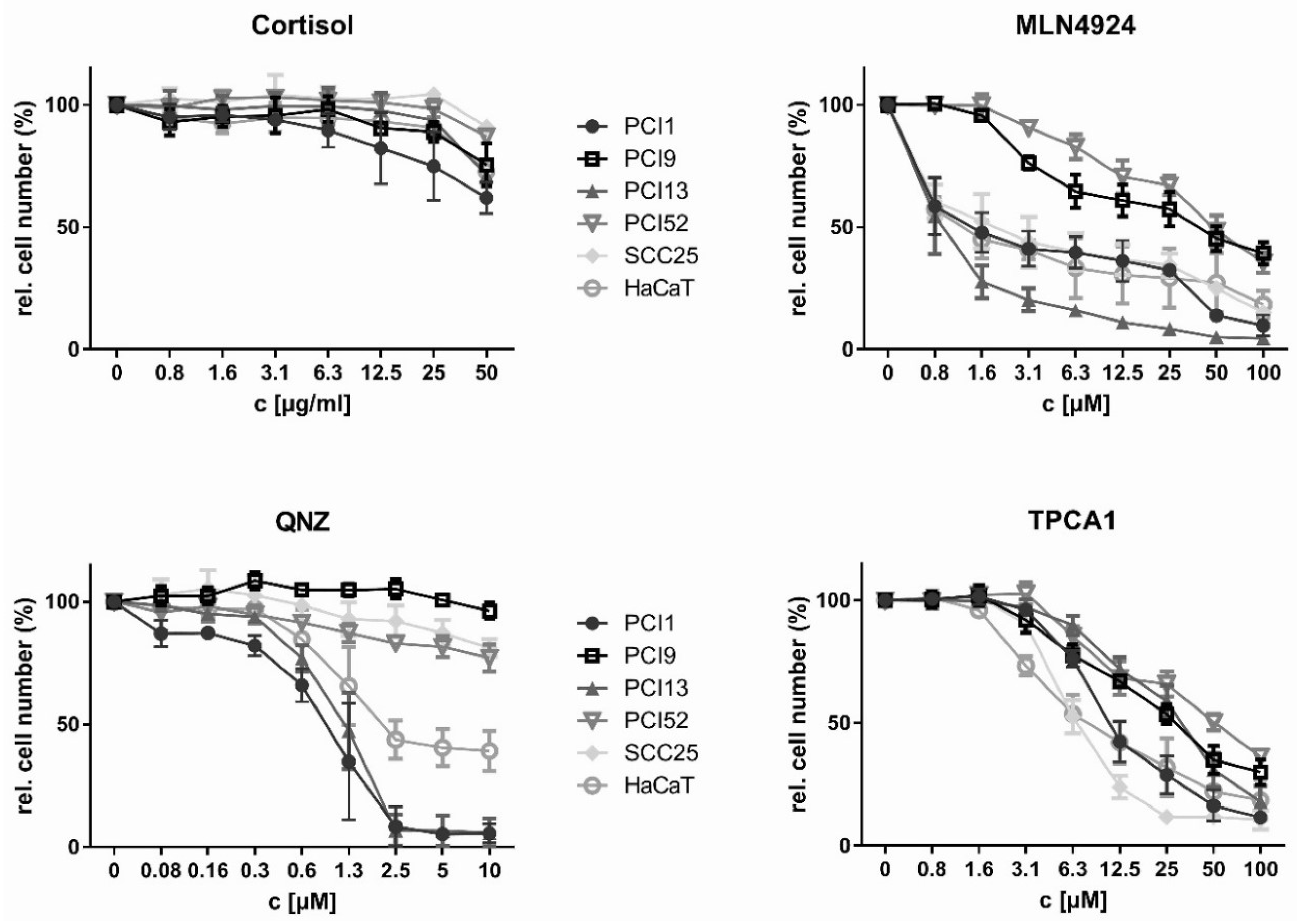
2.2. Inhibition of HNSCC Cell Proliferation by NFκB Inhibitors
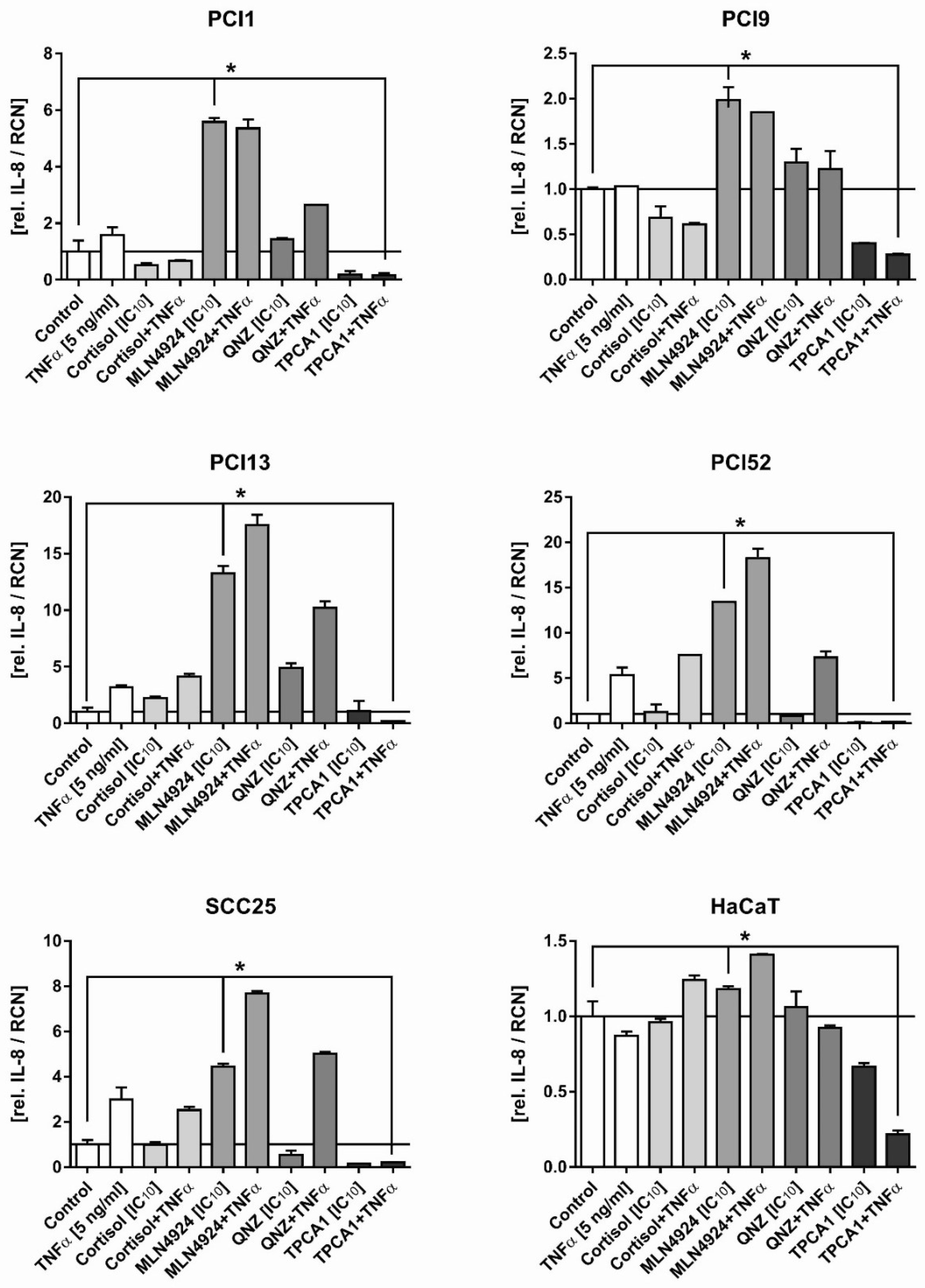
2.3. Inhibition of the Inflammatory Response in HNSCC Cells by NFκB Inhibitors
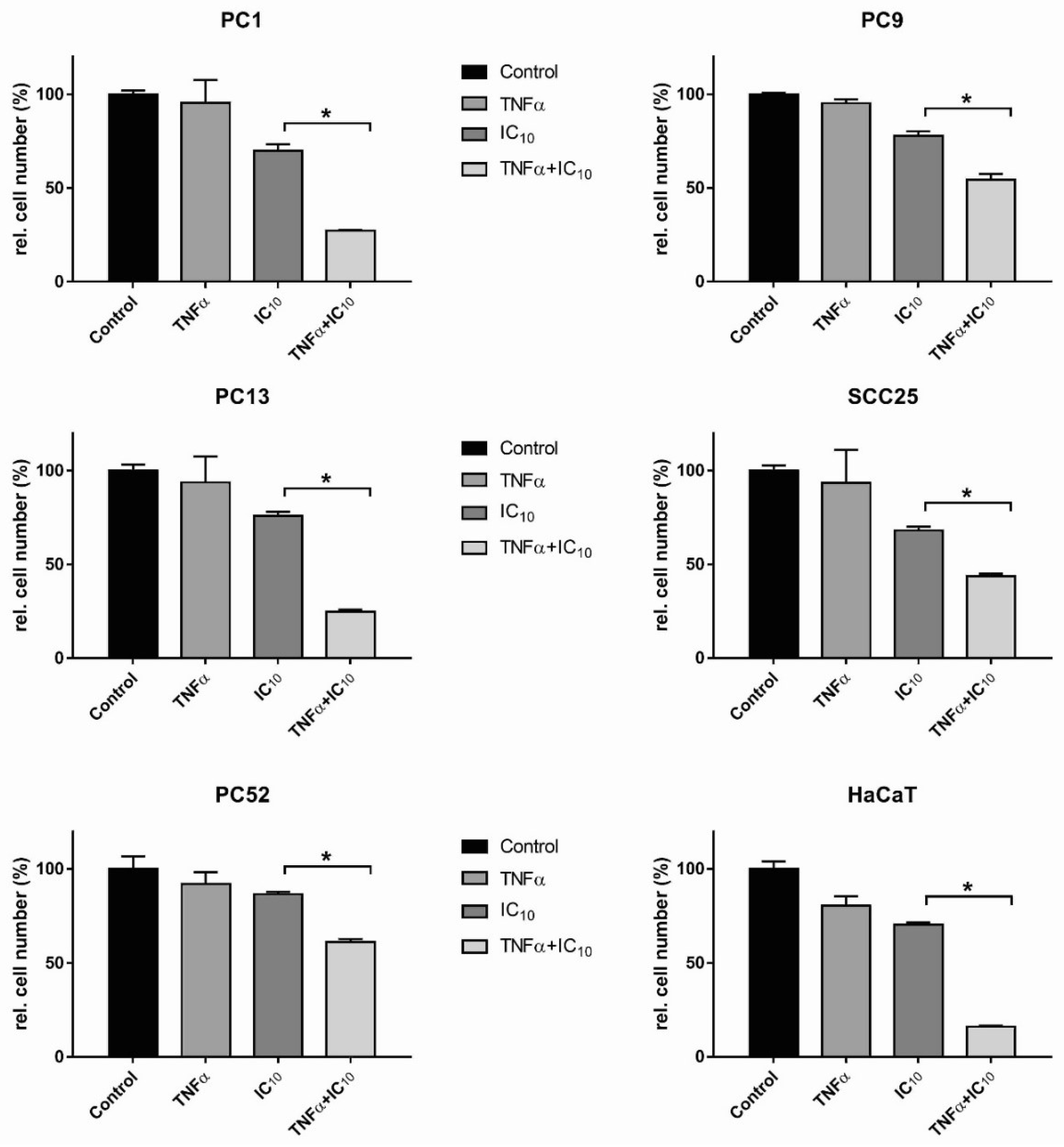
2.4. TNFα Induced HNSCC Cell Death after TPCA1 Stimulation
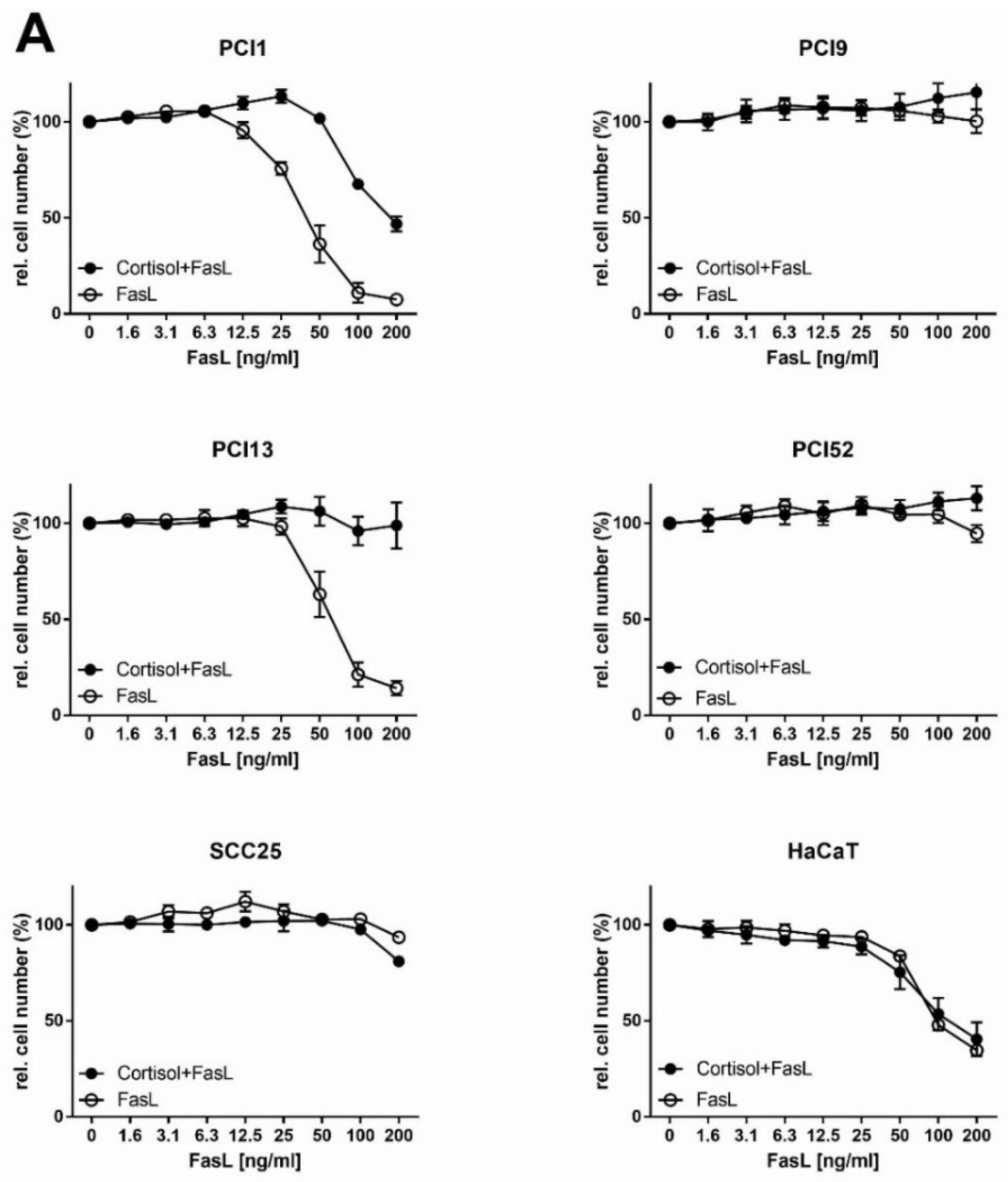
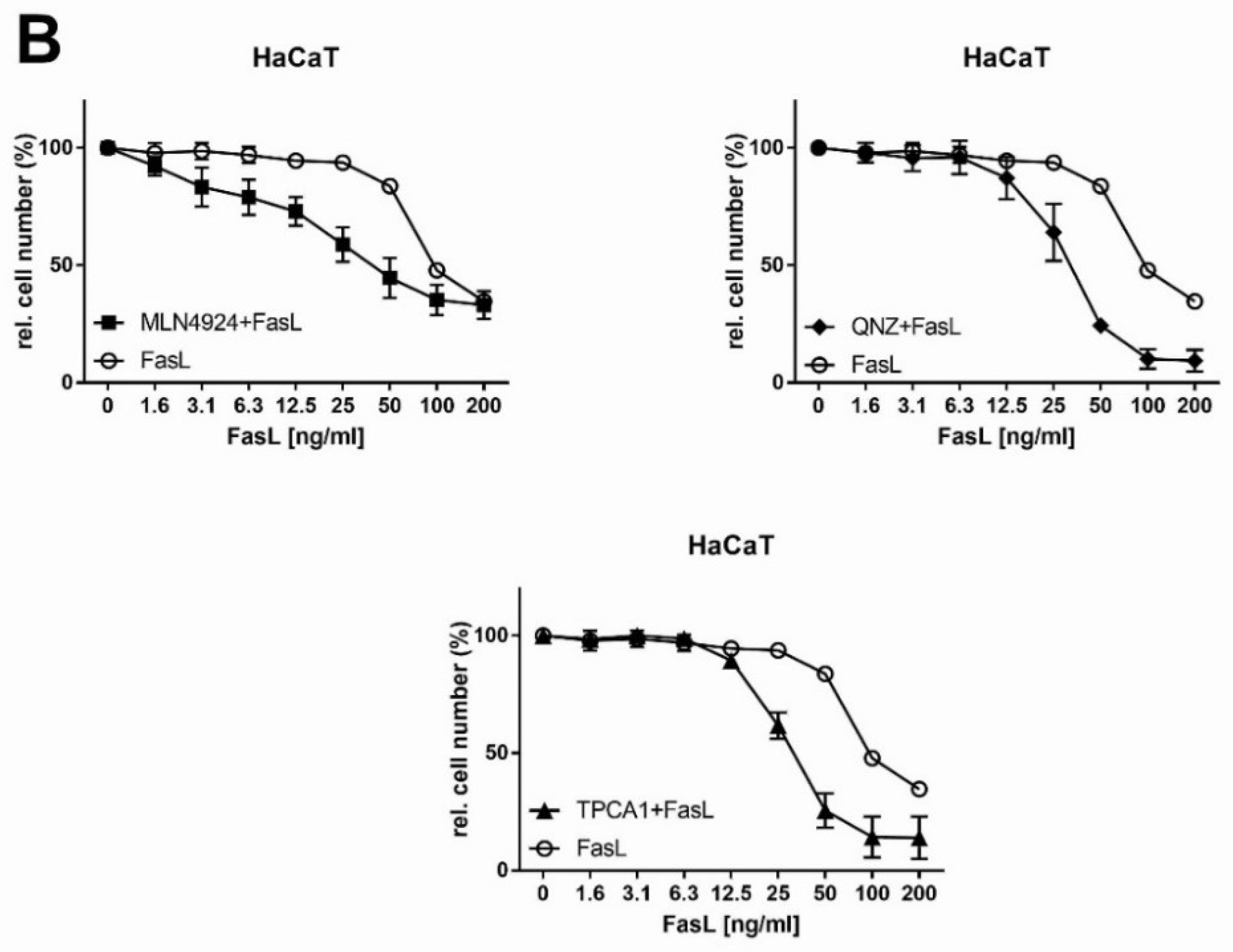
2.5. Analysis of Extrinsic FasL-Induced Apoptosis in Combination with NFκB Inhibitors in HNSCC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Crystal Violet Staining (CytoTox) Assay
4.3. ELISA
4.4. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BAFF | B-cell activating factor |
| CCL-5 | CC-chemokine ligand 5 |
| cFLIP | Cellular FLICE-like inhibitory protein |
| CHX | Cycloheximide |
| COX-2 | Cyclooxygenase-2 |
| CV | Crystal violet |
| HNSCC | Head and neck squamous cell carcinoma |
| IAP | Inhibitor of apoptosis protein |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IKK | IκB kinase |
| IκB | Inhibitory κB |
| IR | Ionising radiation |
| KRAS | Kirsten rat sarcoma |
| LTβ | Lymphotoxin-β |
| MCP-1 | Monocyte chemoattractant protein 1 |
| MMP | Matrix metalloproteinases |
| NAE | NEDD8 activating enzyme |
| NEDD8 | Neural precursor cell expressed developmentally down-regulated 8 |
| NFκB | Nuclear factor kappa light-chain enhancer of activated B-cells |
| NIK | NFκB-inducing kinase |
| TNFα | Tumour necrosis factor α |
| RANKL | Receptor activator of NFκB ligand |
| RCN | Relative cell number |
| VCAM-1 | Vascular cell adhesion protein 1 |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet (Lond. Engl.) 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Chan, A.T.; Ogino, S.; Fuchs, C.S. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N. Engl. J. Med. 2007, 356, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Wang, G.; Ye, T.; Wang, Y. Sulindac, a non-steroidal anti-inflammatory drug, mediates breast cancer inhibition as an immune modulator. Sci. Rep. 2016, 6, 19534. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S17–S27. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Ghosh, S. NF-kappaB: Roles and regulation in different CD4(+) T-cell subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bhimji, S.S. Bortezomib. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2018. [Google Scholar]
- Yamamoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J. Clin. Investig. 2001, 107, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Stark, G.R. Cytokine overexpression and constitutive NFkappaB in cancer. Cell Cycle (Georget. Tex.) 2004, 3, 1114–1117. [Google Scholar] [CrossRef]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J. Clin. Investig. 2001, 107, 241–246. [Google Scholar] [CrossRef]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef]
- Chuang, S.E.; Yeh, P.Y.; Lu, Y.S.; Lai, G.M.; Liao, C.M.; Gao, M.; Cheng, A.L. Basal levels and patterns of anticancer drug-induced activation of nuclear factor-kappaB (NF-kappaB), and its attenuation by tamoxifen, dexamethasone, and curcumin in carcinoma cells. Biochem. Pharmacol. 2002, 63, 1709–1716. [Google Scholar] [CrossRef]
- Jackson-Bernitsas, D.G.; Ichikawa, H.; Takada, Y.; Myers, J.N.; Lin, X.L.; Darnay, B.G.; Chaturvedi, M.M.; Aggarwal, B.B. Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-kappaB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene 2007, 26, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Ondrey, F.G.; Dong, G.; Sunwoo, J.; Chen, Z.; Wolf, J.S.; Crowl-Bancroft, C.V.; Mukaida, N.; Van Waes, C. Constitutive activation of transcription factors NF-(kappa)B, AP-1, and NF-IL6 in human head and neck squamous cell carcinoma cell lines that express pro-inflammatory and pro-angiogenic cytokines. Mol. Carcinog. 1999, 26, 119–129. [Google Scholar] [CrossRef]
- Wolf, J.S.; Chen, Z.; Dong, G.; Sunwoo, J.B.; Bancroft, C.C.; Capo, D.E.; Yeh, N.T.; Mukaida, N.; Van Waes, C. IL (interleukin)-1alpha promotes nuclear factor-kappaB and AP-1-induced IL-8 expression, cell survival, and proliferation in head and neck squamous cell carcinomas. Clin. Cancer Res. 2001, 7, 1812–1820. [Google Scholar]
- Woods, K.V.; Adler-Storthz, K.; Clayman, G.L.; Francis, G.M.; Grimm, E.A. Interleukin-1 regulates interleukin-6 secretion in human oral squamous cell carcinoma in vitro: Possible influence of p53 but not human papillomavirus E6/E7. Cancer Res. 1998, 58, 3142–3149. [Google Scholar] [PubMed]
- Chan, L.P.; Liu, C.; Chiang, F.Y.; Wang, L.F.; Lee, K.W.; Chen, W.T.; Kuo, P.L.; Liang, C.H. IL-8 promotes inflammatory mediators and stimulates activation of p38 MAPK/ERK-NF-kappaB pathway and reduction of JNK in HNSCC. Oncotarget 2017, 8, 56375–56388. [Google Scholar] [CrossRef] [PubMed]
- LoTempio, M.M.; Veena, M.S.; Steele, H.L.; Ramamurthy, B.; Ramalingam, T.S.; Cohen, A.N.; Chakrabarti, R.; Srivatsan, E.S.; Wang, M.B. Curcumin suppresses growth of head and neck squamous cell carcinoma. Clin. Cancer Res. 2005, 11, 6994–7002. [Google Scholar] [CrossRef] [PubMed]
- Kamangar, F.; Dores, G.M.; Anderson, W.F. Patterns of cancer incidence, mortality, and prevalence across five continents: Defining priorities to reduce cancer disparities in different geographic regions of the world. J. Clin. Oncol. 2006, 24, 2137–2150. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Paul, A.; Edwards, J.; Pepper, C.; Mackay, S. Inhibitory-kappaB Kinase (IKK) alpha and Nuclear Factor-kappaB (NFkappaB)-Inducing Kinase (NIK) as Anti-Cancer Drug Targets. Cells 2018, 7, 176. [Google Scholar] [CrossRef]
- Dong, J.; Li, J.; Cui, L.; Wang, Y.; Lin, J.; Qu, Y.; Wang, H. Cortisol modulates inflammatory responses in LPS-stimulated RAW264.7 cells via the NF-kappaB and MAPK pathways. BMC Vet. Res. 2018, 14, 30. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Rauert-Wunderlich, H.; Siegmund, D.; Maier, E.; Giner, T.; Bargou, R.C.; Wajant, H.; Stuhmer, T. The IKK inhibitor Bay 11-7082 induces cell death independent from inhibition of activation of NFkappaB transcription factors. PLoS ONE 2013, 8, e59292. [Google Scholar] [CrossRef] [PubMed]
- Milhollen, M.A.; Traore, T.; Adams-Duffy, J.; Thomas, M.P.; Berger, A.J.; Dang, L.; Dick, L.R.; Garnsey, J.J.; Koenig, E.; Langston, S.P.; et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: Rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood 2010, 116, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Vanderdys, V.; Allak, A.; Guessous, F.; Benamar, M.; Read, P.W.; Jameson, M.J.; Abbas, T. The Neddylation Inhibitor Pevonedistat (MLN4924) Suppresses and Radiosensitizes Head and Neck Squamous Carcinoma Cells and Tumors. Mol. Cancer Ther. 2018, 17, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Tobe, M.; Isobe, Y.; Tomizawa, H.; Nagasaki, T.; Takahashi, H.; Fukazawa, T.; Hayashi, H. Discovery of quinazolines as a novel structural class of potent inhibitors of NF-kappa B activation. Bioorganic Med. Chem. 2003, 11, 383–391. [Google Scholar] [CrossRef]
- Nan, J.; Du, Y.; Chen, X.; Bai, Q.; Wang, Y.; Zhang, X.; Zhu, N.; Zhang, J.; Hou, J.; Wang, Q.; et al. TPCA-1 is a direct dual inhibitor of STAT3 and NF-kappaB and regresses mutant EGFR-associated human non-small cell lung cancers. Mol. Cancer Ther. 2014, 13, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Nagaki, M.; Banno, Y.; Brenner, D.A.; Asano, T.; Nozawa, Y.; Moriwaki, H.; Nakashima, S. Tumor necrosis factor alpha-induced interleukin-8 production via NF-kappaB and phosphatidylinositol 3-kinase/Akt pathways inhibits cell apoptosis in human hepatocytes. Infect. Immun. 2002, 70, 6294–6301. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Leong, K.G.; Karsan, A. Signaling pathways mediated by tumor necrosis factor alpha. Histol. Histopathol. 2000, 15, 1303–1325. [Google Scholar]
- Varfolomeev, E.E.; Ashkenazi, A. Tumor necrosis factor: An apoptosis JuNKie? Cell 2004, 116, 491–497. [Google Scholar] [CrossRef]
- Kreuz, S.; Siegmund, D.; Scheurich, P.; Wajant, H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell. Biol. 2001, 21, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Anany, M.A.; Kreckel, J.; Fullsack, S.; Rosenthal, A.; Otto, C.; Siegmund, D.; Wajant, H. Soluble TNF-like weak inducer of apoptosis (TWEAK) enhances poly(I:C)-induced RIPK1-mediated necroptosis. Cell Death Dis. 2018, 9, 1084. [Google Scholar] [CrossRef] [PubMed]
- Pajak, B.; Gajkowska, B.; Orzechowski, A. Cycloheximide-mediated sensitization to TNF-alpha-induced apoptosis in human colorectal cancer cell line COLO 205; role of FLIP and metabolic inhibitors. J. Physiol. Pharmacol. 2005, 56 (Suppl. 3), 101–118. [Google Scholar] [PubMed]
- El-Mesery, M.; Seher, A.; Stuhmer, T.; Siegmund, D.; Wajant, H. MLN4924 sensitizes monocytes and maturing dendritic cells for TNF-dependent and -independent necroptosis. Br. J. Pharmacol. 2015, 172, 1222–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brands, R.C.; Herbst, F.; Hartmann, S.; Seher, A.; Linz, C.; Kubler, A.C.; Muller-Richter, U.D.A. Cytotoxic effects of SMAC-mimetic compound LCL161 in head and neck cancer cell lines. Clin. Oral Investig. 2016, 20, 2325–2332. [Google Scholar] [CrossRef]
- Brands, R.C.; Scheurer, M.J.J.; Hartmann, S.; Seher, A.; Kubler, A.C.; Muller-Richter, U.D.A. Apoptosis-sensitizing activity of birinapant in head and neck squamous cell carcinoma cell lines. Oncol. Lett. 2018, 15, 4010–4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallarida, R.J. The interaction index: A measure of drug synergism. Pain 2002, 98, 163–168. [Google Scholar] [CrossRef]
- Sandra, F.; Matsuki, N.A.; Takeuchi, H.; Ikebe, T.; Kanematsu, T.; Ohishi, M.; Hirata, M. TNF inhibited the apoptosis by activation of Akt serine/threonine kinase in the human head and neck squamous cell carcinoma. Cell. Signal. 2002, 14, 771–778. [Google Scholar] [CrossRef]
- Wajant, H. The role of TNF in cancer. Results Probl. Cell Differ. 2009, 49, 1–15. [Google Scholar]
- Hedberg, M.L.; Peyser, N.D.; Bauman, J.E.; Gooding, W.E.; Li, H.; Bhola, N.E.; Zhu, T.R.; Zeng, Y.; Brand, T.M.; Kim, M.O.; et al. Use of nonsteroidal anti-inflammatory drugs predicts improved patient survival for PIK3CA-altered head and neck cancer. J. Exp. Med. 2019, 216, 419. [Google Scholar] [CrossRef]
- Thurnher, D.; Bakroeva, M.; Formanek, M.; Knerer, B.; Kornfehl, J. Non-steroidal anti-inflammatory drugs inhibit telomerase activity in head and neck squamous carcinoma cell lines. Head Neck 2001, 23, 1049–1055. [Google Scholar] [CrossRef]
- Thurnher, D.; Bakroeva, M.; Schutz, G.; Pelzmann, M.; Formanek, M.; Knerer, B.; Kornfehl, J. Non-steroidal anti-inflammatory drugs induce apoptosis in head and neck cancer cell lines. Acta Oto-laryngol. 2001, 121, 957–962. [Google Scholar] [CrossRef]
- Zhao, L.; Yue, P.; Lonial, S.; Khuri, F.R.; Sun, S.Y. The NEDD8-activating enzyme inhibitor, MLN4924, cooperates with TRAIL to augment apoptosis through facilitating c-FLIP degradation in head and neck cancer cells. Mol. Cancer Ther. 2011, 10, 2415–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Liang, Y.; Li, L.; Wang, X.; Yan, Z.; Dong, C.; Zeng, M.S.; Zhong, Q.; Liu, X.K.; Yu, J.; et al. The Nedd8-activating enzyme inhibitor MLN4924 (TAK-924/Pevonedistat) induces apoptosis via c-Myc-Noxa axis in head and neck squamous cell carcinoma. Cell Prolif. 2018, 19, e12536. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.P.; Wang, L.F.; Chiang, F.Y.; Lee, K.W.; Kuo, P.L.; Liang, C.H. IL-8 promotes HNSCC progression on CXCR1/2-meidated NOD1/RIP2 signaling pathway. Oncotarget 2016, 7, 61820–61831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagler, C.; El-Mesery, M.; Kubler, A.C.; Muller-Richter, U.D.A.; Stuhmer, T.; Nickel, J.; Muller, T.D.; Wajant, H.; Seher, A. The anti-myeloma activity of bone morphogenetic protein 2 predominantly relies on the induction of growth arrest and is apoptosis-independent. PLoS ONE 2017, 12, e0185720. [Google Scholar] [CrossRef]
- Heo, D.S.; Snyderman, C.; Gollin, S.M.; Pan, S.; Walker, E.; Deka, R.; Barnes, E.L.; Johnson, J.T.; Herberman, R.B.; Whiteside, T.L. Biology, cytogenetics, and sensitivity to immunological effector cells of new head and neck squamous cell carcinoma lines. Cancer Res. 1989, 49, 5167–5175. [Google Scholar] [PubMed]
- Brands, R.C.; Knierim, L.M.; De Donno, F.; Steinacker, V.; Hartmann, S.; Seher, A.; Kubler, A.C.; Muller-Richter, U.D.A. Targeting VEGFR and FGFR in head and neck squamous cell carcinoma in vitro. Oncol. Rep. 2017, 38, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Neckel, N.; Seher, A.; Mutzbauer, G.; Brands, R.C.; Linz, C.; Kubler, A.C.; Muller-Richter, U.D. Erlotinib and gefitinib responsiveness in head and neck cancer cell lines--a comparing analysis with cetuximab. Clin. Oral Investig. 2016, 20, 759–769. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cortisol [µM] | MLN4924 [µM] | QNZ [µM] | TPCA1 [µM] | |||||
|---|---|---|---|---|---|---|---|---|
| IC10 | IC50 | IC10 | IC50 | IC10 | IC50 | IC10 | IC50 | |
| PCI1 | 20 * | >280 ** | 0.2 | 1 | 0.05 | 1 | 4.6 | 9.7 |
| PCI9 | 140 * | >280 ** | 3.1 * | >4.8 * | 1 † | >10 ** | 3.3 | 13.8 |
| PCI13 | 140 * | >280 ** | 0.2 | 0.8 | 0.4 | 1.2 | 5.2 | 24.2 |
| PCI52 | 140 * | >280 ** | 7.3* | >17.4 * | 1 † | >10 ** | 6.3 * | >17 * |
| SCC25 | 140 * | >280 ** | 0.2 | 1.4 | 10 * | >10 ** | 3.5 | 6.1 |
| HaCaT | 140 * | >280 ** | 0.2 | 0.8 | 0.4 | 1.1 | 2 | 5.3 |
| PCI1 | PCI9 | PCI13 | PCI52 | SCC25 | HaCaT | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RCN | ± | RCN | ± | RCN | ± | RCN | ± | RCN | ± | RCN | ± | |
| Cortisol | 62 | 6.6 | 75.5 | 8.8 | 71.3 | 2.8 | 86.9 | 0.7 | 91 | 2.3 | 72.6 | 4.6 |
| MLN4924 | 9.9 | 4.2 | 39.3 | 4.6 | 4.4 | 1.9 | 35.6 | 4.1 | 15.0 | 2.5 | 18.5 | 5.5 |
| QNZ | 5.7 | 3.9 | 96.3 | 3.2 | 6.0 | 5.7 | 77.0 | 5.6 | 81.2 | 3.6 | 39.1 | 8.2 |
| TPCA1 | 11.5 | 2.3 | 29.9 | 5.2 | 18.0 | 6.3 | 36.2 | 2.1 | 10.6 | 3.9 | 18.5 | 8.4 |
| IC50 FasL [ng/mL] | Efficiency IC50 FasLmono/ IC50 FasLkombi) | Interaction Index (y) | |
|---|---|---|---|
| PCI1 | |||
| FasL | 37.3 | - | - |
| Cortisol [IC10] + FasL | >88.8 ± | 0.4 ± | 2.4 *** |
| MLN4924 [IC10] + FasL | 33.6 | 1.1 | 1.1 *** |
| QNZ [IC10] + FasL | 44.3 ± | 0.8 ± | 1.2 *** |
| TPCA1 [IC10] + FasL | 41.9 ± | 0.9 ± | 1.6 *** |
| PCI9 | |||
| FasL | - † | - | - |
| Cortisol [IC10] + FasL | >200 † | 1 | 1.5 *** |
| MLN4924 [IC10] + FasL | >200 † | 1 | 1.7 *** |
| QNZ [IC10] + FasL | >200 † | 1 | 1.1 *** |
| TPCA1 [IC10] + FasL | >200 † | 1 | 1.2 *** |
| PCI13 | |||
| FasL | 55.5 | - | - |
| Cortisol [IC10] + FasL | >200 ± | 0.3 ± | 4.1 *** |
| MLN4924 [IC10] + FasL | >71.9 ± | 0.8 ± | 1.6 *** |
| QNZ [IC10] + FasL | 24.4 | 2.3 | 0.8 * |
| TPCA1 [IC10] + FasL | >55.1 ± | 1 | 1.2 *** |
| PCI52 | |||
| FasL | - † | - | - |
| Cortisol [IC10] + FasL | >200 † | 1 | 1.5 *** |
| MLN4924 [IC10] + FasL | >200 † | 1 | 1.4 *** |
| QNZ [IC10] + FasL | >200 † | 1 | 1.1 *** |
| TPCA1 [IC10] + FasL | >200 † | 1 | 1.4 *** |
| SCC25 | |||
| FasL | - † | - | - |
| Cortisol [IC10] + FasL | >200 † | 1 | 1.5 *** |
| MLN4924 [IC10] + FasL | 39.7 | 5 | 0.3 * |
| QNZ [IC10] + FasL | >200 † | 1 | 2 *** |
| TPCA1 [IC10] + FasL | >200 † | 1 | 1.6 *** |
| HaCaT | |||
| FasL | >68.7 ‡ | - | - |
| Cortisol [IC10] + FasL | >59.1 | 1.2 | 1.4 *** |
| MLN4924 [IC10] + FasL | 17.6 | 3.9 | 0.5 * |
| QNZ [IC10] + FasL | 28.7 | 2.4 | 0.8 * |
| TPCA1 [IC10] + FasL | 27.5 | 2.5 | 0.8 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheurer, M.J.J.; Brands, R.C.; El-Mesery, M.; Hartmann, S.; Müller-Richter, U.D.A.; Kübler, A.C.; Seher, A. The Selection of NFκB Inhibitors to Block Inflammation and Induce Sensitisation to FasL-Induced Apoptosis in HNSCC Cell Lines Is Critical for Their Use as a Prospective Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 1306. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061306
Scheurer MJJ, Brands RC, El-Mesery M, Hartmann S, Müller-Richter UDA, Kübler AC, Seher A. The Selection of NFκB Inhibitors to Block Inflammation and Induce Sensitisation to FasL-Induced Apoptosis in HNSCC Cell Lines Is Critical for Their Use as a Prospective Cancer Therapy. International Journal of Molecular Sciences. 2019; 20(6):1306. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061306
Chicago/Turabian StyleScheurer, Mario Joachim Johannes, Roman Camillus Brands, Mohamed El-Mesery, Stefan Hartmann, Urs Dietmar Achim Müller-Richter, Alexander Christian Kübler, and Axel Seher. 2019. "The Selection of NFκB Inhibitors to Block Inflammation and Induce Sensitisation to FasL-Induced Apoptosis in HNSCC Cell Lines Is Critical for Their Use as a Prospective Cancer Therapy" International Journal of Molecular Sciences 20, no. 6: 1306. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061306