Cellular Cullin RING Ubiquitin Ligases: Druggable Host Dependency Factors of Cytomegaloviruses


{kind=link}
{kind=link}
Abstract
:1. The Human Cytomegalovirus
2. Interferons
3. Posttranslational Modification of Proteins with Ubiquitin (Ubiquitination)
4. The Proteasome
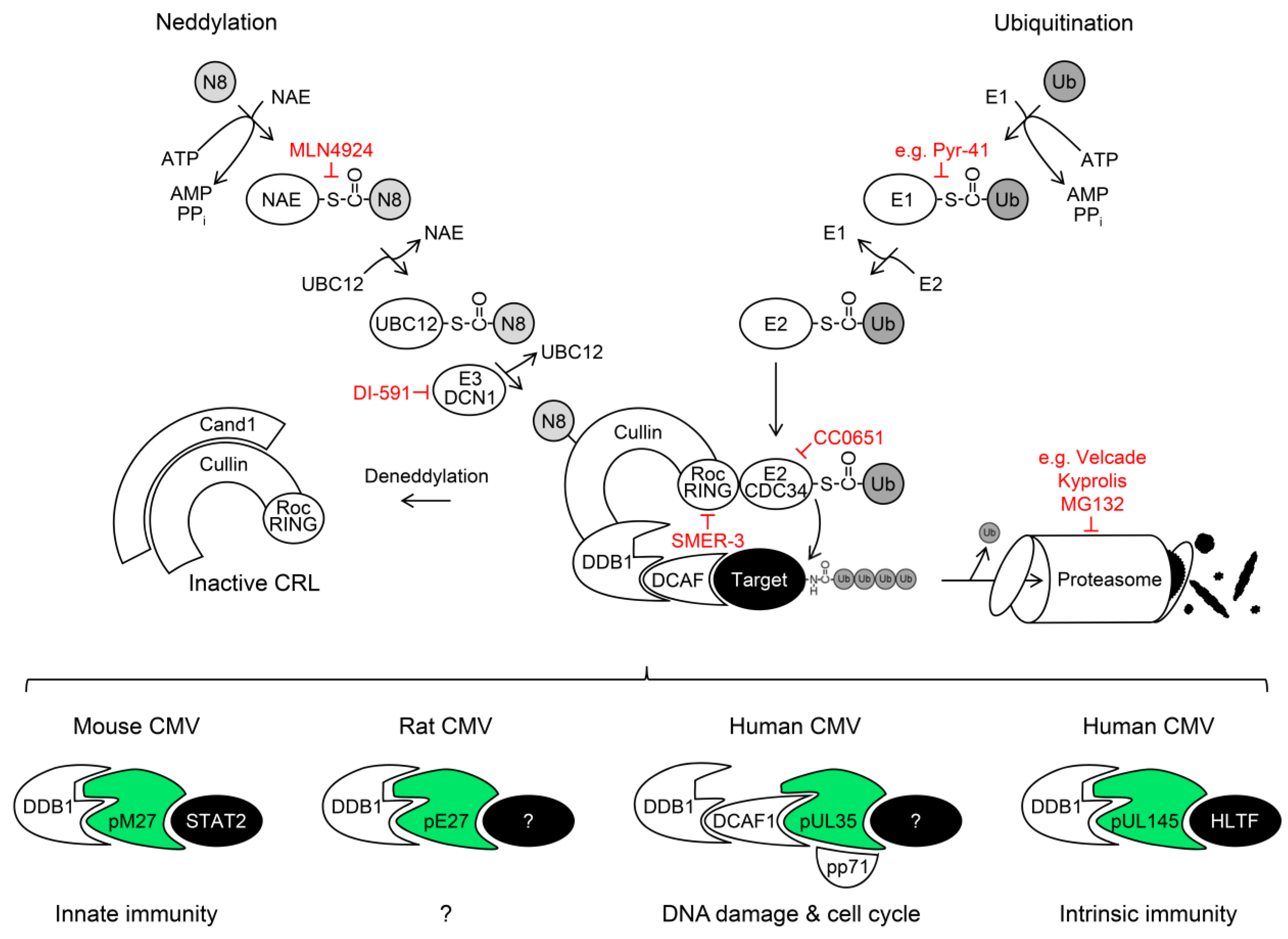
5. Cullin RING Ubiquitin Ligases (CRLs) and Their Regulation by Nedd8 Conjugation
DDB1-Cullin 4A/B-RocA Complexes
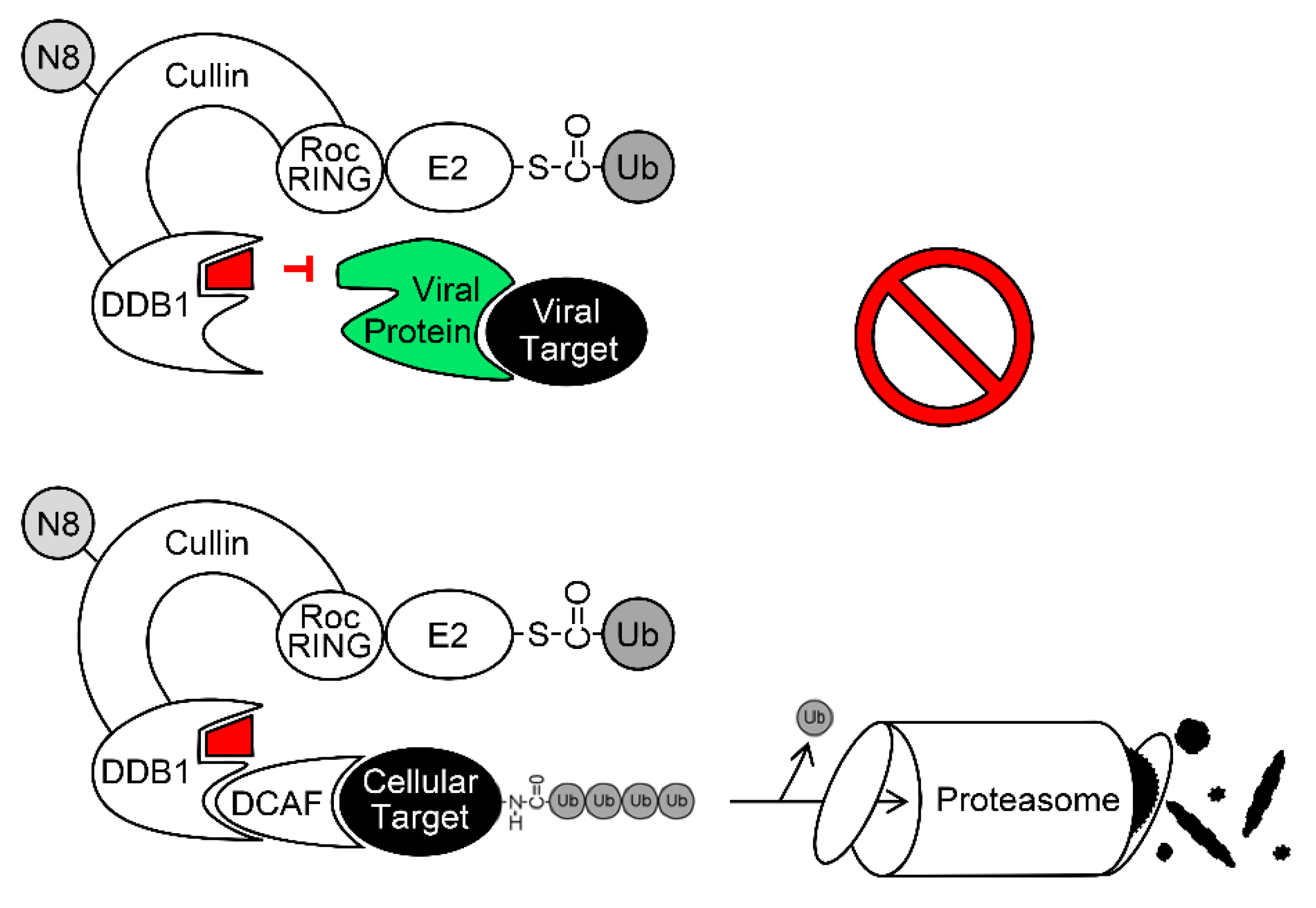
6. Exploitation of DDB1 and CRLs by Viruses
6.1. The DDB1-CRL-Interacting MCMV Protein pM27
6.2. The DDB1-CRL-Interacting HCMV Protein pUL35 and its MCMV Homolog pM35
6.3. The DDB1-CRL-Interacting HCMV Protein pUL145
7. The Ubiquitin-Proteasome System (UPS) as Antiviral Drug Target
8. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| BoHV-1 | bovine herpesvirus 1 |
| CRL | cullin RING ubiquitin ligases |
| CUL | cullin |
| Cul4A | cullin 4A |
| Cul4B | cullin 4B |
| DAA | direct acting antiviral |
| DCAF1 | DDB1- and CUL4-associated factor 1 |
| DDA1 | DET1- and DDB1-Associated 1 |
| DDB1 | DNA-damage binding protein 1 |
| E1 | Ub-activating enzyme |
| E2 | Ub-conjugating enzyme |
| E3 | Ub ligase |
| GCV | Ganciclovir |
| GvHD | graft-versus-host disease |
| HAART | highly active antiretroviral therapy |
| HBV | hepatitis B virus |
| HCMV | human cytomegalovirus |
| HCV | hepatitis C virus |
| HECT | homologous to the E6-associated protein C-terminus |
| HHV-5 | human herpesvirus 5 |
| HIV | human immunodeficiency virus |
| HLTF | helicase-like transcription factor |
| IAA | indirect acting antiviral |
| IFN | interferon |
| IRepG | IFN-repressed gene |
| IRF | IFN-regulatory factor |
| ISG | IFN-stimulated gene |
| ISGF3 | IFN-stimulated gene factor 3 |
| Jak | janus kinase |
| MCMV | mouse cytomegalovirus |
| mDC | myeloid dendritic cell |
| MHV68 | murine gamma herpesvirus 68 |
| MIEP | major immediate early promoter |
| NAE | Nedd8-activating enzyme |
| ORF | open reading frame |
| PCNA | proliferating cell nuclear antigen |
| pDC | plasmacytoid dendritic cell |
| PIV | parainfluenza virus |
| RING | really interesting new gene |
| RL | repeat long |
| RS | repeat short |
| SCF | Skp1/cullin 1/F-box |
| STAT | signal transducer and activator of transcription |
| TP53BP1 | tumor protein p53-binding protein 1 |
| Tyk2 | tyrosine kinase 2 |
| Ub | ubiquitin |
| UL | unique long |
| UPS | ubiquitin-proteasome system |
| US | unique short |
References
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef]
- Lachmann, R.; Loenenbach, A.; Waterboer, T.; Brenner, N.; Pawlita, M.; Michel, A.; Thamm, M.; Poethko-Muller, C.; Wichmann, O.; Wiese-Posselt, M. Cytomegalovirus (CMV) seroprevalence in the adult population of Germany. PLoS ONE 2018, 13, e0200267. [Google Scholar] [CrossRef]
- Rafailidis, P.I.; Mourtzoukou, E.G.; Varbobitis, I.C.; Falagas, M.E. Severe cytomegalovirus infection in apparently immunocompetent patients: A systematic review. Virol. J. 2008, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.W.; Teich, S.A. Cytomegalovirus infection in patients with HIV infection. Mt. Sinai J. Med. 1999, 66, 113–124. [Google Scholar] [PubMed]
- Gianella, S.; Letendre, S. Cytomegalovirus and HIV: A Dangerous Pas de Deux. J. Infect. Dis. 2016, 214 (Suppl. 2), S67–S74. [Google Scholar] [CrossRef] [Green Version]
- Humar, A.; Snydman, D. AST Infectious Diseases Community of Practice. Cytomegalovirus in solid organ transplant recipients. Am. J. Transplant. 2009, 9 (Suppl. 4), S78–S86. [Google Scholar] [CrossRef]
- Schmidt-Hieber, M.; Labopin, M.; Beelen, D.; Volin, L.; Ehninger, G.; Finke, J.; Socie, G.; Schwerdtfeger, R.; Kroger, N.; Ganser, A.; et al. CMV serostatus still has an important prognostic impact in de novo acute leukemia patients after allogeneic stem cell transplantation: A report from the Acute Leukemia Working Party of EBMT. Blood 2013, 122, 3359–3364. [Google Scholar] [CrossRef]
- Cannon, M.J.; Davis, K.F. Washing our hands of the congenital cytomegalovirus disease epidemic. BMC Public Health 2005, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.J.; Westbrook, K.; Levis, D.; Schleiss, M.R.; Thackeray, R.; Pass, R.F. Awareness of and behaviors related to child-to-mother transmission of cytomegalovirus. Prev. Med. 2012, 54, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Boppana, S.B. Vaccination against the human cytomegalovirus. Vaccine 2018. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, M.R. Cytomegalovirus vaccines under clinical development. J. Virus Erad. 2016, 2, 198–207. [Google Scholar]
- Schleiss, M.R.; Permar, S.R.; Plotkin, S.A. Progress toward Development of a Vaccine against Congenital Cytomegalovirus Infection. Clin. Vaccine Immunol. 2017, 24, e00268-17. [Google Scholar] [CrossRef]
- Chee, M.S.; Bankier, A.T.; Beck, S.; Bohni, R.; Brown, C.M.; Cerny, R.; Horsnell, T.; Hutchison, C.A., 3rd; Kouzarides, T.; Martignetti, J.A.; et al. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 1990, 154, 125–169. [Google Scholar]
- Brizic, I.; Lisnic, B.; Brune, W.; Hengel, H.; Jonjic, S. Cytomegalovirus Infection: Mouse Model. Curr. Protoc. Immunol. 2018. [Google Scholar] [CrossRef]
- Balazs, Z.; Tombacz, D.; Szucs, A.; Csabai, Z.; Megyeri, K.; Petrov, A.N.; Snyder, M.; Boldogkoi, Z. Long-Read Sequencing of Human Cytomegalovirus Transcriptome Reveals RNA Isoforms Carrying Distinct Coding Potentials. Sci. Rep. 2017, 7, 15989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erhard, F.; Halenius, A.; Zimmermann, C.; L’Hernault, A.; Kowalewski, D.J.; Weekes, M.P.; Stevanovic, S.; Zimmer, R.; Dolken, L. Improved Ribo-seq enables identification of cryptic translation events. Nat. Methods 2018, 15, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W.; et al. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.; Hein, M.Y.; Huang, S.X.; Ma, M.; Shen, B.; Qian, S.B.; Hengel, H.; et al. Decoding human cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodson, E.M.; Ladhani, M.; Webster, A.C.; Strippoli, G.F.; Craig, J.C. Antiviral medications for preventing cytomegalovirus disease in solid organ transplant recipients. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danziger-Isakov, L.; Mark Baillie, G. Hematologic complications of anti-CMV therapy in solid organ transplant recipients. Clin. Transplant. 2009, 23, 295–304. [Google Scholar] [CrossRef]
- Izzedine, H.; Launay-Vacher, V.; Deray, G. Antiviral drug-induced nephrotoxicity. Am. J. Kidney Dis. 2005, 45, 804–817. [Google Scholar] [CrossRef]
- Klug, S.; Lewandowski, C.; Merker, H.J.; Stahlmann, R.; Wildi, L.; Neubert, D. In vitro and in vivo studies on the prenatal toxicity of five virustatic nucleoside analogues in comparison to aciclovir. Arch. Toxicol. 1991, 65, 283–291. [Google Scholar] [CrossRef]
- Gohring, K.; Hamprecht, K.; Jahn, G. Antiviral Drug- and Multidrug Resistance in Cytomegalovirus Infected SCT Patients. Comput. Struct. Biotechnol. J. 2015, 13, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lurain, N.S.; Chou, S. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef] [PubMed]
- Marty, F.M.; Winston, D.J.; Chemaly, R.F.; Mullane, K.M.; Shore, T.B.; Papanicolaou, G.A.; Chittick, G.; Brundage, T.M.; Wilson, C.; Morrison, M.E.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 3 Trial of Oral Brincidofovir for Cytomegalovirus Prophylaxis in Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, J. Cytomegalovirus: Time for a requiem? Blood 2008, 111, 5265–5266. [Google Scholar] [CrossRef]
- Marty, F.M.; Ljungman, P.; Papanicolaou, G.A.; Winston, D.J.; Chemaly, R.F.; Strasfeld, L.; Young, J.A.; Rodriguez, T.; Maertens, J.; Schmitt, M.; et al. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: A phase 3, double-blind, placebo-controlled, randomised trial. Lancet Infect. Dis. 2011, 11, 284–292. [Google Scholar] [CrossRef]
- Papanicolaou, G.A.; Silveira, F.P.; Langston, A.A.; Pereira, M.R.; Avery, R.K.; Uknis, M.; Wijatyk, A.; Wu, J.; Boeckh, M.; Marty, F.M.; et al. Maribavir for Refractory or Resistant Cytomegalovirus Infections in Hematopoietic-cell or Solid-organ Transplant Recipients: A Randomized, Dose-ranging, Double-blind, Phase 2 Study. Clin. Infect. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef] [PubMed]
- Chou, S. Rapid In Vitro Evolution of Human Cytomegalovirus UL56 Mutations That Confer Letermovir Resistance. Antimicrob. Agents Chemother. 2015, 59, 6588–6593. [Google Scholar] [CrossRef] [Green Version]
- Frietsch, J.J.; Michel, D.; Stamminger, T.; Hunstig, F.; Birndt, S.; Schnetzke, U.; Scholl, S.; Hochhaus, A.; Hilgendorf, I. In Vivo Emergence of UL56 C325Y Cytomegalovirus Resistance to Letermovir in a Patient with Acute Myeloid Leukemia after Hematopoietic Cell Transplantation. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019001. [Google Scholar] [CrossRef]
- Chou, S.; Satterwhite, L.E.; Ercolani, R.J. New Locus of Drug Resistance in the Human Cytomegalovirus UL56 Gene Revealed by In Vitro Exposure to Letermovir and Ganciclovir. Antimicrob. Agents Chemother. 2018, 62, e00922-18. [Google Scholar] [CrossRef]
- Hodowanec, A.C.; Pikis, A.; Komatsu, T.E.; Sampson, M.R.; Younis, I.R.; O’Rear, J.J.; Singer, M.E. Treatment and Prevention of CMV Disease in Transplant Recipients: Current Knowledge and Future Perspectives. J. Clin. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef]
- Majoros, A.; Platanitis, E.; Kernbauer-Holzl, E.; Rosebrock, F.; Muller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef]
- Karaghiosoff, M.; Neubauer, H.; Lassnig, C.; Kovarik, P.; Schindler, H.; Pircher, H.; McCoy, B.; Bogdan, C.; Decker, T.; Brem, G.; et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity 2000, 13, 549–560. [Google Scholar] [CrossRef]
- Le-Trilling, V.T.K.; Wohlgemuth, K.; Ruckborn, M.U.; Jagnjic, A.; Maassen, F.; Timmer, L.; Katschinski, B.; Trilling, M. STAT2-Dependent Immune Responses Ensure Host Survival despite the Presence of a Potent Viral Antagonist. J. Virol. 2018, 92, e00296-18. [Google Scholar] [CrossRef]
- Matsumoto, M.; Tanaka, N.; Harada, H.; Kimura, T.; Yokochi, T.; Kitagawa, M.; Schindler, C.; Taniguchi, T. Activation of the transcription factor ISGF3 by interferon-gamma. Biol. Chem. 1999, 380, 699–703. [Google Scholar] [CrossRef]
- Takaoka, A.; Mitani, Y.; Suemori, H.; Sato, M.; Yokochi, T.; Noguchi, S.; Tanaka, N.; Taniguchi, T. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science 2000, 288, 2357–2360. [Google Scholar] [CrossRef]
- Trilling, M.; Bellora, N.; Rutkowski, A.J.; de Graaf, M.; Dickinson, P.; Robertson, K.; Prazeres da Costa, O.; Ghazal, P.; Friedel, C.C.; Alba, M.M.; et al. Deciphering the modulation of gene expression by type I and II interferons combining 4sU-tagging, translational arrest and in silico promoter analysis. Nucleic Acids Res. 2013, 41, 8107–8125. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, A.; Trilling, M.; Wagner, M.; Wilborn, M.; Bubic, I.; Jonjic, S.; Koszinowski, U.; Hengel, H. A cytomegaloviral protein reveals a dual role for STAT2 in IFN-{gamma} signaling and antiviral responses. J. Exp. Med. 2005, 201, 1543–1553. [Google Scholar] [CrossRef]
- Kueck, T.; Cassella, E.; Holler, J.; Kim, B.; Bieniasz, P.D. The aryl hydrocarbon receptor and interferon gamma generate antiviral states via transcriptional repression. Elife 2018, 7, e38867. [Google Scholar] [CrossRef]
- Megger, D.A.; Philipp, J.; Le-Trilling, V.T.K.; Sitek, B.; Trilling, M. Deciphering of the Human Interferon-Regulated Proteome by Mass Spectrometry-Based Quantitative Analysis Reveals Extent and Dynamics of Protein Induction and Repression. Front. Immunol. 2017, 8, 1139. [Google Scholar] [CrossRef]
- Glasgow, L.A.; Hanshaw, J.B.; Merigan, T.C.; Petralli, J.K. Interferon and cytomegalovirus in vivo and in vitro. Proc. Soc. Exp. Biol. Med. 1967, 125, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- McDowell, G.S.; Philpott, A. Non-canonical ubiquitylation: Mechanisms and consequences. Int. J. Biochem. Cell Biol. 2013, 45, 1833–1842. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Li, X.; Gygi, S.P.; Harper, J.W. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 2007, 447, 1135–1138. [Google Scholar] [CrossRef]
- Pelzer, C.; Kassner, I.; Matentzoglu, K.; Singh, R.K.; Wollscheid, H.P.; Scheffner, M.; Schmidtke, G.; Groettrup, M. UBE1L2, a novel E1 enzyme specific for ubiquitin. J. Biol. Chem. 2007, 282, 23010–23014. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef]
- Emmerich, C.H.; Schmukle, A.C.; Walczak, H. The emerging role of linear ubiquitination in cell signaling. Sci. Signal. 2011, 4, re5. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kitagaki, J.; Dai, R.M.; Tsai, Y.C.; Lorick, K.L.; Ludwig, R.L.; Pierre, S.A.; Jensen, J.P.; Davydov, I.V.; Oberoi, P.; et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007, 67, 9472–9481. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.; Stock, D.; Jap, B.; Zwickl, P.; Baumeister, W.; Huber, R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995, 268, 533–539. [Google Scholar] [CrossRef]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Fenteany, G.; Standaert, R.F.; Lane, W.S.; Choi, S.; Corey, E.J.; Schreiber, S.L. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995, 268, 726–731. [Google Scholar] [CrossRef]
- Tsubuki, S.; Saito, Y.; Tomioka, M.; Ito, H.; Kawashima, S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J. Biochem. 1996, 119, 572–576. [Google Scholar] [CrossRef]
- Teicher, B.A.; Ara, G.; Herbst, R.; Palombella, V.J.; Adams, J. The proteasome inhibitor PS-341 in cancer therapy. Clin. Cancer Res. 1999, 5, 2638–2645. [Google Scholar]
- Kisselev, A.F.; Groettrup, M. Subunit specific inhibitors of proteasomes and their potential for immunomodulation. Curr. Opin. Chem. Biol. 2014, 23, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef]
- Basler, M.; Kirk, C.J.; Groettrup, M. The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol. 2013, 25, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Miller, Z.; Ao, L.; Kim, K.B.; Lee, W. Inhibitors of the immunoproteasome: Current status and future directions. Curr. Pharm. Des. 2013, 19, 4140–4151. [Google Scholar] [CrossRef]
- Ho, Y.K.; Bargagna-Mohan, P.; Wehenkel, M.; Mohan, R.; Kim, K.B. LMP2-specific inhibitors: Chemical genetic tools for proteasome biology. Chem. Biol. 2007, 14, 419–430. [Google Scholar] [CrossRef]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Kovacev, J.; Pan, Z.Q. Priming and extending: A UbcH5/Cdc34 E2 handoff mechanism for polyubiquitination on a SCF substrate. Mol. Cell 2010, 37, 784–796. [Google Scholar] [CrossRef]
- Liu, J.; Furukawa, M.; Matsumoto, T.; Xiong, Y. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol. Cell 2002, 10, 1511–1518. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Enchev, R.I.; Schulman, B.A.; Peter, M. Protein neddylation: Beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lu, J.; Liu, L.; Bernard, D.; Yang, C.Y.; Fernandez-Salas, E.; Chinnaswamy, K.; Layton, S.; Stuckey, J.; Yu, Q.; et al. A potent small-molecule inhibitor of the DCN1-UBC12 interaction that selectively blocks cullin 3 neddylation. Nat. Commun. 2017, 8, 1150. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, D.F.; Tang, X.; Pelletier, B.; Orlicky, S.; Xie, W.; Plantevin, V.; Neculai, D.; Chou, Y.C.; Ogunjimi, A.; Al-Hakim, A.; et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell 2011, 145, 1075–1087. [Google Scholar] [CrossRef]
- Dualan, R.; Brody, T.; Keeney, S.; Nichols, A.F.; Admon, A.; Linn, S. Chromosomal localization and cDNA cloning of the genes (DDB1 and DDB2) for the p127 and p48 subunits of a human damage-specific DNA binding protein. Genomics 1995, 29, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Keeney, S.; Chang, G.J.; Linn, S. Characterization of a human DNA damage binding protein implicated in xeroderma pigmentosum E. J. Biol. Chem. 1993, 268, 21293–21300. [Google Scholar] [PubMed]
- Shiyanov, P.; Nag, A.; Raychaudhuri, P. Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J. Biol. Chem. 1999, 274, 35309–35312. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, X.; Garbutt, K.C.; Zhou, P.; Zheng, N. Structure of DDB1 in complex with a paramyxovirus V protein: Viral hijack of a propeller cluster in ubiquitin ligase. Cell 2006, 124, 105–117. [Google Scholar] [CrossRef]
- Cang, Y.; Zhang, J.; Nicholas, S.A.; Bastien, J.; Li, B.; Zhou, P.; Goff, S.P. Deletion of DDB1 in mouse brain and lens leads to p53-dependent elimination of proliferating cells. Cell 2006, 127, 929–940. [Google Scholar] [CrossRef]
- Cang, Y.; Zhang, J.; Nicholas, S.A.; Kim, A.L.; Zhou, P.; Goff, S.P. DDB1 is essential for genomic stability in developing epidermis. Proc. Natl. Acad. Sci. USA 2007, 104, 2733–2737. [Google Scholar] [CrossRef] [Green Version]
- Wakasugi, M.; Matsuura, K.; Nagasawa, A.; Fu, D.; Shimizu, H.; Yamamoto, K.; Takeda, S.; Matsunaga, T. DDB1 gene disruption causes a severe growth defect and apoptosis in chicken DT40 cells. Biochem. Biophys. Res. Commun. 2007, 364, 771–777. [Google Scholar] [CrossRef]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef]
- Li, T.; Robert, E.I.; van Breugel, P.C.; Strubin, M.; Zheng, N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat. Struct. Mol. Biol. 2010, 17, 105–111. [Google Scholar] [CrossRef]
- Emanuele, M.J.; Elia, A.E.; Xu, Q.; Thoma, C.R.; Izhar, L.; Leng, Y.; Guo, A.; Chen, Y.N.; Rush, J.; Hsu, P.W.; et al. Global identification of modular cullin-RING ligase substrates. Cell 2011, 147, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. CRL4Cdt2: Master coordinator of cell cycle progression and genome stability. Cell Cycle 2011, 10, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.; Fruh, K. Viral modulators of cullin RING ubiquitin ligases: Culling the host defense. Sci. Stke 2006, 2006, pe21. [Google Scholar] [CrossRef]
- Mahon, C.; Krogan, N.J.; Craik, C.S.; Pick, E. Cullin E3 ligases and their rewiring by viral factors. Biomolecules 2014, 4, 897–930. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.A.; Lee, T.H.; Butel, J.S.; Slagle, B.L. Hepatitis B virus X protein interferes with cellular DNA repair. J. Virol. 1998, 72, 266–272. [Google Scholar] [PubMed]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed]
- Van Breugel, P.C.; Robert, E.I.; Mueller, H.; Decorsiere, A.; Zoulim, F.; Hantz, O.; Strubin, M. Hepatitis B virus X protein stimulates gene expression selectively from extrachromosomal DNA templates. Hepatology 2012, 56, 2116–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Vu, T.; Novince, Z.; Guerrero-Santoro, J.; Rapic-Otrin, V.; Gronenborn, A.M. HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. J. Biol. Chem. 2010, 285, 37333–37341. [Google Scholar] [CrossRef]
- Belzile, J.P.; Duisit, G.; Rougeau, N.; Mercier, J.; Finzi, A.; Cohen, E.A. HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 2007, 3, e85. [Google Scholar] [CrossRef]
- Hrecka, K.; Gierszewska, M.; Srivastava, S.; Kozaczkiewicz, L.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. USA 2007, 104, 11778–11783. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chiang, S.F.; Lin, T.Y.; Chiou, S.H.; Chow, K.C. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PLoS ONE 2012, 7, e33657. [Google Scholar] [CrossRef]
- Laguette, N.; Bregnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef]
- Lahouassa, H.; Blondot, M.L.; Chauveau, L.; Chougui, G.; Morel, M.; Leduc, M.; Guillonneau, F.; Ramirez, B.C.; Schwartz, O.; Margottin-Goguet, F. HIV-1 Vpr degrades the HLTF DNA translocase in T cells and macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 5311–5316. [Google Scholar] [CrossRef] [Green Version]
- Romani, B.; Baygloo, N.S.; Hamidi-Fard, M.; Aghasadeghi, M.R.; Allahbakhshi, E. HIV-1 Vpr Protein Induces Proteasomal Degradation of Chromatin-associated Class I HDACs to Overcome Latent Infection of Macrophages. J. Biol. Chem. 2016, 291, 2696–2711. [Google Scholar] [CrossRef] [PubMed]
- Romani, B.; Shaykh Baygloo, N.; Aghasadeghi, M.R.; Allahbakhshi, E. HIV-1 Vpr Protein Enhances Proteasomal Degradation of MCM10 DNA Replication Factor through the Cul4-DDB1[VprBP] E3 Ubiquitin Ligase to Induce G2/M Cell Cycle Arrest. J. Biol. Chem. 2015, 290, 17380–17389. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Ehrlich, E.; Yu, X.F. DDB1 and Cul4A are required for human immunodeficiency virus type 1 Vpr-induced G2 arrest. J. Virol. 2007, 81, 10822–10830. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Singh, S.; Jung, H.Y.; Yang, G.; Jun, S.; Sastry, K.J.; Park, J.I. HIV-1 Vpr protein inhibits telomerase activity via the EDD-DDB1-VPRBP E3 ligase complex. J. Biol. Chem. 2013, 288, 15474–15480. [Google Scholar] [CrossRef]
- Ahn, J.; Hao, C.; Yan, J.; DeLucia, M.; Mehrens, J.; Wang, C.; Gronenborn, A.M.; Skowronski, J. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J. Biol. Chem. 2012, 287, 12550–12558. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Sharova, N.; Wu, Y.; Zhu, X.; Stranska, R.; Kaushik, R.; Sharkey, M.; Stevenson, M. Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 2008, 4, e1000057. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Swanson, S.K.; Manel, N.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008, 4, e1000059. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Guo, H.; Gao, Q.; Markham, R.; Yu, X.F. Variation of two primate lineage-specific residues in human SAMHD1 confers resistance to N terminus-targeted SIV Vpx proteins. J. Virol. 2014, 88, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Guo, H.; Liu, X.; Zhang, H.; Qian, L.; Luo, K.; Markham, R.B.; Yu, X.F. A first-in-class NAE inhibitor, MLN4924, blocks lentiviral infection in myeloid cells by disrupting neddylation-dependent Vpx-mediated SAMHD1 degradation. J. Virol. 2014, 88, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, J.; Poole, E.; Young, D.F.; Goodbourn, S.; Randall, R.E. The p127 subunit (DDB1) of the UV-DNA damage repair binding protein is essential for the targeted degradation of STAT1 by the V protein of the paramyxovirus simian virus 5. J. Virol. 2002, 76, 11379–11386. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.Y.; Paterson, R.G.; Richardson, C.D.; Lamb, R.A. The V protein of the paramyxovirus SV5 interacts with damage-specific DNA binding protein. Virology 1998, 249, 189–200. [Google Scholar] [CrossRef]
- Ulane, C.M.; Horvath, C.M. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 2002, 304, 160–166. [Google Scholar] [CrossRef]
- Afroz, S.; Garg, R.; Fodje, M.; van Drunen Littel-van den Hurk, S. The Major Tegument Protein of Bovine Herpesvirus 1, VP8, Interacts with DNA Damage Response Proteins and Induces Apoptosis. J. Virol. 2018, 92, e00773-18. [Google Scholar] [CrossRef]
- Vasilenko, N.L.; Snider, M.; Labiuk, S.L.; Lobanov, V.A.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. Bovine herpesvirus-1 VP8 interacts with DNA damage binding protein-1 (DDB1) and is monoubiquitinated during infection. Virus Res. 2012, 167, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Pickering, M.T.; Cho, N.H.; Chang, H.; Volkert, M.R.; Kowalik, T.F.; Jung, J.U. Deregulation of DNA damage signal transduction by herpesvirus latency-associated M2. J. Virol. 2006, 80, 5862–5874. [Google Scholar] [CrossRef] [PubMed]
- Trilling, M.; Le, V.T.; Fiedler, M.; Zimmermann, A.; Bleifuss, E.; Hengel, H. Identification of DNA-damage DNA-binding protein 1 as a conditional essential factor for cytomegalovirus replication in interferon-gamma-stimulated cells. PLoS Pathog. 2011, 7, e1002069. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, C.D.; Megger, D.A.; Hotter, D.; Ruckborn, M.U.; Eilbrecht, M.; Rashidi-Alavijeh, J.; Howe, S.; Heinrichs, S.; Sauter, D.; Sitek, B.; et al. A Mass Spectrometry-Based Profiling of Interactomes of Viral DDB1- and Cullin Ubiquitin Ligase-Binding Proteins Reveals NF-kappaB Inhibitory Activity of the HIV-2-Encoded Vpx. Front. Immunol. 2018, 9, 2978. [Google Scholar] [CrossRef] [PubMed]
- Le-Trilling, V.T.; Megger, D.A.; Katschinski, B.; Landsberg, C.D.; Ruckborn, M.U.; Tao, S.; Krawczyk, A.; Bayer, W.; Drexler, I.; Tenbusch, M.; et al. Broad and potent antiviral activity of the NAE inhibitor MLN4924. Sci. Rep. 2016, 6, 19977. [Google Scholar] [CrossRef] [Green Version]
- Salsman, J.; Jagannathan, M.; Paladino, P.; Chan, P.K.; Dellaire, G.; Raught, B.; Frappier, L. Proteomic profiling of the human cytomegalovirus UL35 gene products reveals a role for UL35 in the DNA repair response. J. Virol. 2012, 86, 806–820. [Google Scholar] [CrossRef]
- Nightingale, K.; Lin, K.M.; Ravenhill, B.J.; Davies, C.; Nobre, L.; Fielding, C.A.; Ruckova, E.; Fletcher-Etherington, A.; Soday, L.; Nichols, H.; et al. High-Definition Analysis of Host Protein Stability during Human Cytomegalovirus Infection Reveals Antiviral Factors and Viral Evasion Mechanisms. Cell Host Microbe 2018, 24, 447–460.e11. [Google Scholar] [CrossRef]
- Abenes, G.; Lee, M.; Haghjoo, E.; Tong, T.; Zhan, X.; Liu, F. Murine cytomegalovirus open reading frame M27 plays an important role in growth and virulence in mice. J. Virol. 2001, 75, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Abenes, G.; Zhan, X.; Dunn, W.; Haghjoo, E.; Tong, T.; Tam, A.; Chan, K.; Liu, F. Genetic analyses of gene function and pathogenesis of murine cytomegalovirus by transposon-mediated mutagenesis. J. Clin. Virol. 2002, 25 (Suppl. 2), S111–S122. [Google Scholar] [CrossRef]
- Zhan, X.; Lee, M.; Abenes, G.; Von Reis, I.; Kittinunvorakoon, C.; Ross-Macdonald, P.; Snyder, M.; Liu, F. Mutagenesis of murine cytomegalovirus using a Tn3-based transposon. Virology 2000, 266, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Zimmermann, A.; Basler, M.; Groettrup, M.; Hengel, H. A cytomegalovirus inhibitor of gamma interferon signaling controls immunoproteasome induction. J. Virol. 2004, 78, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Le, V.T.; Trilling, M.; Zimmermann, A.; Hengel, H. Mouse cytomegalovirus inhibits beta interferon (IFN-beta) gene expression and controls activation pathways of the IFN-beta enhanceosome. J. Gen. Virol. 2008, 89, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Doring, M.; Lessin, I.; Frenz, T.; Spanier, J.; Kessler, A.; Tegtmeyer, P.; Dag, F.; Thiel, N.; Trilling, M.; Lienenklaus, S.; et al. M27 expressed by cytomegalovirus counteracts effective type I interferon induction of myeloid cells but not of plasmacytoid dendritic cells. J. Virol. 2014, 88, 13638–13650. [Google Scholar] [CrossRef]
- Barchet, W.; Cella, M.; Odermatt, B.; Asselin-Paturel, C.; Colonna, M.; Kalinke, U. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J. Exp. Med. 2002, 195, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Unterstab, G.; Ludwig, S.; Anton, A.; Planz, O.; Dauber, B.; Krappmann, D.; Heins, G.; Ehrhardt, C.; Wolff, T. Viral targeting of the interferon-{beta}-inducing Traf family member-associated NF-{kappa}B activator (TANK)-binding kinase-1. Proc. Natl. Acad. Sci. USA 2005, 102, 13640–13645. [Google Scholar] [CrossRef]
- Reitsma, J.M.; Savaryn, J.P.; Faust, K.; Sato, H.; Halligan, B.D.; Terhune, S.S. Antiviral inhibition targeting the HCMV kinase pUL97 requires pUL27-dependent degradation of Tip60 acetyltransferase and cell-cycle arrest. Cell Host Microbe 2011, 9, 103–114. [Google Scholar] [CrossRef]
- Le, V.T.; Trilling, M.; Wilborn, M.; Hengel, H.; Zimmermann, A. Human cytomegalovirus interferes with signal transducer and activator of transcription (STAT) 2 protein stability and tyrosine phosphorylation. J. Gen. Virol. 2008, 89, 2416–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, A.; Zhu, J.; Hai, R.; Haghjoo, E.; Tong, T.; Zhan, X.; Lu, S.; Liu, F. Murine cytomegalovirus with a transposon insertional mutation at open reading frame M35 is defective in growth in vivo. J. Virol. 2003, 77, 7746–7755. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.; Goncalves Magalhaes, V.; Lemmermann, N.A.W.; Juranic Lisnic, V.; Stempel, M.; Bussey, K.A.; Reimer, E.; Podlech, J.; Lienenklaus, S.; Reddehase, M.J.; et al. The murine cytomegalovirus M35 protein antagonizes type I IFN induction downstream of pattern recognition receptors by targeting NF-kappaB mediated transcription. PLoS Pathog. 2017, 13, e1006382. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Biegalke, B.J. The human cytomegalovirus UL35 gene encodes two proteins with different functions. J. Virol. 2002, 76, 2460–2468. [Google Scholar] [CrossRef] [PubMed]
- Schierling, K.; Buser, C.; Mertens, T.; Winkler, M. Human cytomegalovirus tegument protein ppUL35 is important for viral replication and particle formation. J. Virol. 2005, 79, 3084–3096. [Google Scholar] [CrossRef]
- Schierling, K.; Stamminger, T.; Mertens, T.; Winkler, M. Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer. J. Virol. 2004, 78, 9512–9523. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kalejta, R.F.; Kerry, J.; Semmes, O.J.; O’Connor, C.M.; Khan, Z.; Garcia, B.A.; Shenk, T.; Murphy, E. BclAF1 restriction factor is neutralized by proteasomal degradation and microRNA repression during human cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2012, 109, 9575–9580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salsman, J.; Wang, X.; Frappier, L. Nuclear body formation and PML body remodeling by the human cytomegalovirus protein UL35. Virology 2011, 414, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maschkowitz, G.; Gartner, S.; Hofmann-Winkler, H.; Fickenscher, H.; Winkler, M. Interaction of Human Cytomegalovirus Tegument Proteins ppUL35 and ppUL35A with Sorting Nexin 5 Regulates Glycoprotein B (gpUL55) Localization. J. Virol. 2018, 92, e00013-18. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Lu, Y.; Ruan, Q.; Ji, Y.; He, R.; Qi, Y.; Ma, Y.; Huang, Y. Human cytomegalovirus UL145 gene is highly conserved among clinical strains. J. Biosci. 2007, 32, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hu, J.J.; Yan, C.F.; Su, H.H.; Ding, J.C.; Guo, Y.Y.; Ye, N.; Zhang, S.Q.; Zhang, X.Z.; Zhou, S.F. Characterization of human cytomegalovirus UL145 and UL136 genes in low-passage clinical isolates from infected Chinese infants. Med. Sci. Monit. 2011, 17, CR423–CR431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Ma, Y.; Sun, Z.; Qi, Y.; Ji, Y.; He, R.; Li, M.; Ruan, Q. Transcriptional features and transcript structure of UL145 in different strains of human cytomegalovirus. J. Med. Virol. 2011, 83, 2151–2156. [Google Scholar] [CrossRef]
- Qi, Y.; Ma, Y.; He, R.; Wang, N.; Ruan, Q.; Ji, Y.; Li, M.; Sun, Z.; Ren, G. Characterization of 3′ termini of human cytomegalovirus UL138-UL145 transcripts in a clinical strain. Microbiol. Immunol. 2011, 55, 95–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motegi, A.; Liaw, H.J.; Lee, K.Y.; Roest, H.P.; Maas, A.; Wu, X.; Moinova, H.; Markowitz, S.D.; Ding, H.; Hoeijmakers, J.H.; et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. USA 2008, 105, 12411–12416. [Google Scholar] [CrossRef] [Green Version]
- Unk, I.; Hajdu, I.; Fatyol, K.; Hurwitz, J.; Yoon, J.H.; Prakash, L.; Prakash, S.; Haracska, L. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc. Natl. Acad. Sci. USA 2008, 105, 3768–3773. [Google Scholar] [CrossRef]
- Achar, Y.J.; Balogh, D.; Haracska, L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc. Natl. Acad. Sci. USA 2011, 108, 14073–14078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blastyak, A.; Hajdu, I.; Unk, I.; Haracska, L. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol. Cell. Biol. 2010, 30, 684–693. [Google Scholar] [CrossRef]
- Ding, H.; Descheemaeker, K.; Marynen, P.; Nelles, L.; Carvalho, T.; Carmo-Fonseca, M.; Collen, D.; Belayew, A. Characterization of a helicase-like transcription factor involved in the expression of the human plasminogen activator inhibitor-1 gene. DNA Cell Biol. 1996, 15, 429–442. [Google Scholar] [CrossRef]
- Mahajan, M.C.; Weissman, S.M. DNA-dependent adenosine triphosphatase (helicaselike transcription factor) activates beta-globin transcription in K562 cells. Blood 2002, 99, 348–356. [Google Scholar] [CrossRef]
- Schleker, S.; Trilling, M. Data-warehousing of protein-protein interactions indicates that pathogens preferentially target hub and bottleneck proteins. Front. Microbiol. 2013, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Kaspari, M.; Tavalai, N.; Stamminger, T.; Zimmermann, A.; Schilf, R.; Bogner, E. Proteasome inhibitor MG132 blocks viral DNA replication and assembly of human cytomegalovirus. FEBS Lett. 2008, 582, 666–672. [Google Scholar] [CrossRef] [Green Version]
- Prosch, S.; Priemer, C.; Hoflich, C.; Liebenthaf, C.; Babel, N.; Kruger, D.H.; Volk, H.D. Proteasome inhibitors: A novel tool to suppress human cytomegalovirus replication and virus-induced immune modulation. Antivir. Ther. 2003, 8, 555–567. [Google Scholar] [PubMed]
- Tran, K.; Mahr, J.A.; Spector, D.H. Proteasome subunits relocalize during human cytomegalovirus infection, and proteasome activity is necessary for efficient viral gene transcription. J. Virol. 2010, 84, 3079–3093. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Perel, G.; Bliss, J.; Thomas, C.M. Carfilzomib (Kyprolis): A Novel Proteasome Inhibitor for Relapsed and/or Refractory Multiple Myeloma. Pharm. Ther. 2016, 41, 303–307. [Google Scholar]
- Basler, M.; Lauer, C.; Beck, U.; Groettrup, M. The proteasome inhibitor bortezomib enhances the susceptibility to viral infection. J. Immunol. 2009, 183, 6145–6150. [Google Scholar] [CrossRef]
- Raaben, M.; Grinwis, G.C.; Rottier, P.J.; de Haan, C.A. The proteasome inhibitor Velcade enhances rather than reduces disease in mouse hepatitis coronavirus-infected mice. J. Virol. 2010, 84, 7880–7885. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, K.; Kim, B.S.; Lee, H.J.; Kim, H.; Lee, N.R.; Nam, S.H.; Kwon, J.H.; Kim, H.J.; Sohn, S.K.; et al. Bortezomib and the increased incidence of herpes zoster in patients with multiple myeloma. Clin. Lymphoma Myeloma 2008, 8, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, F.; Mengarelli, A.; Giannotti, F.; Tendas, A.; Anaclerico, B.; Porrini, R.; Picardi, A.; Cerchiara, E.; Dentamaro, T.; Chierichini, A.; et al. High incidence of post-transplant cytomegalovirus reactivations in myeloma patients undergoing autologous stem cell transplantation after treatment with bortezomib-based regimens: A survey from the Rome transplant network. Transpl. Infect. Dis. 2014, 16, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Kelly, K.R.; Smith, P.G.; Garnsey, J.J.; Mahalingam, D.; Medina, E.; Oberheu, K.; Padmanabhan, S.; O’Dwyer, M.; Nawrocki, S.T.; et al. Inhibition of NEDD8-activating enzyme: A novel approach for the treatment of acute myeloid leukemia. Blood 2010, 115, 3796–3800. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.J.; Jakubowiak, A.J.; O’Connor, O.A.; Orlowski, R.Z.; Harvey, R.D.; Smith, M.R.; Lebovic, D.; Diefenbach, C.; Kelly, K.; Hua, Z.; et al. Phase I Study of the Novel Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (MLN4924) in Patients with Relapsed/Refractory Multiple Myeloma or Lymphoma. Clin. Cancer Res. 2016, 22, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Nekorchuk, M.D.; Sharifi, H.J.; Furuya, A.K.; Jellinger, R.; de Noronha, C.M. HIV relies on neddylation for ubiquitin ligase-mediated functions. Retrovirology 2013, 10, 138. [Google Scholar] [CrossRef]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Kota, K.P.; Whitehouse, C.A.; Bavari, S. Protein Kinase R Degradation Is Essential for Rift Valley Fever Virus Infection and Is Regulated by SKP1-CUL1-F-box (SCF)FBXW11-NSs E3 Ligase. PLoS Pathog. 2016, 12, e1005437. [Google Scholar] [CrossRef]
- Sun, H.; Yao, W.; Wang, K.; Qian, Y.; Chen, H.; Jung, Y.S. Inhibition of neddylation pathway represses influenza virus replication and pro-inflammatory responses. Virology 2018, 514, 230–239. [Google Scholar] [CrossRef]
- Chang, P.J.; Chen, L.W.; Chen, L.Y.; Hung, C.H.; Shih, Y.J.; Wang, S.S. Effects of the NEDD8-Activating Enzyme Inhibitor MLN4924 on Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2017, 91, e00505-17. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.J.; Wood, J.J.; Jackson, B.R.; Baquero-Perez, B.; Whitehouse, A. NEDDylation is essential for Kaposi’s sarcoma-associated herpesvirus latency and lytic reactivation and represents a novel anti-KSHV target. PLoS Pathog. 2015, 11, e1004771. [Google Scholar] [CrossRef]
- Sekiba, K.; Otsuka, M.; Ohno, M.; Yamagami, M.; Kishikawa, T.; Seimiya, T.; Suzuki, T.; Tanaka, E.; Ishibashi, R.; Funato, K.; et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, is a potent inhibitor of hepatitis B virus. Hepatology 2018. [Google Scholar] [CrossRef]
- Bulatov, E.; Ciulli, A. Targeting Cullin-RING E3 ubiquitin ligases for drug discovery: Structure, assembly and small-molecule modulation. Biochem. J. 2015, 467, 365–386. [Google Scholar] [CrossRef]
- Wertz, I.E.; Wang, X. From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26, 156–177. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.C.; Xu, Q.; Chou, D.M.; Zhao, Z.; Elledge, S.J. Global protein stability profiling in mammalian cells. Science 2008, 322, 918–923. [Google Scholar] [CrossRef]
- Jones, J.; Wu, K.; Yang, Y.; Guerrero, C.; Nillegoda, N.; Pan, Z.Q.; Huang, L. A targeted proteomic analysis of the ubiquitin-like modifier nedd8 and associated proteins. J. Proteome Res. 2008, 7, 1274–1287. [Google Scholar] [CrossRef] [PubMed]
- Xirodimas, D.P.; Saville, M.K.; Bourdon, J.C.; Hay, R.T.; Lane, D.P. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 2004, 118, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Xirodimas, D.P.; Sundqvist, A.; Nakamura, A.; Shen, L.; Botting, C.; Hay, R.T. Ribosomal proteins are targets for the NEDD8 pathway. EMBO Rep. 2008, 9, 280–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghajan, M.; Jonai, N.; Flick, K.; Fu, F.; Luo, M.; Cai, X.; Ouni, I.; Pierce, N.; Tang, X.; Lomenick, B.; et al. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat. Biotechnol. 2010, 28, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Sekiba, K.; Otsuka, M.; Ohno, M.; Yamagami, M.; Kishikawa, T.; Suzuki, T.; Ishibashi, R.; Seimiya, T.; Tanaka, E.; Koike, K. Inhibition of HBV Transcription From cccDNA with Nitazoxanide by Targeting the HBx-DDB1 Interaction. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Schwefel, D.; Boucherit, V.C.; Christodoulou, E.; Walker, P.A.; Stoye, J.P.; Bishop, K.N.; Taylor, I.A. Molecular determinants for recognition of divergent SAMHD1 proteins by the lentiviral accessory protein Vpx. Cell Host Microbe 2015, 17, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Schwefel, D.; Groom, H.C.; Boucherit, V.C.; Christodoulou, E.; Walker, P.A.; Stoye, J.P.; Bishop, K.N.; Taylor, I.A. Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature 2014, 505, 234–238. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, T.; Le-Trilling, V.T.K.; Trilling, M. Cellular Cullin RING Ubiquitin Ligases: Druggable Host Dependency Factors of Cytomegaloviruses. Int. J. Mol. Sci. 2019, 20, 1636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071636
Becker T, Le-Trilling VTK, Trilling M. Cellular Cullin RING Ubiquitin Ligases: Druggable Host Dependency Factors of Cytomegaloviruses. International Journal of Molecular Sciences. 2019; 20(7):1636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071636
Chicago/Turabian StyleBecker, Tanja, Vu Thuy Khanh Le-Trilling, and Mirko Trilling. 2019. "Cellular Cullin RING Ubiquitin Ligases: Druggable Host Dependency Factors of Cytomegaloviruses" International Journal of Molecular Sciences 20, no. 7: 1636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071636