5-HT3 Antagonist Ondansetron Increases apoE Secretion by Modulating the LXR-ABCA1 Pathway

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
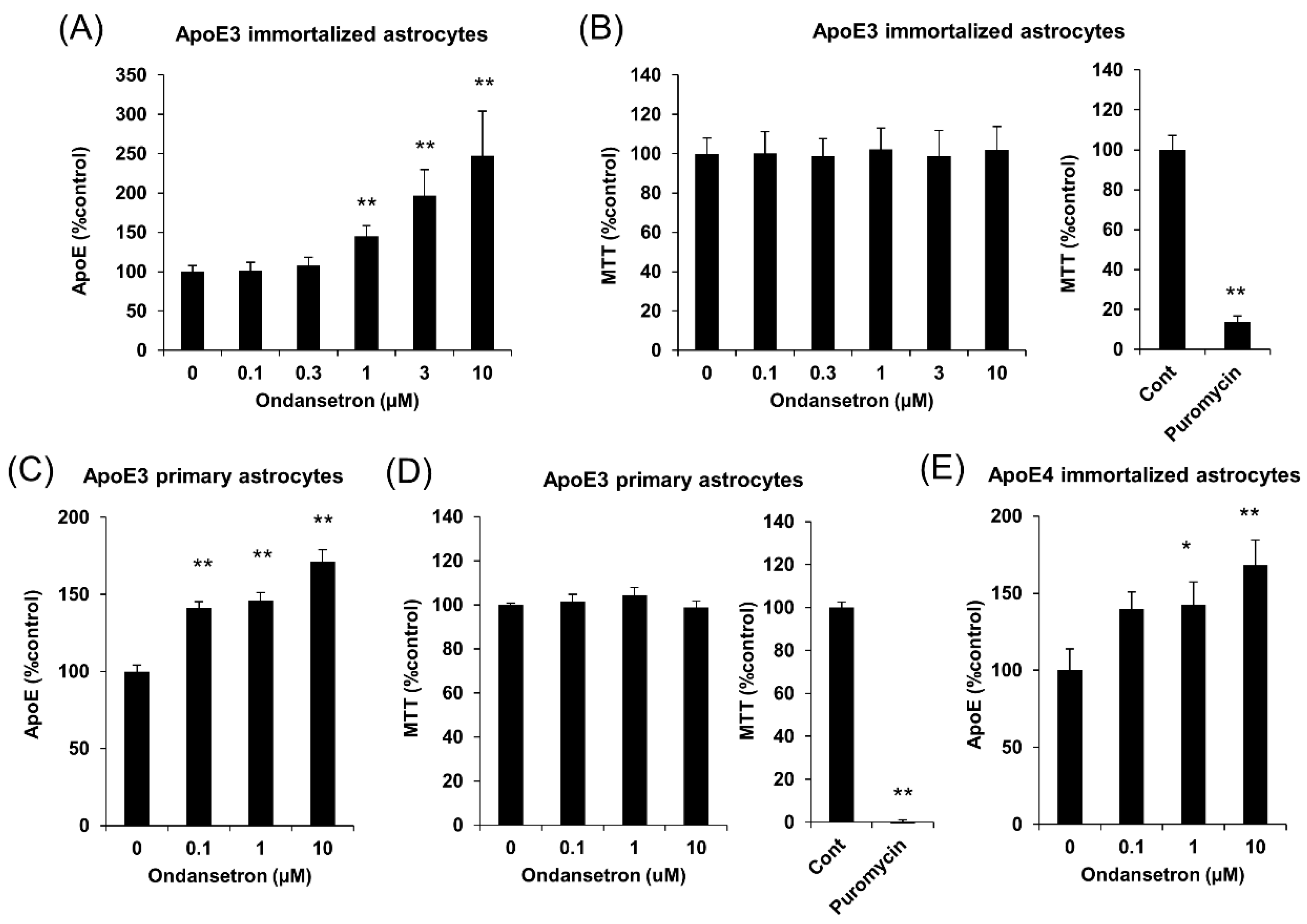
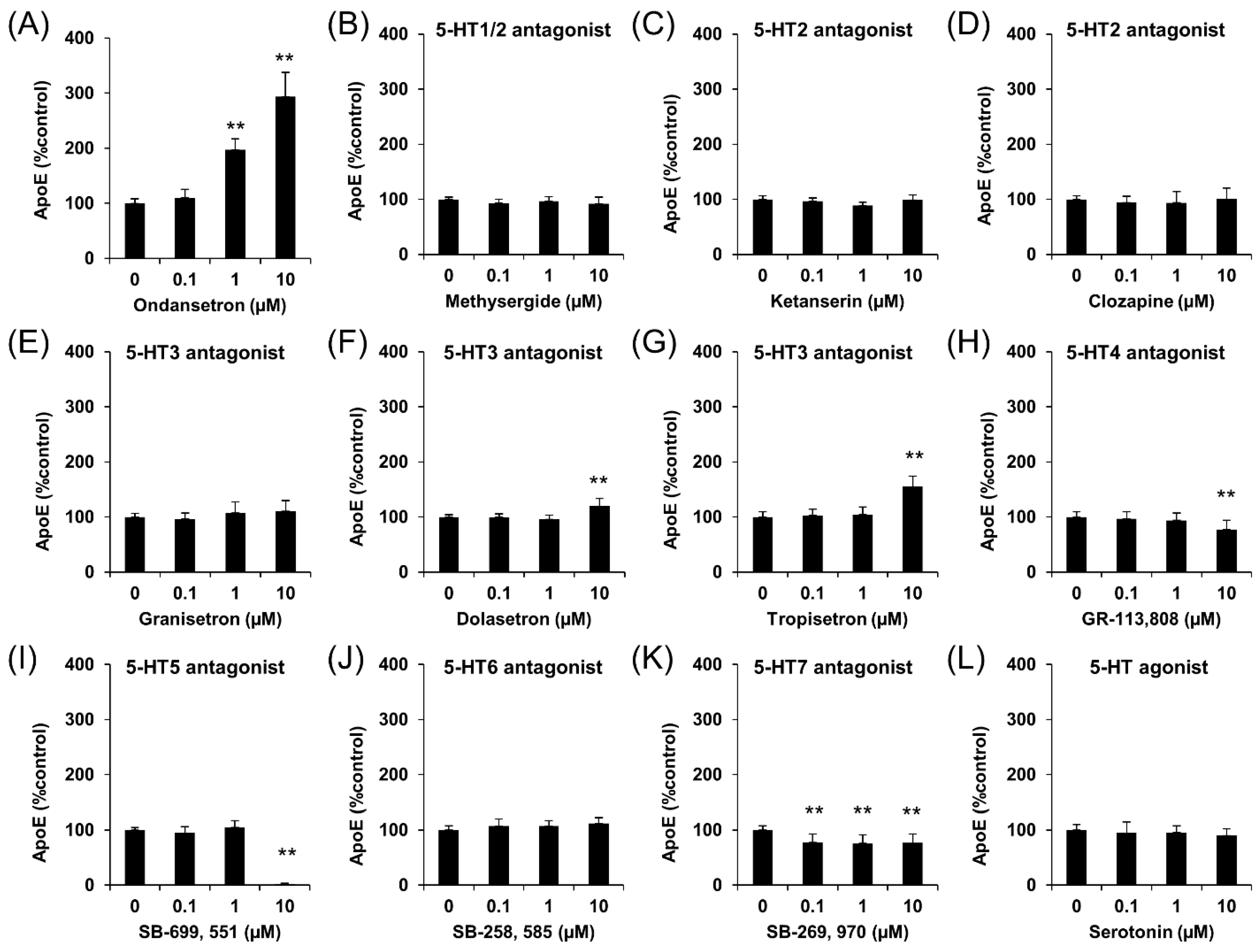
2.1. OS Increases apoE Secretion from Murine Astrocytes
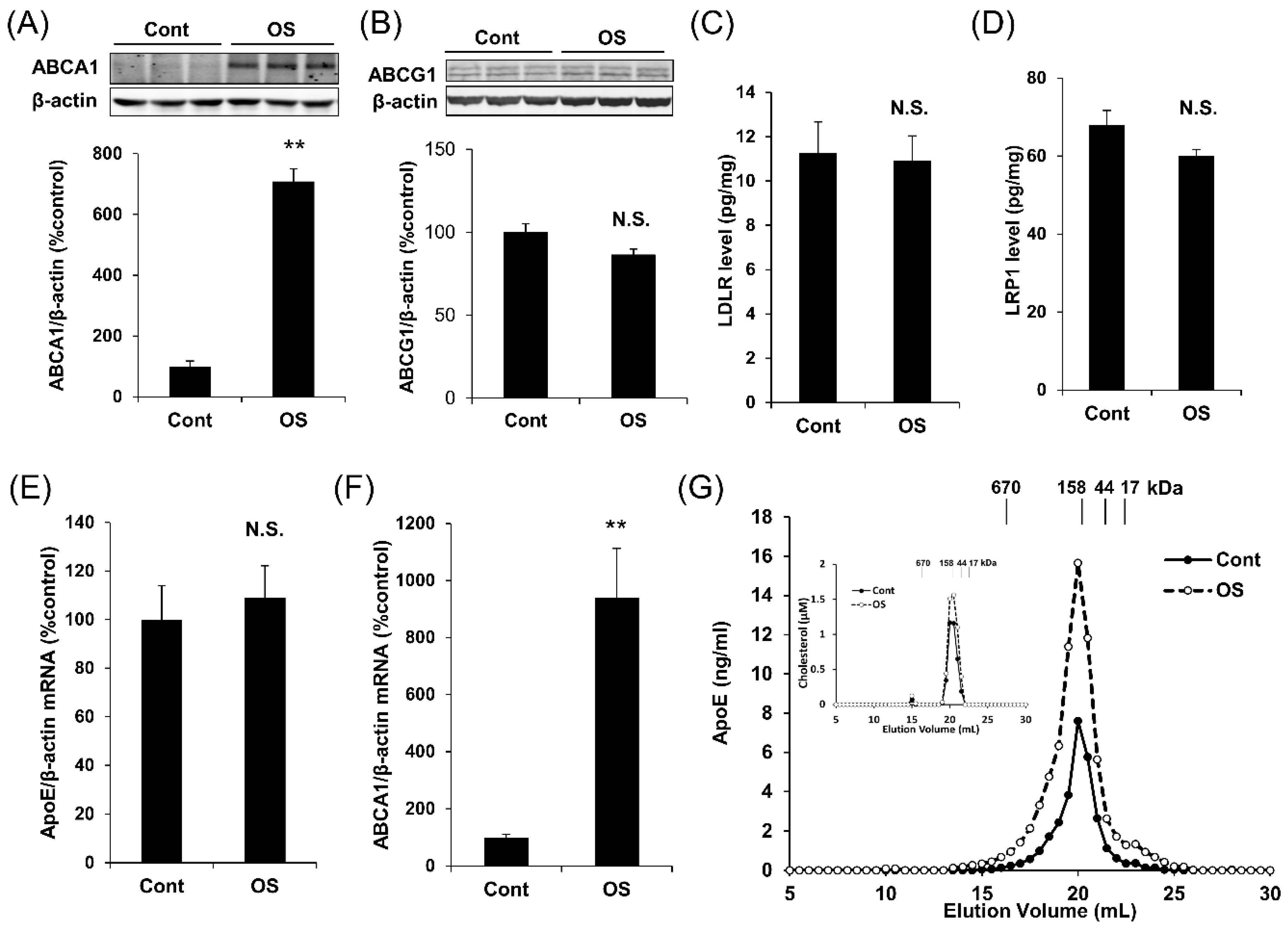
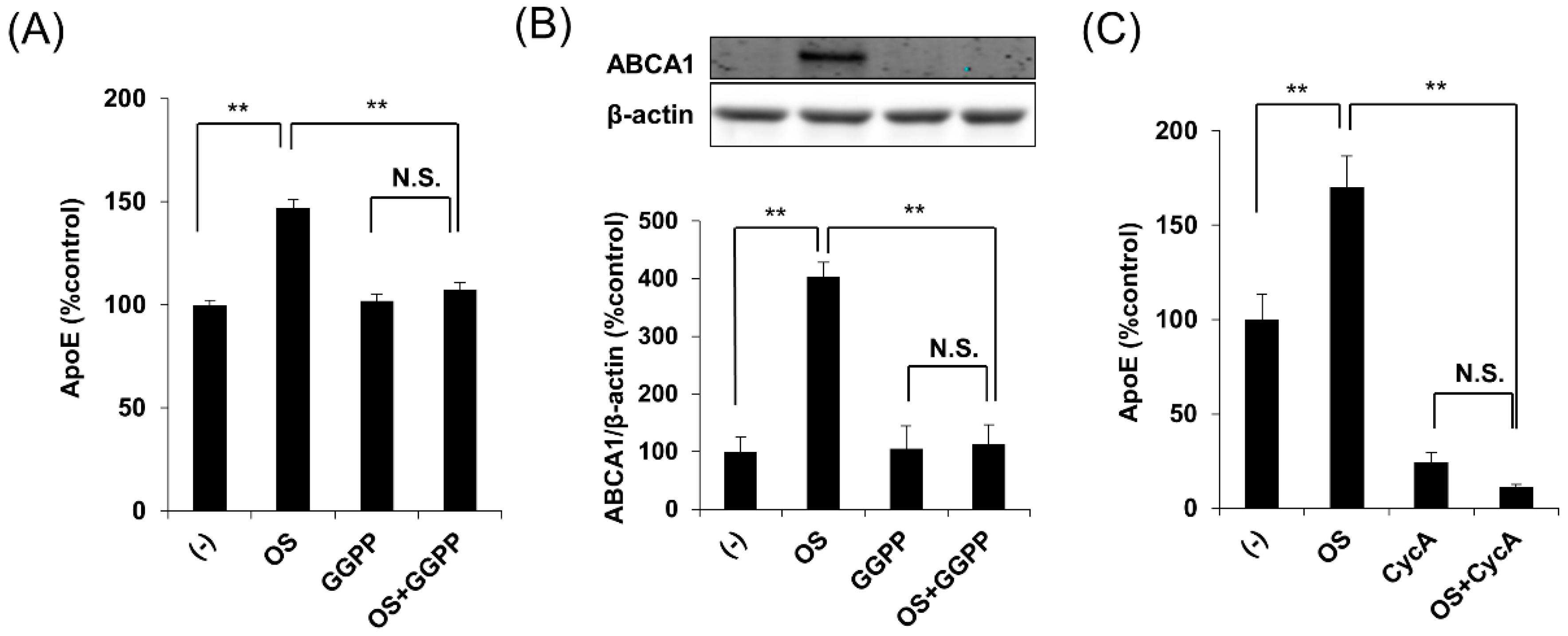
2.2. OS Increases apoE Secretion through LXR-ABCA1 Pathway
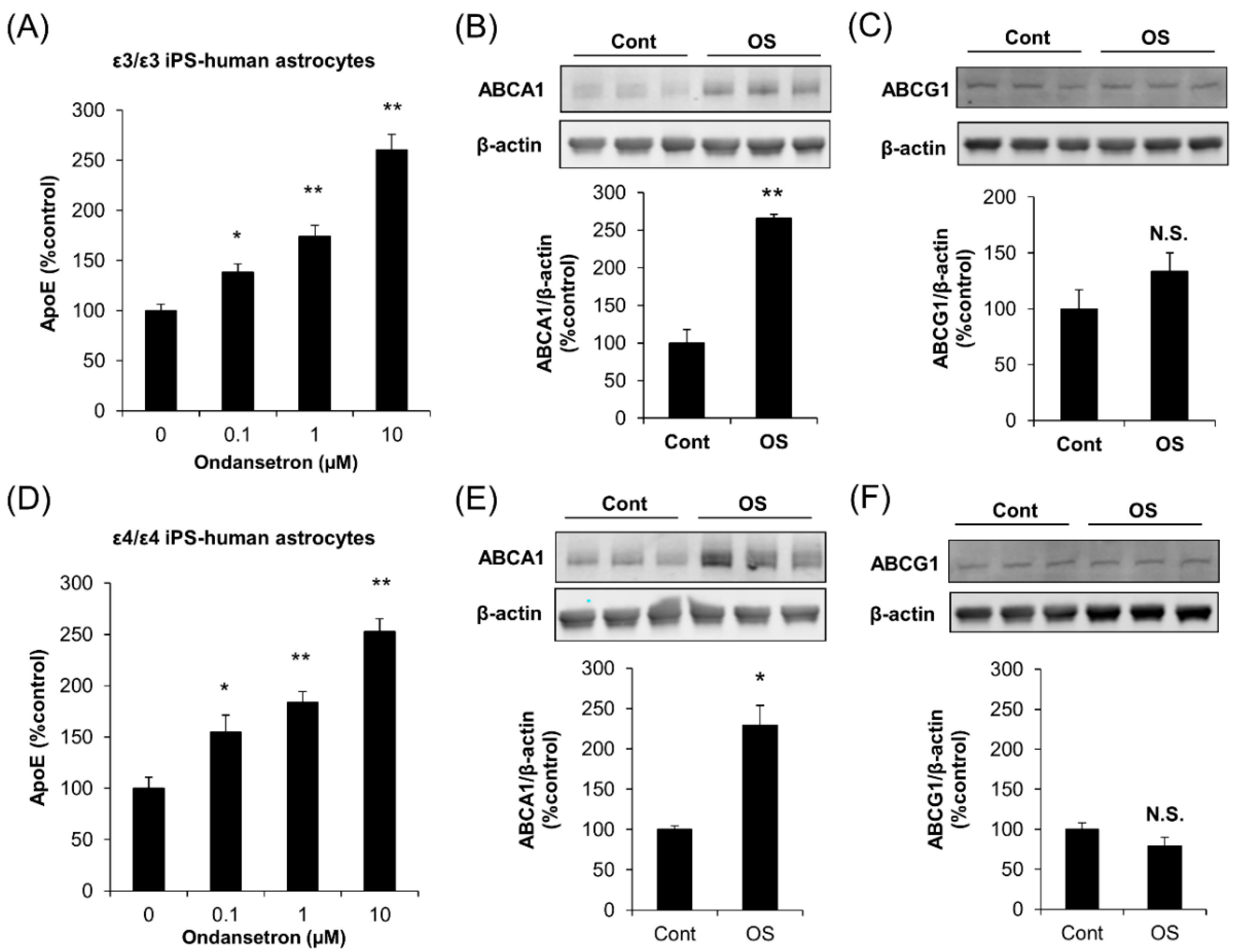
2.3. OS Increases apoE Secretion from Human Astrocytes
2.4. Effects of OS on apoE Levels In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Immortalized Astrocyte Cultures
4.3. Primary Astrocyte Cultures
4.4. Human Astrocyte Cultures
4.5. Biochemical Analyses
4.6. In Vivo Effects of Tested Compounds
4.7. Cytotoxicity Assays
4.8. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Xu, H.; Bu, G. apoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Riddell, D.R.; Zhou, H.; Atchison, K.; Warwick, H.K.; Atkinson, P.J.; Jefferson, J.; Xu, L.; Aschmies, S.; Kirksey, Y.; Hu, Y.; et al. Impact of apolipoprotein E (apoE) polymorphism on brain apoE levels. J. Neurosci. 2008, 28, 11445–11453. [Google Scholar] [CrossRef]
- Cruchaga, C.; Kauwe, J.S.K.; Nowotny, P.; Bales, K.; Pickering, E.H.; Mayo, K.; Bertelsen, S.; Hinrichs, A.; Fagan, A.M.; Alzheimer’s Disease Neuroimaging Initiative; et al. Cerebrospinal fluid APOE levels: An endophenotype for genetic studies for Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 4558–4571. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, J.T.; Wang, H.F.; Jiang, T.; Tan, C.C.; Meng, X.F.; Soares, H.D.; Tan, L. Meta-analysis of peripheral blood apolipoprotein E levels in Alzheimer’s disease. PLoS ONE 2014, 9, e89041. [Google Scholar] [CrossRef]
- Rasmussen, K.L.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Plasma levels of apolipoprotein E and risk of dementia in the general population. Ann. Neurol. 2015, 77, 301–311. [Google Scholar] [CrossRef]
- Zelcer, N.; Khanlou, N.; Clare, R.; Jiang, Q.; Reed-Geaghan, E.G.; Landreth, G.E.; Vinters, H.V.; Tontonoz, P. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 10601–10606. [Google Scholar] [CrossRef]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. apoE promotes the proteolytic degradation of Aβ. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. apoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef]
- Tousi, B. The emerging role of bexarotene in the treatment of Alzheimer’s disease: Current evidence. Neuropsychiatry Dis. Treat. 2015, 11, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug. Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.M.; Koster, K.P.; Luo, J.; Lee, S.H.; Wang, Y.T.; Collins, N.C.; Ben Aissa, M.; Thatcher, G.R.; LaDu, M.J. Amyloid-β pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J. Biol. Chem. 2014, 289, 30538–30555. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fu, Y.; Liu, C.C.; Shinohara, M.; Nielsen, H.M.; Dong, Q.; Kanekiyo, T.; Bu, G. Retinoic acid isomers facilitate apolipoprotein E production and lipidation in astrocytes through the retinoid X receptor/retinoic acid receptor pathway. J. Biol. Chem. 2014, 289, 11282–11292. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Kaplan, R.; Menke, J.G.; MacNaul, K.; Chen, Y.; Sparrow, C.P.; Zhou, G.; Wright, S.D.; Cai, T.-Q. Dual Mechanisms of ABCA1 Regulation by Geranylgeranyl Pyrophosphate. J. Biol. Chem. 2001, 276, 48702–48708. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.S.; Goto, T.; Takahashi, N.; Egawa, K.; Takahashi, H.; Jheng, H.F.; Kim, Y.I.; Kawada, T. Geranylgeranyl pyrophosphate performs as an endogenous regulator of adipocyte function via suppressing the LXR pathway. Biochem. Biophys. Res. Commun. 2016, 478, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, W.; Peng, D.Q.; Settle, M.; Brubaker, G.; Morton, R.E.; Smith, J.D. Cyclosporin A traps ABCA1 at the plasma membrane and inhibits ABCA1-mediated lipid efflux to apolipoprotein A-I. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2155–2161. [Google Scholar] [CrossRef]
- Nagao, K.; Maeda, M.; Manucat, N.B.; Ueda, K. Cyclosporine A and PSC833 inhibit ABCA1 function via direct binding. Biochim. Biophys. Acta 2013, 1831, 398–406. [Google Scholar] [CrossRef]
- Roychoudhury, M.; Kulkarni, S.K. Antianxiety profile of ondansetron, a selective 5-HT3 antagonist, in a novel animal model. Methods Find. Exp. Clin. Pharm. 1997, 19, 107–111. [Google Scholar]
- Gupta, D.; Radhakrishnan, M.; Kurhe, Y. Ondansetron, a 5HT3 receptor antagonist reverses depression and anxiety-like behavior in streptozotocin-induced diabetic mice: Possible implication of serotonergic system. Eur. J. Pharm. 2014, 744, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Imaki, J.; Mae, Y.; Takahashi, T.; Tatara, A. Serotonergic modulation of extrapyramidal motor disorders in mice and rats: Role of striatal 5-HT3 and 5-HT6 receptors. Neuropharmacology 2011, 60, 201–208. [Google Scholar] [CrossRef]
- Christofaki, M.; Papaioannou, A. Ondansetron: A review of pharmacokinetics and clinical experience in postoperative nausea and vomiting. Expert Opin. Drug Metab. Toxicol. 2014, 10, 437–444. [Google Scholar] [CrossRef]
- Thompson, A.J.; Lummis, S.C. 5-HT3 receptors. Curr. Pharm. Des. 2006, 12, 3615–3630. [Google Scholar] [CrossRef]
- Gan, T.J. Selective Serotonin 5-HT3 Receptor Antagonists for Postoperative Nausea and Vomiting. CNS Drugs 2005, 19, 225–238. [Google Scholar] [CrossRef]
- Liu, F.C.; Liou, J.T.; Liao, H.R.; Mao, C.C.; Yang, P.; Day, Y.J. The anti-aggregation effects of ondansetron on platelets involve IP3 signaling and MAP kinase pathway, but not 5-HT3-dependent pathway. Thromb. Res. 2012, 130, 18. [Google Scholar] [CrossRef]
- Tang, S.L.; Chen, W.J.; Yin, K.; Zhao, G.J.; Mo, Z.C.; Lv, Y.C.; Ouyang, X.P.; Yu, X.H.; Kuang, H.J.; Jiang, Z.S.; et al. PAPP-A negatively regulates ABCA1, ABCG1 and SR-B1 expression by inhibiting LXRα through the IGF-I-mediated signaling pathway. Atherosclerosis 2012, 222, 344–354. [Google Scholar] [CrossRef]
- Suon, S.; Zhao, J.; Villarreal, S.A.; Anumula, N.; Liu, M.; Carangia, L.M.; Renger, J.J.; Zerbinatti, C.V. Systemic treatment with liver X receptor agonists raises apolipoprotein E, cholesterol, and amyloid-β peptides in the cerebral spinal fluid of rats. Mol. Neurodegener. 2010, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Tyers, M.B.; Freeman, A.J. Mechanism of the anti-emetic activity of 5-HT3 receptor antagonists. Oncology 1992, 49, 263–268. [Google Scholar] [CrossRef]
- Simpson, K.H.; Murphy, P.; Colthup, P.V.; Whelan, P. Concentration of ondansetron in cerebrospinal fluid following oral dosing in volunteers. Psychopharmacology 1992, 109, 497–498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Kang, W.H.; Li, Q.; Wang, X.Y.; Yao, S.M.; Ma, A.Q. Beneficial effects of ondansetron as an adjunct to haloperidol for chronic, treatment-resistant schizophrenia: A double-blind, randomized, placebo-controlled study. Schizophr. Res. 2006, 88, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Sirota, P.; Mosheva, T.; Shabtay, H.; Giladi, N.; Korczyn, A.D. Use of the selective serotonin 3 receptor antagonist ondansetron in the treatment of neuroleptic-induced tardive dyskinesia. Am. J. Psychiatry 2000, 157, 287–289. [Google Scholar] [CrossRef]
- Kurhe, Y.; Mahesh, R. Ondansetron attenuates co-morbid depression and anxiety associated with obesity by inhibiting the biochemical alterations and improving serotonergic neurotransmission. Pharm. Biochem. Behav. 2015, 136, 107–116. [Google Scholar] [CrossRef]
- Krupkova, M.; Sedova, L.; Liska, F.; Krenova, D.; Kren, V.; Seda, O. Differential effects of 5-HT3 receptor antagonist on lipid profile in spontaneously hypertensive rat and chromosome 8 congenic strain. Neuro Endocrinol. Lett. 2012, 2, 43–49. [Google Scholar]
- Dysken, M.; Kuskowski, M.; Love, S. Ondansetron in the treatment of cognitive decline in Alzheimer dementia. Am. J. Geriatr. Psychiatry 2002, 10, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Davis, M.D.; Shinohara, M.; Younkin, S.G.; Kanekiyo, T.; Martens, Y.A.; Bu, G.; Graff-Radford, N.R.; Wszolek, Z.K. APOE ε4/ε4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet. 2017, 26, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Fujioka, S.; Murray, M.E.; Wojtas, A.; Baker, M.; Rovelet-Lecrux, A.; Rademakers, R.; Das, P.; Parisi, J.E.; Graff-Radford, N.R.; et al. Regional distribution of synaptic markers and APP correlate with distinct clinicopathological features in sporadic and familial Alzheimer’s disease. Brain 2014, 137 Pt 5, 1533–1549. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, M.; Petersen, R.C.; Dickson, D.W.; Bu, G. Brain regional correlation of amyloid-β with synapses and apolipoprotein E in non-demented individuals: Potential mechanisms underlying regional vulnerability to amyloid-β accumulation. Acta Neuropathol. 2013, 125, 535–547. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shinohara, M.; Shinohara, M.; Zhao, J.; Fu, Y.; Liu, C.-C.; Kanekiyo, T.; Bu, G. 5-HT3 Antagonist Ondansetron Increases apoE Secretion by Modulating the LXR-ABCA1 Pathway. Int. J. Mol. Sci. 2019, 20, 1488. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061488
Shinohara M, Shinohara M, Zhao J, Fu Y, Liu C-C, Kanekiyo T, Bu G. 5-HT3 Antagonist Ondansetron Increases apoE Secretion by Modulating the LXR-ABCA1 Pathway. International Journal of Molecular Sciences. 2019; 20(6):1488. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061488
Chicago/Turabian StyleShinohara, Motoko, Mitsuru Shinohara, Jing Zhao, Yuan Fu, Chia-Chen Liu, Takahisa Kanekiyo, and Guojun Bu. 2019. "5-HT3 Antagonist Ondansetron Increases apoE Secretion by Modulating the LXR-ABCA1 Pathway" International Journal of Molecular Sciences 20, no. 6: 1488. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061488