Hydrophobic Amines and Their Guanidine Analogues Modulate Activation and Desensitization of ASIC3

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
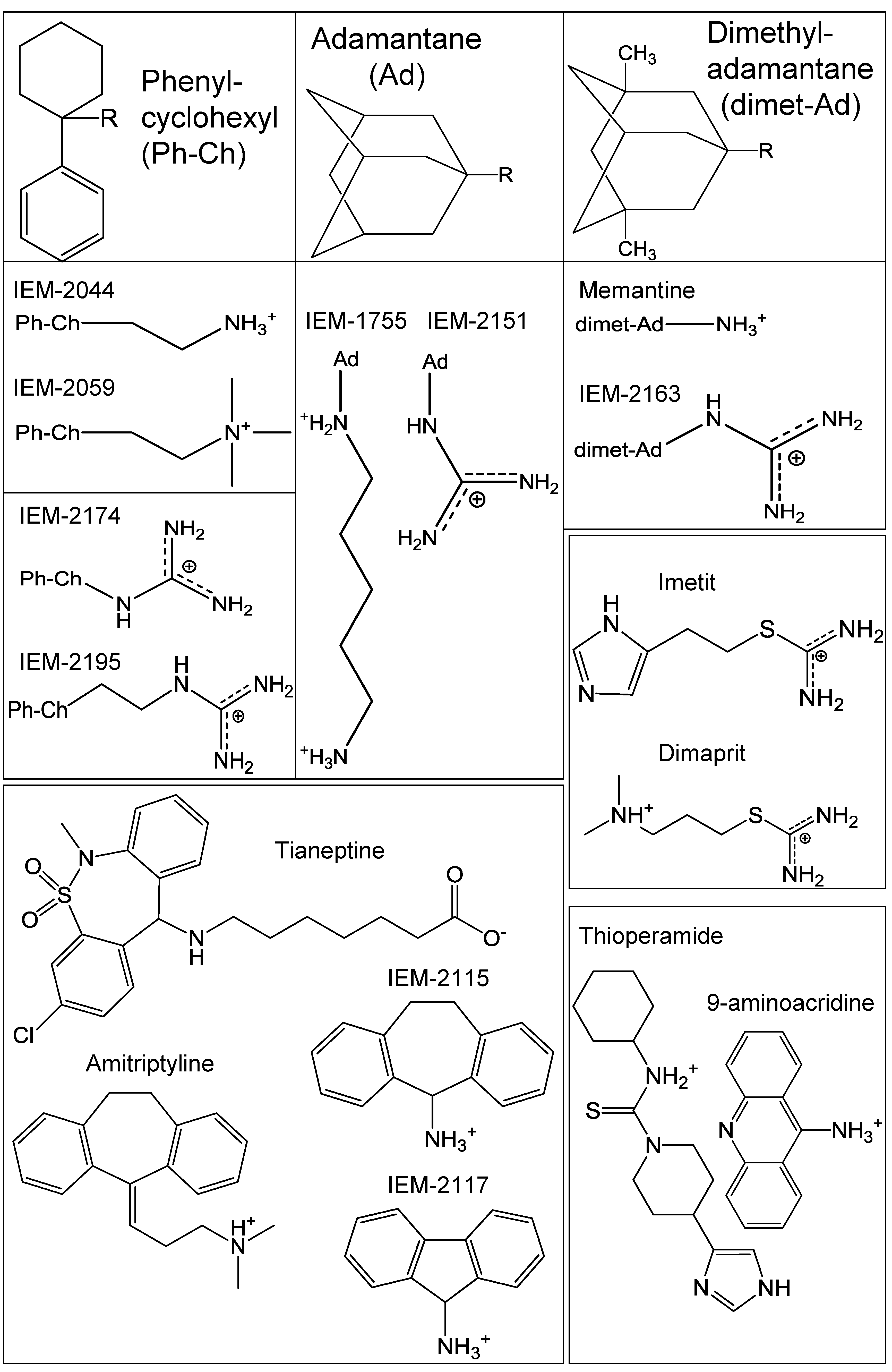
2.1. Drug Selection
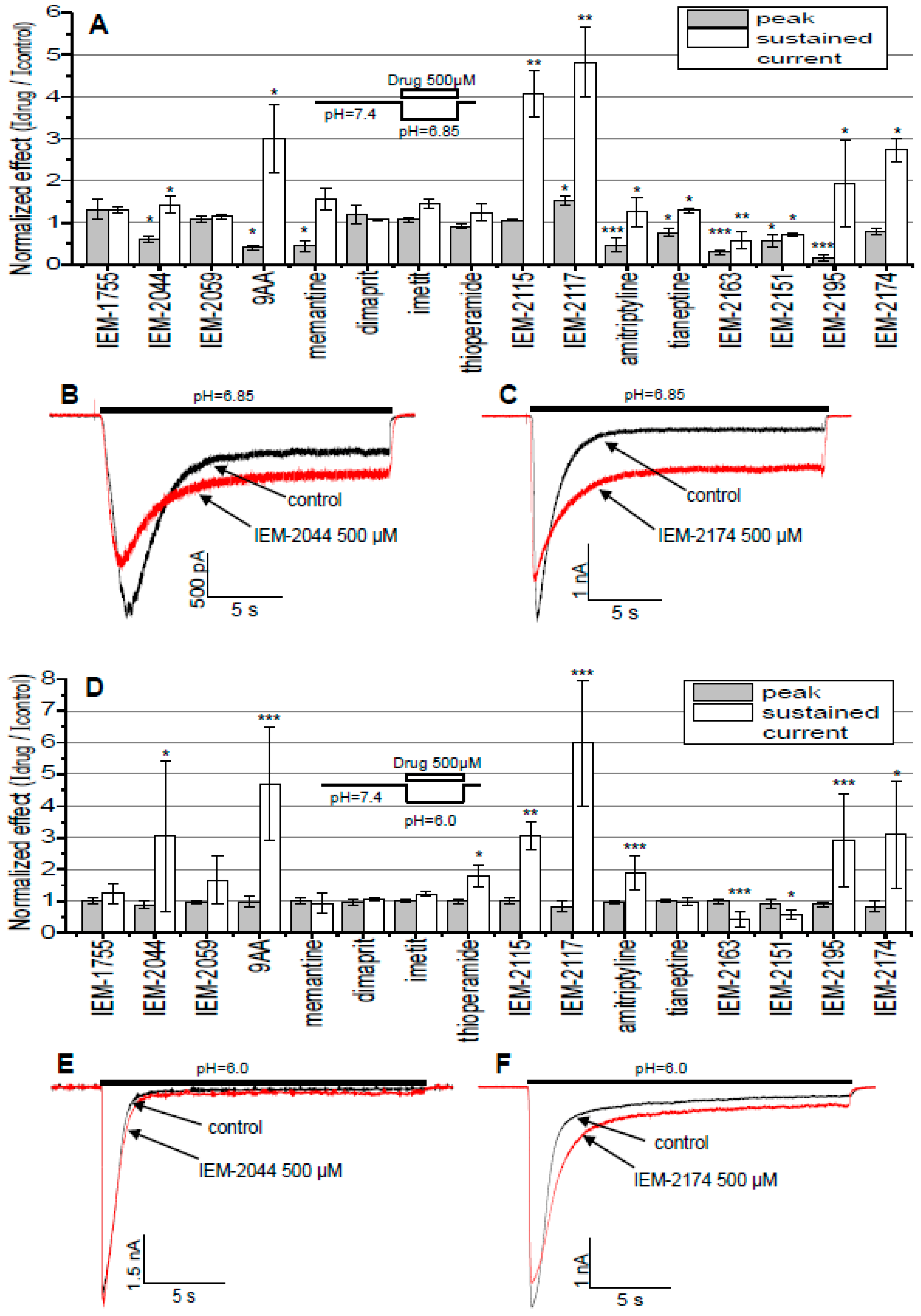
2.2. Estimation of Drug Activities
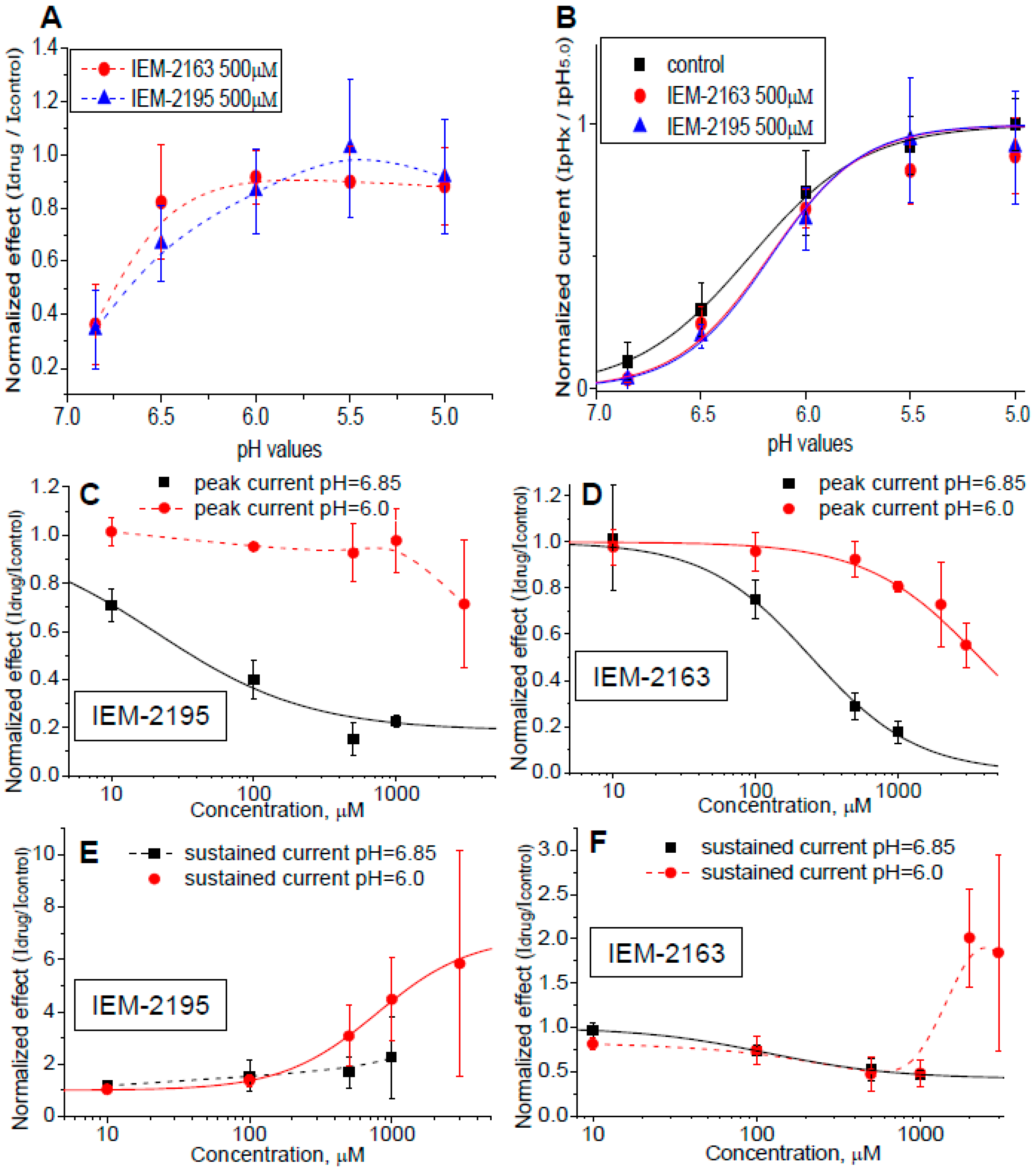
2.3. pH and Concentration Dependencies
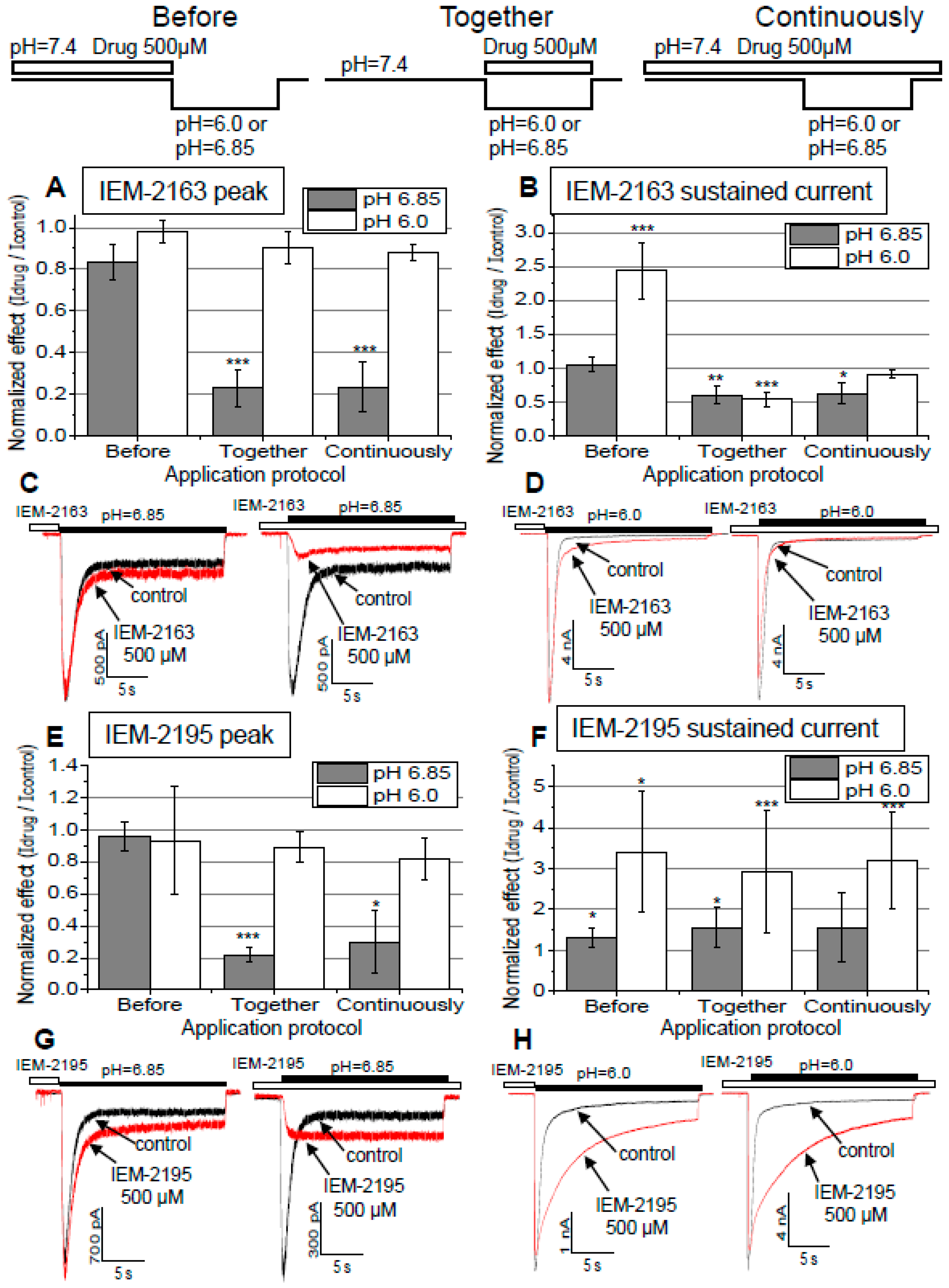
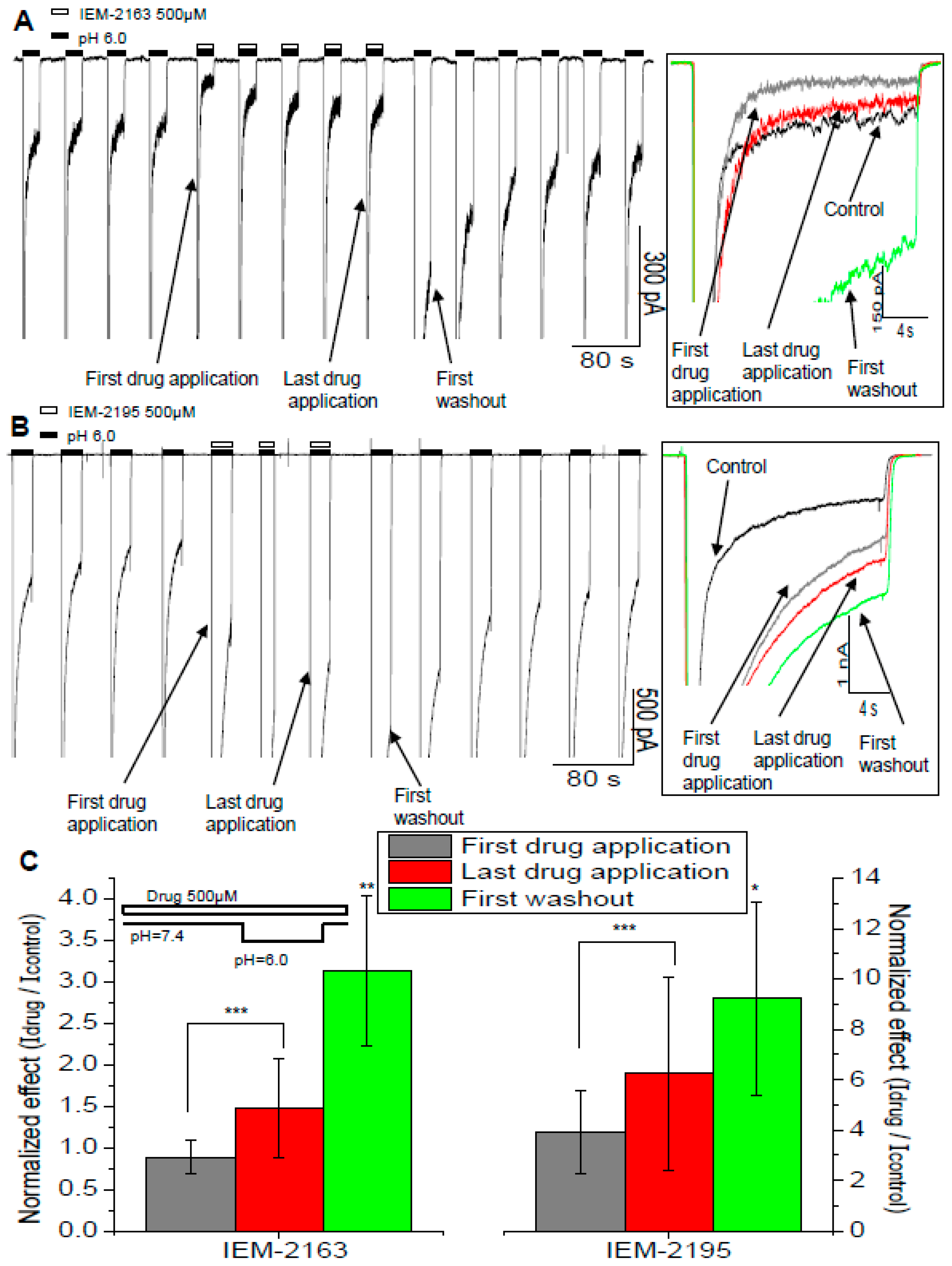
2.4. Dependence of Action on the Application Protocols
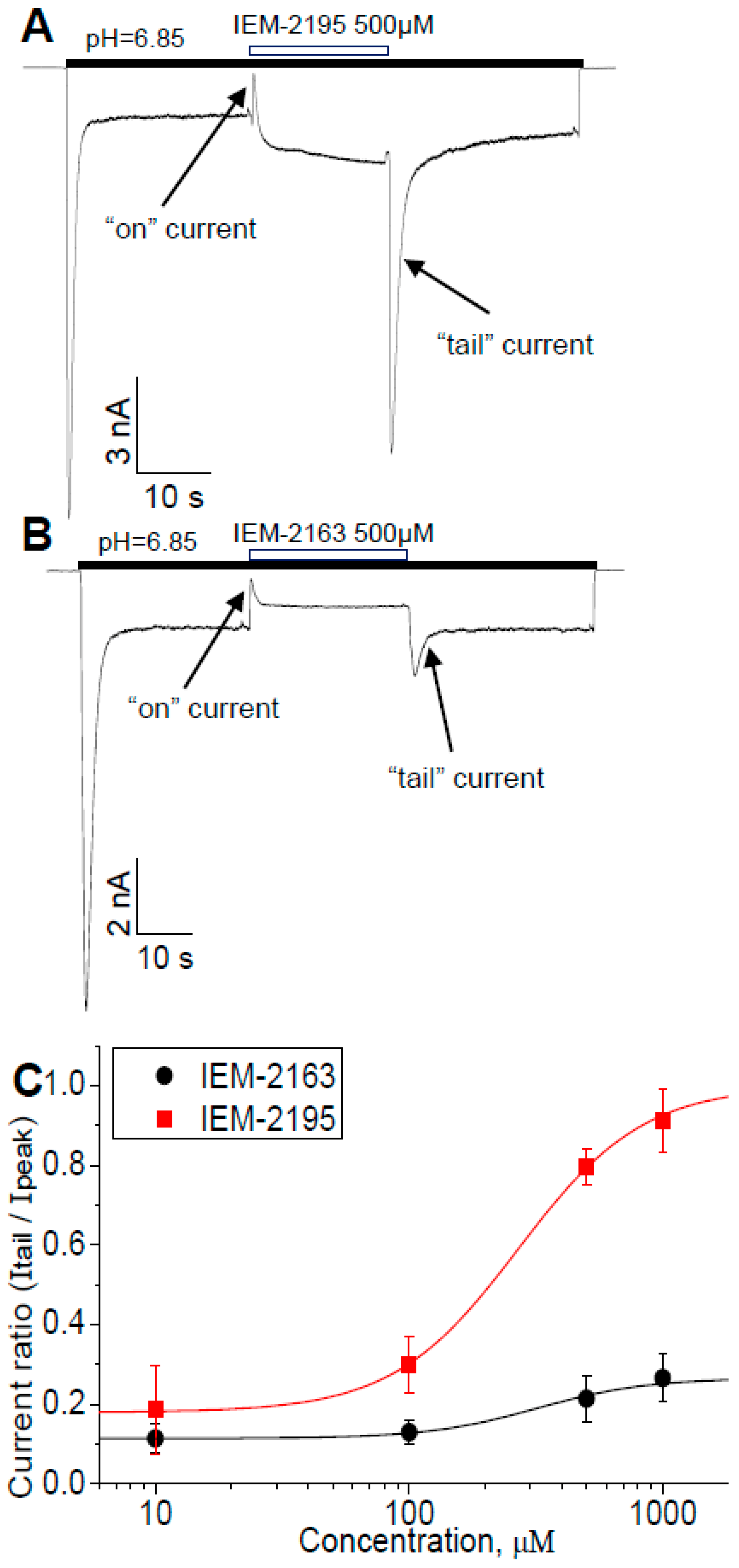
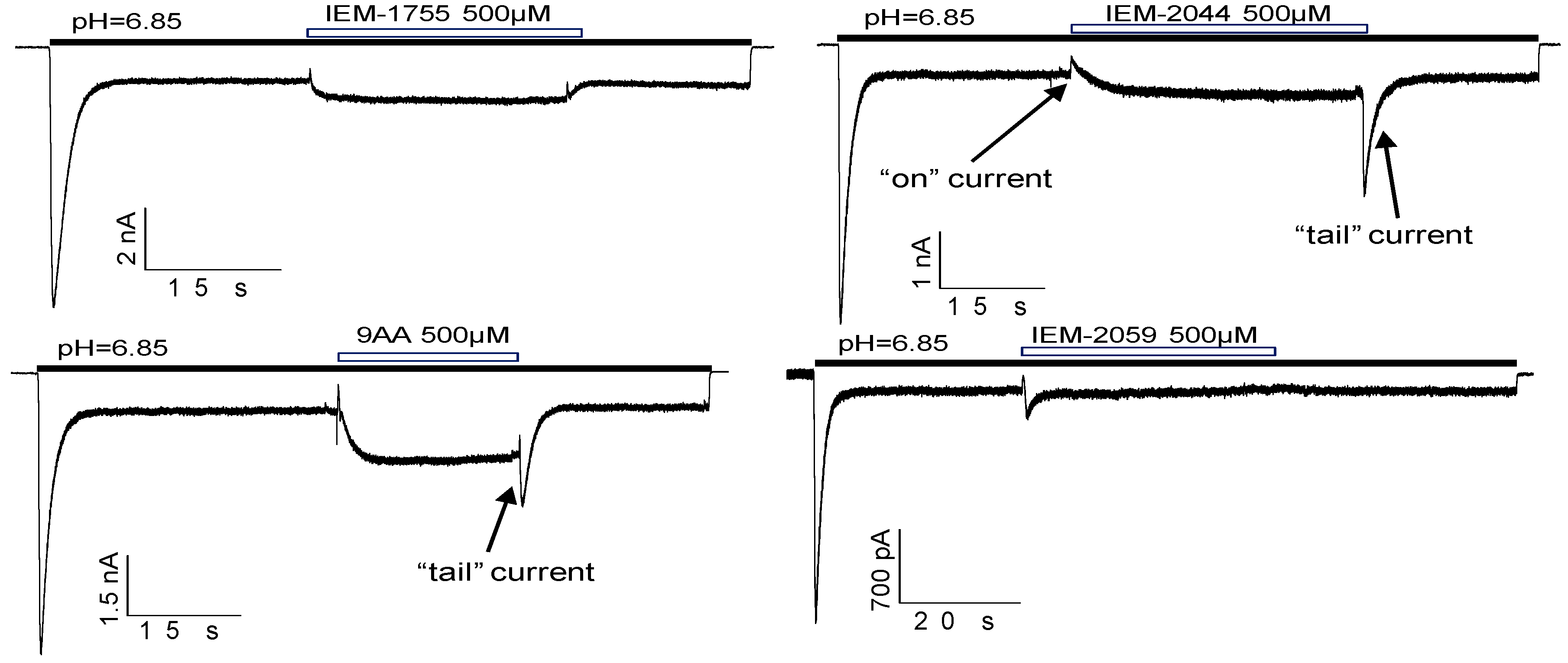
2.5. Kinetics of Action
2.6. Biphasic Drug Effects, when Applied Exclusively to the Sustained Current
3. Discussion
4. Materials and Methods
4.1. Chemicals and Synthesis
4.2. Cell Culture and Transfection
4.3. Drugs and Solutions
4.4. Electrophysiology
4.5. Experimental Protocol
4.6. Data Analysis and Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASIC | Acid-sensing ion channel |
| CHO | Chinese hamster ovary (cells) |
| DEG/ENaC | Degenerin/Epithelial sodium channels |
| GMQ | 2-Guanidine-4-methylquinazoline |
References
- Deval, E.; Lingueglia, E. Acid-Sensing Ion Channels and nociception in the peripheral and central nervous systems. Neuropharmacology 2015, 94, 49–57. [Google Scholar] [CrossRef]
- Kreple, C.J.; Lu, Y.; Taugher, R.J.; Schwager-Gutman, A.L.; Du, J.; Stump, M.; Wang, Y.; Ghobbeh, A.; Fan, R.; Cosme, C.V.; et al. Acid-sensing ion channels contribute to synaptic transmission and inhibit cocaine-evoked plasticity. Nat. Neurosci. 2014, 17, 1083–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Jiang, N.; Li, J.; Ji, Y.-H.; Xiong, Z.-G.; Zha, X. Two aspects of ASIC function: Synaptic plasticity and neuronal injury. Neuropharmacology 2015, 94, 42–48. [Google Scholar] [CrossRef]
- Kweon, H.-J.; Suh, B.-C. Acid-sensing ion channels (ASICs): therapeutic targets for neurological diseases and their regulation. BMB Rep. 2013, 46, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.-G.; Xu, T.-L. The role of ASICS in cerebral ischemia. Wiley Interdiscip. Rev. Membr. Transp. Signal 2012, 1, 655–662. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-H.; Sun, W.-H.; Chen, C.-C. Genetic exploration of the role of acid-sensing ion channels. Neuropharmacology 2015, 94, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Yagi, J.; Wenk, H.N.; Naves, L.A.; McCleskey, E.W. Sustained currents through ASIC3 ion channels at the modest pH changes that occur during myocardial ischemia. Circ. Res. 2006, 99, 501–509. [Google Scholar] [CrossRef]
- Deval, E.; Gasull, X.; Noël, J.; Salinas, M.; Baron, A.; Diochot, S.; Lingueglia, E. Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol. Ther. 2010, 128, 549–558. [Google Scholar] [CrossRef]
- Jones, N.G.; Slater, R.; Cadiou, H.; McNaughton, P.; McMahon, S.B. Acid-induced pain and its modulation in humans. J. Neurosci. 2004, 24, 10974–10979. [Google Scholar] [CrossRef]
- Dubé, G.R.; Lehto, S.G.; Breese, N.M.; Baker, S.J.; Wang, X.; Matulenko, M.A.; Honoré, P.; Stewart, A.O.; Moreland, R.B.; Brioni, J.D. Electrophysiological and in vivo characterization of A-317567, a novel blocker of acid sensing ion channels. Pain 2005, 117, 88–96. [Google Scholar] [CrossRef]
- Rocha-González, H.I.; Herrejon-Abreu, E.B.; López-Santillán, F.J.; García-López, B.E.; Murbartián, J.; Granados-Soto, V. Acid increases inflammatory pain in rats: effect of local peripheral ASICs inhibitors. Eur. J. Pharmacol. 2009, 603, 56–61. [Google Scholar] [CrossRef]
- Price, M.P.; McIlwrath, S.L.; Xie, J.; Cheng, C.; Qiao, J.; Tarr, D.E.; Sluka, K.A.; Brennan, T.J.; Lewin, G.R.; Welsh, M.J. The DRASIC Cation Channel Contributes to the Detection of Cutaneous Touch and Acid Stimuli in Mice. Neuron 2001, 32, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Jang, J.H.; Price, M.P.; Gautam, M.; Benson, C.J.; Gong, H.; Welsh, M.J.; Brennan, T.J. Simultaneous Disruption of Mouse ASIC1a, ASIC2 and ASIC3 Genes Enhances Cutaneous Mechanosensitivity. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Deval, E.; Noël, J.; Lay, N.; Alloui, A.; Diochot, S.; Friend, V.; Jodar, M.; Lazdunski, M.; Lingueglia, E. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J. 2008, 27, 3047–3055. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, S.P.; Benson, C.J.; Adelman, J.P.; McCleskey, E.W. Acid-sensing ion channel 3 matches the acid-gated current in cardiac ischemia-sensing neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 711–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, T.; Chen, J.; Harding, A.M.S.; Price, M.P.; Lu, Y.; Abboud, F.M.; Benson, C.J. ASIC2a and ASIC3 heteromultimerize to form pH-sensitive channels in mouse cardiac dorsal root ganglia neurons. Circ. Res. 2009, 105, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Edelmayer, R.M.; Wei, X.; De Felice, M.; Porreca, F.; Dussor, G. Dural afferents express acid-sensing ion channels: a role for decreased meningeal pH in migraine headache. Pain 2011, 152, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Izumi, M.; Ikeuchi, M.; Ji, Q.; Tani, T. Local ASIC3 modulates pain and disease progression in a rat model of osteoarthritis. J. Biomed. Sci. 2012, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Walder, R.Y.; Rasmussen, L.A.; Rainier, J.D.; Light, A.R.; Wemmie, J.A.; Sluka, K.A. ASIC1 and ASIC3 Play Different Roles in the Development of Hyperalgesia Following Inflammatory Muscle Injury. J. Pain 2010, 11, 210–218. [Google Scholar] [CrossRef]
- Baron, A.; Lingueglia, E. Pharmacology of acid-sensing ion channels - Physiological and therapeutical perspectives. Neuropharmacology 2015, 94, 19–35. [Google Scholar] [CrossRef]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-G.; Yu, Y.; Huang, C.; Cao, H.; Xu, T.-L. Nonproton Ligand Sensing Domain Is Required for Paradoxical Stimulation of Acid-sensing Ion Channel 3 (ASIC3) Channels by Amiloride. J. Biol. Chem. 2011, 286, 42635–42646. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, Z.; Li, W.-G.; Cao, H.; Feng, E.-G.; Yu, F.; Liu, H.; Jiang, H.; Xu, T.-L. A nonproton ligand sensor in the acid-sensing ion channel. Neuron 2010, 68, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Besson, T.; Lingueglia, E.; Salinas, M. Pharmacological modulation of Acid-Sensing Ion Channels 1a and 3 by amiloride and 2-guanidine-4-methylquinazoline (GMQ). Neuropharmacology 2017, 125, 429–440. [Google Scholar] [CrossRef]
- Askwith, C.C.; Cheng, C.; Ikuma, M.; Benson, C.; Price, M.P.; Welsh, M.J. Neuropeptide FF and FMRFamide Potentiate Acid-Evoked Currents from Sensory Neurons and Proton-Gated DEG/ENaC Channels. Neuron 2000, 26, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Li, W.-G.; Yu, Y.; Zhang, Z.-D.; Cao, H.; Xu, T.-L. ASIC3 Channels Integrate Agmatine and Multiple Inflammatory Signals through the Nonproton Ligand Sensing Domain. Mol. Pain 2010, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, W.-G.; Yu, Y.; Xiao, X.; Cheng, J.; Zeng, W.-Z.; Peng, Z.; Xi Zhu, M.; Xu, T.-L. Serotonin facilitates peripheral pain sensitivity in a manner that depends on the nonproton ligand sensing domain of ASIC3 channel. J. Neurosci. 2013, 33, 4265–4279. [Google Scholar] [CrossRef]
- Diochot, S.; Baron, A.; Rash, L.D.; Deval, E.; Escoubas, P.; Scarzello, S.; Salinas, M.; Lazdunski, M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 2004, 23, 1516–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmakov, D.I.; Kozlov, S.A.; Andreev, Y.A.; Koshelev, S.G.; Sanamyan, N.P.; Sanamyan, K.E.; Dyachenko, I.A.; Bondarenko, D.A.; Murashev, A.N.; Mineev, K.S.; et al. Sea anemone peptide with uncommon β-hairpin structure inhibits acid-sensing ion channel 3 (ASIC3) and reveals analgesic activity. J. Biol. Chem. 2013, 288, 23116–23127. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Chesler, A.T.; Sharif-Naeini, R.; Medzihradszky, K.F.; Zhou, S.; King, D.; Sánchez, E.E.; Burlingame, A.L.; Basbaum, A.I.; Julius, D. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature 2011, 479, 410–414. [Google Scholar] [CrossRef] [Green Version]
- Tikhonova, T.B.; Nagaeva, E.I.; Barygin, O.I.; Potapieva, N.N.; Bolshakov, K.V.; Tikhonov, D.B. Monoamine NMDA receptor channel blockers inhibit and potentiate native and recombinant proton-gated ion channels. Neuropharmacology 2015, 89, 1–10. [Google Scholar] [CrossRef]
- Shteinikov, V.Y.; Barygin, O.I.; Gmiro, V.E.; Tikhonov, D.B. Multiple modes of action of hydrophobic amines and their guanidine analogues on ASIC1a. Eur. J. Pharmacol. 2019, 844, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Nagaeva, E.I.; Tikhonova, T.B.; Magazanik, L.G.; Tikhonov, D.B. Histamine selectively potentiates acid-sensing ion channel 1a. Neurosci. Lett. 2016, 632, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, M.; Komarova, M.; Tikhonova, T.; Korosteleva, A.; Potapjeva, N.; Tikhonov, D.B. Modulation of proton-gated channels by antidepressants. ACS Chem. Neurosci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shteinikov, V.Y.; Korosteleva, A.S.; Tikhonova, T.B.; Potapieva, N.N.; Tikhonov, D.B. Ligands of histamine receptors modulate acid-sensing ion channels. Biochem. Biophys. Res. Commun. 2017, 490, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Bolshakov, K.V.; Kim, K.H.; Potapjeva, N.N.; Gmiro, V.E.; Tikhonov, D.B.; Usherwood, P.N.R.; Mellor, I.R.; Magazanik, L.G. Design of antagonists for NMDA and AMPA receptors. Neuropharmacology 2005, 49, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Magazanik, L.G.; Buldakova, S.L.; Samoilova, M.V.; Gmiro, V.E.; Mellor, I.R.; Usherwood, P.N. Block of open channels of recombinant AMPA receptors and native AMPA/kainate receptors by adamantane derivatives. J. Physiol. (Lond.) 1997, 505, 655–663. [Google Scholar] [CrossRef]
- Bormann, J. Memantine is a potent blocker of N-methyl-d-aspartate (NMDA) receptor channels. Eur. J. Pharmacol. 1989, 166, 591–592. [Google Scholar] [CrossRef]
- Benveniste, M.; Mayer, M.L. Trapping of glutamate and glycine during open channel block of rat hippocampal neuron NMDA receptors by 9-aminoacridine. J. Physiol. (Lond.) 1995, 483 (Pt 2), 367–384. [Google Scholar] [CrossRef]
- Pereira, V.S.; Hiroaki-Sato, V.A. A brief history of antidepressant drug development: from tricyclics to beyond ketamine. Acta Neuropsychiatrica 2018, 30, 307–322. [Google Scholar] [CrossRef]
- Wagstaff, A.J.; Ormrod, D.; Spencer, C.M. Tianeptine: a review of its use in depressive disorders. Mol. Diag. Ther. 2001, 15, 231–259. [Google Scholar] [CrossRef] [PubMed]
- Arrang, J.M.; Garbarg, M.; Lancelot, J.C.; Lecomte, J.M.; Pollard, H.; Robba, M.; Schunack, W.; Schwartz, J.C. Highly potent and selective ligands for histamine H3-receptors. Nature 1987, 327, 117–123. [Google Scholar] [CrossRef]
- Garbarg, M.; Arrang, J.M.; Rouleau, A.; Ligneau, X.; Tuong, M.D.; Schwartz, J.C.; Ganellin, C.R. S-[2-(4-imidazolyl)ethyl]isothiourea, a highly specific and potent histamine H3 receptor agonist. J. Pharmacol. Exp. Ther. 1992, 263, 304–310. [Google Scholar]
- Alijevic, O.; Hammoud, H.; Vaithia, A.; Trendafilov, V.; Bollenbach, M.; Schmitt, M.; Bihel, F.; Kellenberger, S. Heteroarylguanidines as Allosteric Modulators of ASIC1a and ASIC3 Channels. ACS Chem. Neurosci. 2018, 9, 1357–1365. [Google Scholar] [CrossRef]
- Chen, X.; Kalbacher, H.; Grunder, S. The tarantula toxin psalmotoxin 1 inhibits acid-sensing ion channel (ASIC) 1a by increasing its apparent H+ affinity. J. Gen. Physiol. 2005, 126, 71–79. [Google Scholar] [CrossRef]
- Yoder, N.; Yoshioka, C.; Gouaux, E. Gating mechanisms of acid-sensing ion channels. Nature 2018, 555, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Vullo, S.; Bonifacio, G.; Roy, S.; Johner, N.; Bernèche, S.; Kellenberger, S. Conformational dynamics and role of the acidic pocket in ASIC pH-dependent gating. Proc. Natl. Acad. Sci. USA 2017, 114, 3768–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alijevic, O.; Kellenberger, S. Subtype-specific modulation of acid-sensing ion channel (ASIC) function by 2-guanidine-4-methylquinazoline. J. Biol. Chem. 2012, 287, 36059–36070. [Google Scholar] [CrossRef] [PubMed]
- Reeh, P.W.; Steen, K.H. Tissue acidosis in nociception and pain. Prog. Brain Res. 1996, 113, 143–151. [Google Scholar]
- Yan, G.X.; Kléber, A.G. Changes in extracellular and intracellular pH in ischemic rabbit papillary muscle. Circ. Res. 1992, 71, 460–470. [Google Scholar] [CrossRef]
- Street, D.; Bangsbo, J.; Juel, C. Interstitial pH in human skeletal muscle during and after dynamic graded exercise. J. Physiol. 2001, 537, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.C.; Park, S.S.; Subieta, A.R.; Brennan, T.J. Changes in tissue pH and temperature after incision indicate acidosis may contribute to postoperative pain. Anesthesiology 2004, 101, 468–475. [Google Scholar] [CrossRef]
- McVicar, N.; Li, A.X.; Gonçalves, D.F.; Bellyou, M.; Meakin, S.O.; Prado, M.A.; Bartha, R. Quantitative tissue pH measurement during cerebral ischemia using amine and amide concentration-independent detection (AACID) with MRI. J. Cereb. Blood Flow Metab. 2014, 34, 690–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinas, M.; Lazdunski, M.; Lingueglia, E. Structural elements for the generation of sustained currents by the acid pain sensor ASIC3. J. Biol. Chem. 2009, 284, 31851–31859. [Google Scholar] [CrossRef] [PubMed]
- Maddox, V.H.; Godefroi, E.F.; Parcell, R.F. The Synthesis of Phencyclidine and Other 1-Arylcyclohexylamines. J. Med. Chem. 1965, 8, 230–235. [Google Scholar] [CrossRef]
- Kalir, A.; Edery, H.; Pelah, Z.; Balderman, D.; Porath, G. 1-Phenylcycloalkylamine derivatives. II. Synthesis and pharmacological activity. J. Med. Chem. 1969, 12, 473–477. [Google Scholar] [CrossRef]
- Geluk, H.W.; Schut, J.; Schlatmann, J.L.M.A. Synthesis and antiviral properties of 1-adamantylguanidine. A modified method for preparing tert-alkylguanidines. J. Med. Chem. 1969, 12, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Thurkauf, A.; De Costa, B.; Yamaguchi, S.; Mattson, M.V.; Jacobson, A.E.; Rice, K.C.; Rogawski, M.A. Synthesis and anticonvulsant activity of 1-phenylcyclohexylamine analogs. J. Med. Chem. 1990, 33, 1452–1458. [Google Scholar] [CrossRef]
- Staruschenko, A.; Dorofeeva, N.A.; Bolshakov, K.V.; Stockand, J.D. Subunit-dependent cadmium and nickel inhibition of acid-sensing ion channels. Dev. Neurobiol. 2007, 67, 97–107. [Google Scholar] [CrossRef]
- Hesselager, M.; Timmermann, D.B.; Ahring, P.K. pH dependency and desensitization kinetics of heterologously expressed combinations of acid-sensing ion channel subunits. J. Biol. Chem. 2004, 279, 11006–11015. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shteinikov, V.Y.; Potapieva, N.N.; Gmiro, V.E.; Tikhonov, D.B. Hydrophobic Amines and Their Guanidine Analogues Modulate Activation and Desensitization of ASIC3. Int. J. Mol. Sci. 2019, 20, 1713. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071713
Shteinikov VY, Potapieva NN, Gmiro VE, Tikhonov DB. Hydrophobic Amines and Their Guanidine Analogues Modulate Activation and Desensitization of ASIC3. International Journal of Molecular Sciences. 2019; 20(7):1713. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071713
Chicago/Turabian StyleShteinikov, Vasilii Y, Natalia N Potapieva, Valery E Gmiro, and Denis B Tikhonov. 2019. "Hydrophobic Amines and Their Guanidine Analogues Modulate Activation and Desensitization of ASIC3" International Journal of Molecular Sciences 20, no. 7: 1713. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071713