Calcium Release from Endoplasmic Reticulum Involves Calmodulin-Mediated NADPH Oxidase-Derived Reactive Oxygen Species Production in Endothelial Cells

 , and
, and
Abstract
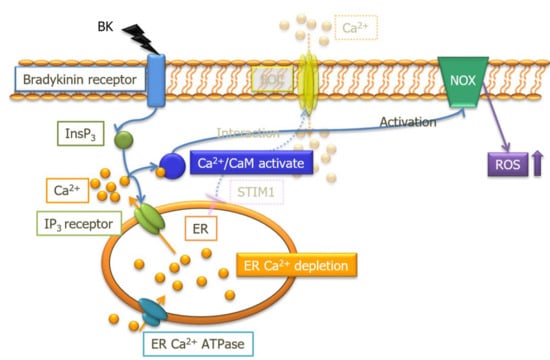
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Bradykinin Increased Cytosolic Ca2+ Concentration
2.2. Bradykinin Increased Cytosolic ROS Production
2.3. Bradykinin-Induced Cytosolic ROS Production was Caused by NOX Activation
2.4. Endoplasmic Reticulum Ca2+ and BK-Induced NOX-Derived ROS Production
2.5. The Ca2+/CaM Pathway Regulated BK-Induced NOX-Derived ROS Production
2.6. The Ca2+/CaM Pathway Regulated BK-Induced NOX-Derived ROS Production
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Measurement of Endothelial Ca2+ Concentration and ROS Production
4.3. Phos-tag™ SDS-PAGE Western Blotting
4.4. Chemicals
4.5. Statistics Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CaM | calmodulin |
| CaMK | Ca2+/calmodulin-dependent protein kinase |
| ER | endoplasmic reticulum |
| GPCR | G-protein coupled receptor |
| NOX | nicotinamide adenine dinucleotide phosphate oxidases |
| PAECs | porcine aortic endothelial cells |
| ROS | Reactive oxygen species |
| SOCE | store-operated Ca2+ entry |
References
- Guzik, T.J.; Harrison, D.G. Vascular nadph oxidases as drug targets for novel antioxidant strategies. Drug Discov. Today 2006, 11, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: Nadph oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef]
- Salazar, G. Nadph oxidases and mitochondria in vascular senescence. Int. J. Mol. Sci. 2018, 19, 1327. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Shah, A.M. Nadph oxidase and endothelial cell function. Clin. Sci. 2005, 109, 217–226. [Google Scholar] [CrossRef]
- D’Autreaux, B.; Toledano, M.B. Ros as signalling molecules: Mechanisms that generate specificity in ros homeostasis. Nat. Rev. Mol. Cell Biol 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Mansego, M.L.; Redon, J.; Martinez-Hervas, S.; Real, J.T.; Martinez, F.; Blesa, S.; Gonzalez-Albert, V.; Saez, G.T.; Carmena, R.; Chaves, F.J. Different impacts of cardiovascular risk factors on oxidative stress. Int. J. Mol. Sci. 2011, 12, 6146–6163. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function--role of nitric oxide synthase uncoupling and nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef]
- Choi, D.H.; Lee, J. A mini-review of the nadph oxidases in vascular dementia: Correlation with noxs and risk factors for vad. Int. J. Mol. Sci. 2017, 18, 2500. [Google Scholar] [CrossRef]
- Li, J.M.; Shah, A.M. Endothelial cell superoxide generation: Regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1014–R1030. [Google Scholar] [CrossRef] [PubMed]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. Nadph oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. Nadph oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed]
- Schramm, A.; Matusik, P.; Osmenda, G.; Guzik, T.J. Targeting nadph oxidases in vascular pharmacology. Vascul. Pharmacol. 2012, 56, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Burger, D.; Paravicini, T.M.; Chignalia, A.Z.; Yusuf, H.; Almasri, M.; He, Y.; Callera, G.E.; He, G.; Krause, K.H.; et al. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (nox5) regulation by angiotensin ii and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ. Res. 2010, 106, 1363–1373. [Google Scholar] [CrossRef]
- Pandey, D.; Gratton, J.P.; Rafikov, R.; Black, S.M.; Fulton, D.J. Calcium/calmodulin-dependent kinase ii mediates the phosphorylation and activation of nadph oxidase 5. Mol. Pharmacol. 2011, 80, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Odagiri, K.; Katoh, H.; Kawashima, H.; Tanaka, T.; Ohtani, H.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J. Mol. Cell Cardiol. 2009, 46, 989–997. [Google Scholar] [CrossRef]
- Takahashi, R.; Watanabe, H.; Kakizawa, H.; Zhang, X.X.; Hayashi, H.; Ohno, R. Regulation of bradykinin-stimulated cation entry into endothelial cells by tyrosine kinase. Jpn Circ. J. 1997, 61, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Watanabe, H.; Zhang, X.X.; Kakizawa, H.; Hayashi, H.; Ohno, R. Roles of inhibitors of myosin light chain kinase and tyrosine kinase on cation influx in agonist-stimulated endothelial cells. Biochem Biophys Res. Commun. 1997, 235, 657–662. [Google Scholar] [CrossRef]
- Brechard, S.; Tschirhart, E.J. Regulation of superoxide production in neutrophils: Role of calcium influx. J. Leukoc. Biol. 2008, 84, 1223–1237. [Google Scholar] [CrossRef]
- Trebak, M.; Ginnan, R.; Singer, H.A.; Jourd’heuil, D. Interplay between calcium and reactive oxygen/nitrogen species: An essential paradigm for vascular smooth muscle signaling. Antioxid. Redox Signal. 2010, 12, 657–674. [Google Scholar] [CrossRef]
- Gallois, A.; Bueb, J.L.; Tschirhart, E. Effect of sk&f 96365 on extracellular Ca2+ -dependent O2− production in neutrophil-like hl-60 cells. Eur. J. Pharmacol. 1998, 361, 293–298. [Google Scholar]
- Pozzan, T.; Lew, D.P.; Wollheim, C.B.; Tsien, R.Y. Is cytosolic ionized calcium regulating neutrophil activation? Science 1983, 221, 1413–1415. [Google Scholar] [CrossRef]
- Foyouzi-Youssefi, R.; Petersson, F.; Lew, D.P.; Krause, K.H.; Nusse, O. Chemoattractant-induced respiratory burst: Increases in cytosolic Ca2+ concentrations are essential and synergize with a kinetically distinct second signal. Biochem J. 1997, 322, 709–718. [Google Scholar] [CrossRef]
- Freay, A.; Johns, A.; Adams, D.J.; Ryan, U.S.; Van Breemen, C. Bradykinin and inositol 1,4,5-trisphosphate-stimulated calcium release from intracellular stores in cultured bovine endothelial cells. Pflugers Arch. 1989, 414, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Mendelowitz, D.; Bacal, K.; Kunze, D.L. Bradykinin-activated calcium influx pathway in bovine aortic endothelial cells. Am. J. Physiol. 1992, 262, H942–H948. [Google Scholar] [CrossRef]
- Takeuchi, K.; Watanabe, H.; Tran, Q.K.; Ozeki, M.; Sumi, D.; Hayashi, T.; Iguchi, A.; Ignarro, L.J.; Ohashi, K.; Hayashi, H. Nitric oxide: Inhibitory effects on endothelial cell calcium signaling, prostaglandin i2 production and nitric oxide synthase expression. Cardiovasc. Res. 2004, 62, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Watanabe, H.; Takahashi, R.; Kosuge, K.; Umemura, K.; Hayashi, H.; Ohashi, K.; Ohno, R. Inhibitory effect of insulin on bradykinin-induced venodilation. J. Hypertens 2000, 18, 287–292. [Google Scholar] [CrossRef]
- Pinheiro, A.R.; Paramos-de-Carvalho, D.; Certal, M.; Costa, C.; Magalhaes-Cardoso, M.T.; Ferreirinha, F.; Costa, M.A.; Correia-de-Sa, P. Bradykinin-induced Ca2+ signaling in human subcutaneous fibroblasts involves atp release via hemichannels leading to p2y12 receptors activation. Cell Commun. Signal. 2013, 11, 70. [Google Scholar] [CrossRef]
- Wei, J.; Takeuchi, K.; Watanabe, H. Linoleic acid attenuates endothelium-derived relaxing factor production by suppressing camp-hydrolyzing phosphodiesterase activity. Circ. J. 2013, 77, 2823–2830. [Google Scholar] [CrossRef]
- Duerrschmidt, N.; Wippich, N.; Goettsch, W.; Broemme, H.J.; Morawietz, H. Endothelin-1 induces nad(p)h oxidase in human endothelial cells. Biochem. Biophys. Res. Commun. 2000, 269, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Shah, A.M. Mechanism of endothelial cell nadph oxidase activation by angiotensin ii. Role of the p47phox subunit. J. Biol. Chem. 2003, 278, 12094–12100. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Wang, H.H.; Wu, C.Y.; Yang, C.M. Reactive oxygen species-dependent c-fos/activator protein 1 induction upregulates heme oxygenase-1 expression by bradykinin in brain astrocytes. Antioxid. Redox Signal. 2010, 13, 1829–1844. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Hsieh, H.L.; Shih, R.H.; Chi, P.L.; Cheng, S.E.; Chen, J.C.; Yang, C.M. Nadph oxidase 2-derived reactive oxygen species signal contributes to bradykinin-induced matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Cell Commun. Signal. 2012, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schroder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular nadph oxidases but an antioxidant. Hypertension 2008, 51, 211–217. [Google Scholar] [CrossRef]
- Altenhofer, S.; Kleikers, P.W.; Radermacher, K.A.; Scheurer, P.; Rob Hermans, J.J.; Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H. The nox toolbox: Validating the role of nadph oxidases in physiology and disease. Cell Mol. Life Sci. 2012, 69, 2327–2343. [Google Scholar] [CrossRef]
- Yao, X.; Garland, C.J. Recent developments in vascular endothelial cell transient receptor potential channels. Circ. Res. 2005, 97, 853–863. [Google Scholar] [CrossRef]
- Tran, Q.K.; Ohashi, K.; Watanabe, H. Calcium signalling in endothelial cells. Cardiovasc. Res. 2000, 48, 13–22. [Google Scholar] [CrossRef]
- Serrander, L.; Jaquet, V.; Bedard, K.; Plastre, O.; Hartley, O.; Arnaudeau, S.; Demaurex, N.; Schlegel, W.; Krause, K.H. Nox5 is expressed at the plasma membrane and generates superoxide in response to protein kinase c activation. Biochimie 2007, 89, 1159–1167. [Google Scholar] [CrossRef]
- Tirone, F.; Cox, J.A. Nadph oxidase 5 (nox5) interacts with and is regulated by calmodulin. FEBS Lett. 2007, 581, 1202–1208. [Google Scholar] [CrossRef]
- Chen, F.; Yu, Y.; Haigh, S.; Johnson, J.; Lucas, R.; Stepp, D.W.; Fulton, D.J. Regulation of nadph oxidase 5 by protein kinase c isoforms. PLoS ONE 2014, 9, e88405. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.R.; He, B.J.; Grumbach, I.M.; Anderson, M.E. Camkii in the cardiovascular system: Sensing redox states. Physiol. Rev. 2011, 91, 889–915. [Google Scholar] [CrossRef]
- Koledova, V.V.; Khalil, R.A. Ca2+, calmodulin, and cyclins in vascular smooth muscle cell cycle. Circ. Res. 2006, 98, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Usatyuk, P.V.; Gorshkova, I.A.; Garcia, J.G.; Natarajan, V. Regulation of nadph oxidase in vascular endothelium: The role of phospholipases, protein kinases, and cytoskeletal proteins. Antioxid. Redox Signal. 2009, 11, 841–860. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family nadph oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Banfi, B.; Tirone, F.; Durussel, I.; Knisz, J.; Moskwa, P.; Molnar, G.Z.; Krause, K.H.; Cox, J.A. Mechanism of Ca2+ activation of the nadph oxidase 5 (nox5). J. Biol. Chem. 2004, 279, 18583–18591. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Kuhne, W.; Spahr, R.; Schwartz, P.; Piper, H.M. Macromolecule permeability of coronary and aortic endothelial monolayers under energy depletion. Am. J. Physiol. 1991, 260, H1344–H1352. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakurada, R.; Odagiri, K.; Hakamata, A.; Kamiya, C.; Wei, J.; Watanabe, H. Calcium Release from Endoplasmic Reticulum Involves Calmodulin-Mediated NADPH Oxidase-Derived Reactive Oxygen Species Production in Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 1644. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071644
Sakurada R, Odagiri K, Hakamata A, Kamiya C, Wei J, Watanabe H. Calcium Release from Endoplasmic Reticulum Involves Calmodulin-Mediated NADPH Oxidase-Derived Reactive Oxygen Species Production in Endothelial Cells. International Journal of Molecular Sciences. 2019; 20(7):1644. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071644
Chicago/Turabian StyleSakurada, Ryugo, Keiichi Odagiri, Akio Hakamata, Chiaki Kamiya, Jiazhang Wei, and Hiroshi Watanabe. 2019. "Calcium Release from Endoplasmic Reticulum Involves Calmodulin-Mediated NADPH Oxidase-Derived Reactive Oxygen Species Production in Endothelial Cells" International Journal of Molecular Sciences 20, no. 7: 1644. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071644