CAR-T with License to Kill Solid Tumors in Search of a Winning Strategy

 and
and 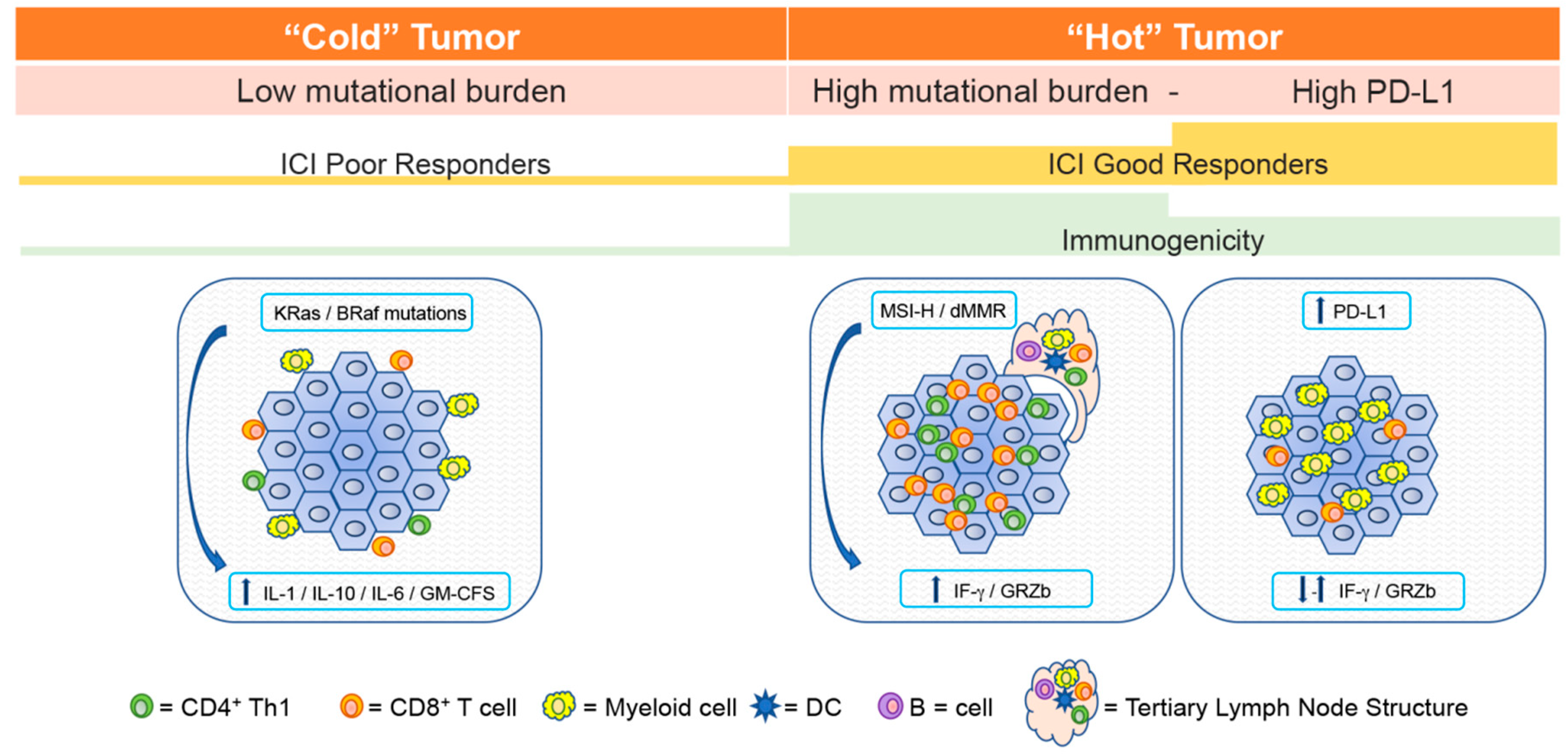{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lesson from Immune Checkpoint Inhibitors
2.1. Tumor Cell Biology and Mutational Burden
2.2. “Cold “and “Hot” Tumors
3. Lesson from Car-T Cell Therapy
3.1. Antigen Loss and Immune Escape
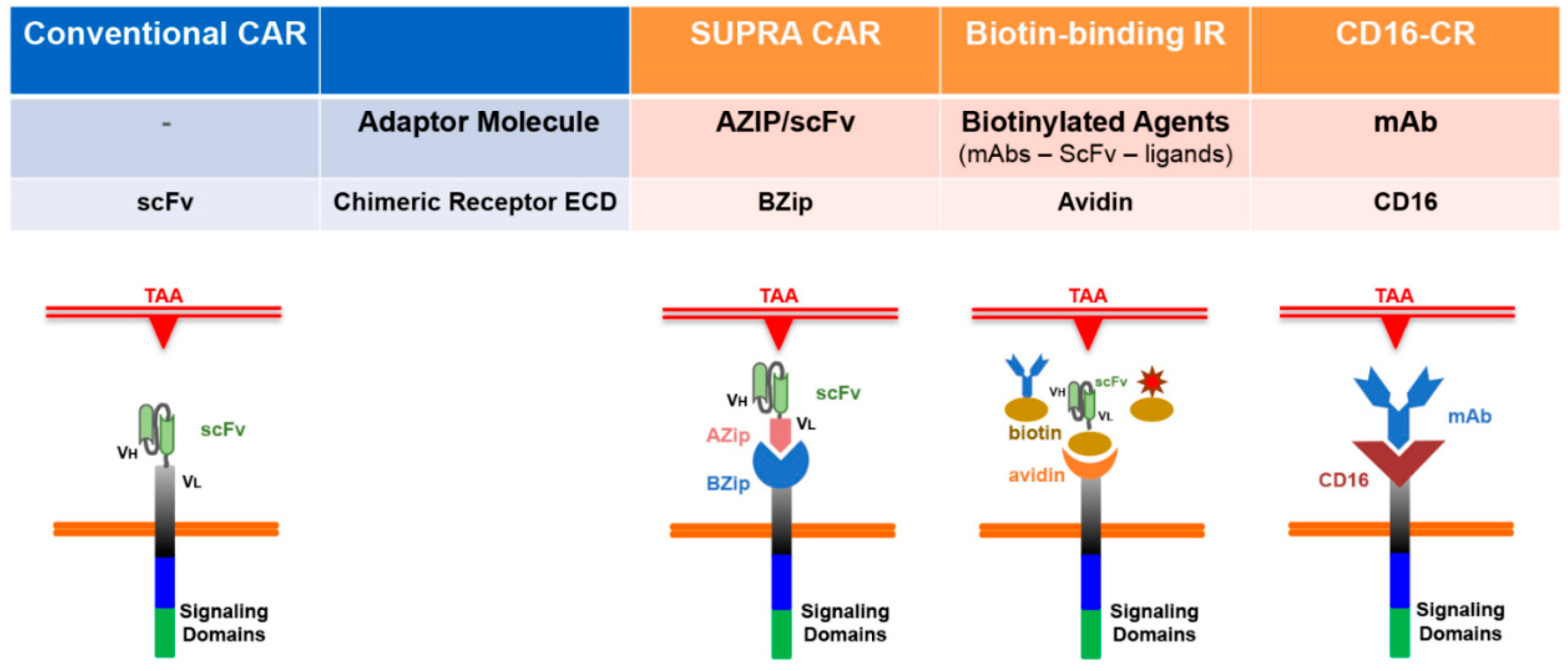
3.2. Combining Humoral and Cellular Immune Responses
3.3. CD16-CR and Therapeutic mAbs: “Like Two Peas in a Pod”
4. Fitness of the Immune System
4.1. Interplay between Metronomic Chemotherapy and Immunotherapy
4.2. Anti-Angiogenetic Effects
4.3. Immune System
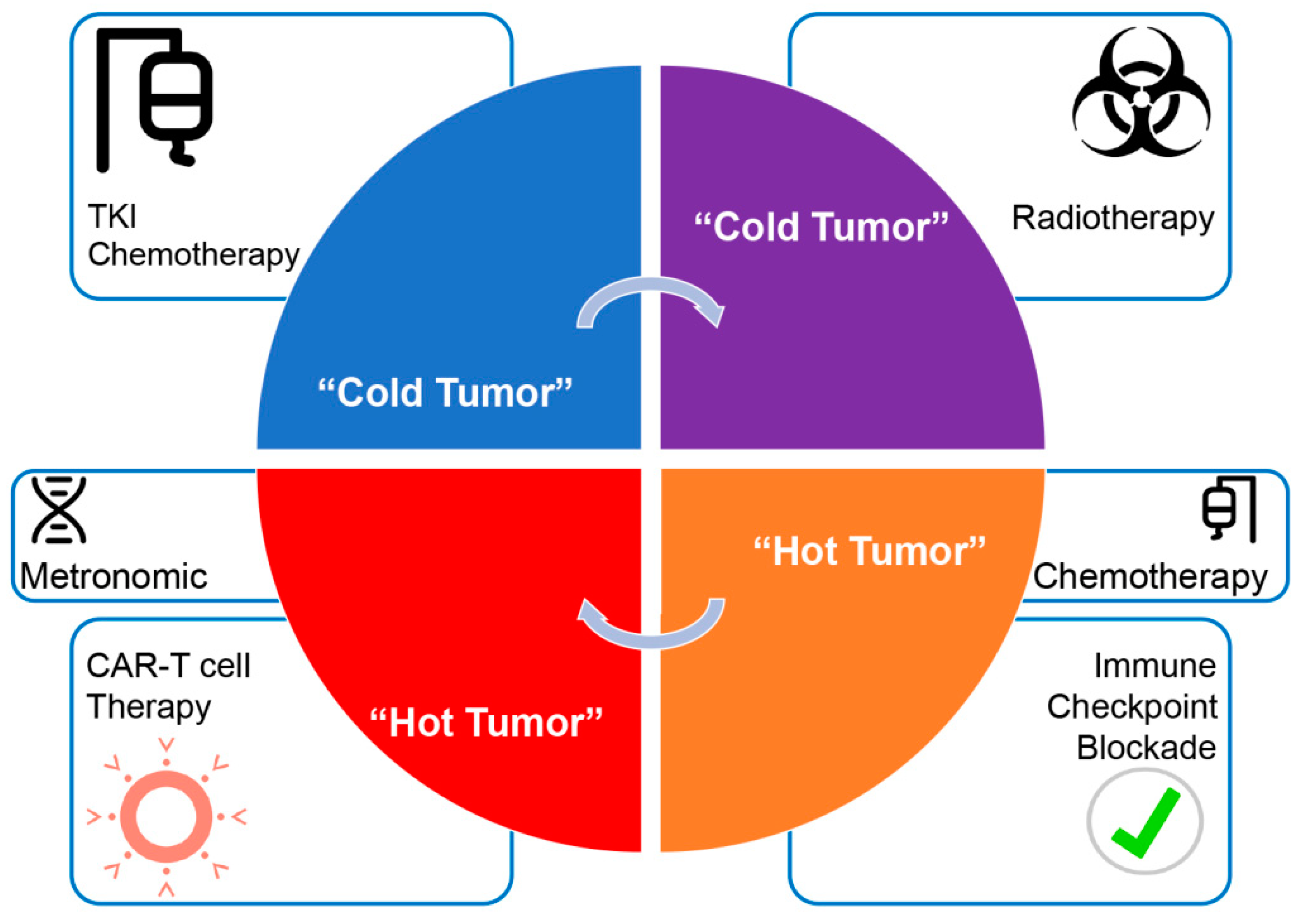
4.4. The Right Therapy, for the Right Patient, at the Right Moment
4.5. Timing of Administration of Immunotherapy
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Gross, G.; Waks, T.; Eshar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Heyman, B.; Yang, Y. Chimeric Antigen Receptor T Cell Therapy for Solid Tumors: Current Status, Obstacles and Future Strategies. Cancers 2019, 11, 191. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Mussai, F.; Egan, S.; Hunter, S.; Webber, H.; Fisher, J.; Wheat, R.; McConville, C.; Sbirkov, Y.; Wheeler, K.; Bendle, G.; et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. 2015, 75, 3043–3053. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.S.; Karanikas, V.; Evers, S. The where.; the when.; and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef]
- Gajewski, T.F. The next hurdle in cancer immunotherapy: Overcoming the non-T-cell-inflamed tumor microenvironment. Semin. Oncol. 2015, 42, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Wood, R.D. Quality control by DNA repair. Science 1999, 286, 1897–1905. [Google Scholar] [CrossRef]
- Stoler, D.L.; Chen, N.; Basik, M.; Kahlenberg, M.S.; Rodriguez-Bigas, M.A.; Petrelli, N.J.; Anderson, G.R. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc. Natl. Acad. Sci. USA 1999, 96, 15121–15126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidoboni, M.; Gafà, R.; Viel, A.; Doglioni, C.; Russo, A.; Santini, A.; Del Tin, L.; Macrì, E.; Lanza, G.; Boiocchi, M.; et al. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am. J. Pathol. 2001, 159, 297–304. [Google Scholar] [CrossRef]
- Buckowitz, A.; Knaebel, H.P.; Benner, A.; Bläker, H.; Gebert, J.; Kienle, P.; von Knebel Doeberitz, M.; Kloor, M. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br. J. Cancer 2005, 92, 1746–1753. [Google Scholar] [CrossRef] [Green Version]
- Dolcetti, R.; Viel, A.; Doglioni, C.; Russo, A.; Guidoboni, M.; Capozzi, E.; Vecchiato, N.; Macrì, E.; Fornasarig, M.; Boiocchi, M. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am. J. Pathol. 1999, 154, 1805–1813. [Google Scholar] [CrossRef]
- Dung, T.L.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar]
- Ishikawa, T.; Fujita, T.; Suzuki, Y.; Okabe, S.; Yuasa, Y.; Iwai, T.; Kawakami, Y. Tumor-specific immunological recognition of frameshift-mutated peptides in colon cancer with microsatellite instability. Cancer Res. 2003, 63, 5564–5572. [Google Scholar]
- Saeterdal, I.; Bjørheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Møller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 13255–13260. [Google Scholar] [CrossRef] [Green Version]
- Nakata, B.; Wang, Y.Q.; Yashiro, M.; Nishioka, N.; Tanaka, H.; Ohira, M.; Ishikawa, T.; Nishino, H.; Hirakawa, K. Prognostic value of microsatellite instability in resectable pancreatic cancer. Clin. Cancer Res. 2002, 8, 2536–2540. [Google Scholar]
- Duval, A.; Raphael, M.; Brennetot, C.; Poirel, H.; Buhard, O.; Aubry, A.; Martin, A.; Krimi, A.; Leblond, V.; Gabarre, J.; et al. The mutator pathway is a feature of immunodeficiency-related lymphomas. Proc. Natl. Acad. Sci. USA 2004, 101, 5002–5007. [Google Scholar] [CrossRef]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Peter, S.; Creelan, B.; Hellmann, M.D.; Socinski, M.A.; Reck, M.; Bhagavatheeswaran, P.; Chang, H.; Geese, W.J.; Paz-Ares, L.; Carbone, D.P. Abstract CT082—Impact of tumor mutation burden on the efficacy of first-line nivolumab in stage iv or recurrent non-small cell lung cancer: An exploratory analysis of CheckMate 026. In Proceedings of the AACR Annual Meeting 2017, Washington, DC, USA, 1–5 April 2017. [Google Scholar] [CrossRef]
- Passardi, A.; Canale, M.; Valgiusti, M.; Ulivi, P. Immune checkpoints as a target for colorectal cancer treatment. Int. J. Mol. Sci. 2017, 18, 1324. [Google Scholar] [CrossRef]
- Leal, A.D.; Paludo, J.; Finnes, H.D.; Grothey, A. Response to pembrolizumab in patients with mismatch repair deficient (dMMR) colorectal cancer (CRC). J. Clin. Oncol. 2017, 35, 3558. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Van den Eynde, M.; Mlecnik, B.; Bindea, G.; Fredriksen, T.; Church, S.E.; Lafontaine, L.; Haicheur, N.; Marliot, F.; Angelova, M.; Vasaturo, A.; et al. The Link between the Multiverse of Immune Microenvironments in Metastases and the Survival of Colorectal Cancer Patients. Cancer Cell 2018, 34, 1012–1026.e3. [Google Scholar] [CrossRef]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Teng, F.; Kong, L.; Meng, X.; Yang, J.; Yu, J. Radiotherapy combined with immune checkpoint blockade immunotherapy: Achievements and challenges. Cancer Lett. 2015, 365, 23–29. [Google Scholar] [CrossRef]
- Ngiow, S.F.; McArthur, G.A.; Smyth, M.J. Radiotherapy complements immune checkpoint blockade. Cancer Cell 2015, 27, 437–438. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef]
- Zhang, J.; Endres, S.; Kobold, S. Enhancing tumor T cell infiltration to enable cancer immunotherapy. Immunotherapy 2019, 11, 201–213. [Google Scholar] [CrossRef]
- Airoldi, I.; Di Carlo, E.; Cocco, C.; Sorrentino, C.; Fais, F.; Cilli, M.; D’Antuono, T.; Colombo, M.P.; Pistoia, V. Lack of Il12rb2 signaling predisposes to spontaneous autoimmunity and malignancy. Blood 2005, 106, 3846–3853. [Google Scholar] [CrossRef] [Green Version]
- Enzler, T.; Gillessen, S.; Manis, J.P.; Ferguson, D.; Fleming, J.; Alt, F.W.; Mihm, M.; Dranoff, G. Deficiencies of GM-CSF and interferon gamma link inflammation and cancer. J. Exp. Med. 2003, 197, 1213–1219. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Proc. Natl. Acad. Sci. USA 2002, 20, 860–867. [Google Scholar] [CrossRef]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751–765.e16. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef]
- Chen, Y.Y. Increasing T Cell Versatility with SUPRA CARs. Cell 2018, 173, 1316–1317. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438. [Google Scholar] [CrossRef]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132–141. [Google Scholar] [CrossRef]
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J., Jr. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef]
- Clémenceau, B.; Congy-Jolivet, N.; Gallot, G.; Vivien, R.; Gaschet, J.; Thibault, G.; Vié, H. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood 2006, 107, 4669–4677. [Google Scholar] [CrossRef] [Green Version]
- Kudo, K.; Imai, C.; Lorenzini, P.; Kamiya, T.; Kono, K.; Davidoff, A.M.; Chng, W.J.; Campana, D. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014, 74, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ochi, F.; Fujiwara, H.; Tanimoto, K.; Asai, H.; Miyazaki, Y.; Okamoto, S.; Mineno, J.; Kuzushima, K.; Shiku, H.; Barrett, J.; et al. Gene-modified human α/β-T cells expressing a chimeric CD16- CD3ζ receptor as adoptively transferable effector cells for anticancer monoclonal antibody therapy. Cancer Immunol. Res. 2014, 2, 249–262. [Google Scholar] [CrossRef]
- D’Aloia, M.M.; Caratelli, S.; Palumbo, C.; Battella, S.; Arriga, R.; Lauro, D.; Palmieri, G.; Sconocchia, G.; Alimandi, M. T lymphocytes engineered to express a CD16-chimeric antigen receptor redirect T-cell immune response against immunoglobulin G-opsonized target cells. Cytotherapy 2016, 18, 278–290. [Google Scholar] [CrossRef]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef]
- Minutolo, N.G.; Hollander, E.E.; Powell, D.J., Jr. The Emergence of Universal Immune Receptor T Cell Therapy for Cancer. Front. Oncol. 2019, 9, 176. [Google Scholar] [CrossRef]
- Ferris, R.L.; Jaffee, E.M.; Ferrone, S. Tumor antigen-targeted monoclonal antibody-based immunotherapy: Clinical response, cellular immunity, and immunoescape. J. Clin. Oncol. 2010, 28, 4390–4399. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.R.; Song, A.; Bergelson, S.; Arroll, T.; Parekh, B.; May, K.; Chung, S.; Strouse, R.; Mire-Sluis, A.; Schenerman, M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Discov. 2011, 10, 101–111. [Google Scholar] [CrossRef]
- Ochoa, M.C.; Minute, L.; Rodriguez, I.; Garasa, S.; Perez-Ruiz, E.; Inoges, S.; Melero, I.; Berraondo, P. Antibody-dependent cell cytotoxicity: Immunotherapy strategies enhancing effector NK cells. Immunol. Cell Biol. 2017, 95, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Fan, X.; Deng, H.; Brezski, R.J.; Rycyzyn, M.; Jordan, R.E.; Strohl, W.R.; Zou, Q.; Zhang, N.; An, Z. Trastuzumab triggers phagocytic killing of high HER2 cancer cells by interaction with FcγRIV on immune cells. J. Immunol. 2015, 194, 4379–4386. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.P.; Lindorfer, M.A. Cytotoxic mechanisms of immunotherapy: Harnessing complement in the action of anti- tumor monoclonal antibodies. Semin. Immunol. 2016, 28, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.M.; Liu, Y.C.; Li, W.; Liu, S.; Liu, H.X.; Li, L.-W.; Ma, R. Nivolumab effectively inhibit platinum- resistant ovarian cancer cells via induction of cell apoptosis and inhibition of ADAM17 expression. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1198–1205. [Google Scholar]
- Alduaij, W.; Ivanov, A.; Honeychurch, J.; Cheadle, E.J.; Potluri, S.; Lim, S.H.; Shimada, K.; Chan, C.H.T.; Tutt, A.; Beers, S.A. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood 2011, 117, 4519–4528. [Google Scholar] [CrossRef]
- Nuti, M.; Bellati, F.; Visconti, V.; Napoletano, C.; Domenici, L.; Caccetta, J.; Zizzari, I.G.; Ruscito, I.; Rahimi, H.; Benedetti-Panici, P.; et al. Immune effects of trastuzumab. J. Cancer 2011, 2, 17–23. [Google Scholar] [CrossRef]
- Bellati, F.; Napoletano, C.; Ruscito, I.; Liberati, M.; Benedetti-Panici, P.; Nuti, M. Cellular Adaptive Immune System Plays a Crucial Role in Trastuzumab Clinical Efficacy. J. Clin. Oncol. 2010, 28, 369–370. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicenter, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Srivastava, R.M.; Lee, S.C.; Andrade Filho, P.A.; Lord, C.A.; Jie, H.B.; Davidson, H.C.; López-Albaitero, A.; Gibson, S.P.; Gooding, W.E.; Ferrone, S.; et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin. Cancer Res. 2013, 19, 1858–1872. [Google Scholar] [CrossRef]
- Strohl, W.R. Current progress in innovative engineered antibodies. Protein Cell 2018, 9, 86–120. [Google Scholar] [CrossRef]
- Bryniarski, K.; Szczepanik, M.; Ptak, M.; Zemelka, M.; Ptak, W. Influence of cyclophosphamide and its metabolic products on the activity of peritoneal macrophages in mice. Pharmacol. Rep. 2009, 61, 550–557. [Google Scholar] [CrossRef]
- Sabnis, G.J.; Macedo, L.F.; Goloubeva, O.; Schayowitz, A.; Brodie, A.M. Stopping treatment can reverse acquired resistance to letrozole. Cancer Res. 2008, 68, 4518–4524. [Google Scholar] [CrossRef] [Green Version]
- Browder, T.; Butterfield, C.E.; Kräling, B.M.; Shi, B.; Marshall, B.; O’Reilly, M.S.; Folkman, J. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000, 60, 1878–1886. [Google Scholar]
- Kerbel, R.S.; Kamen, B.A. The anti-angiogenic basis of metronomic chemotherapy. Nat. Rev. Cancer 2004, 4, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Haba, A.; Matsuno, F.; Seon, B.K. Antiangiogenic therapy of established tumors in human skin/severe combined immunodeficiency mouse chimeras by anti-endoglin (CD105) monoclonal antibodies.; and synergy between anti-endoglin antibody and cyclophosphamide. Cancer Res. 2001, 61, 7846–7854. [Google Scholar] [PubMed]
- Hamano, Y.; Sugimoto, H.; Soubasakos, M.A.; Kieran, M.; Olsen, B.R.; Lawler, J.; Sudhakar, A.; Kalluri, R. Thrombospondin-1 associated with tumor microenvironment contributes to low-dose cyclophosphamide-mediated endothelial cell apoptosis and tumor growth suppression. Cancer Res. 2004, 64, 1570–1574. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Mackall, C.L.; Fleisher, T.A. Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood 1994, 84, 2221–2228. [Google Scholar]
- Andre, N.; Carre, M.; Pasquier, E. Metronomics: Towards personalized chemotherapy? Nat. Rev. Clin. Oncol. 2014, 11, 413–431. [Google Scholar] [CrossRef]
- Lutsiak, M.E.; Semnani, R.T.; De Pascalis, R.; Kashmiri, S.V.; Schlom, J.; Sabzevari, H. Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005, 105, 2862–2868. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef]
- Kaneno, R.; Shurin, G.V.; Kaneno, F.M.; Naiditch, H.; Luo, J.; Shurin, M.R. Chemotherapeutic agents in low noncytotoxic concentrations increase immunogenicity of human colon cancer cells. Cell Oncol. 2011, 34, 97–106. [Google Scholar] [CrossRef]
- Michels, T.; Shurin, G.V.; Naiditch, H.; Sevko, A.; Umansky, V.; Shurin, M.R. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J. Immunotoxicol. 2012, 9, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Kaneno, R.; Shurin, G.V.; Tourkova, I.L.; Shurin, M.R. Chemomodulation of human dendritic cell function by antineoplastic agents in low noncytotoxic concentrations. J. Transl. Med. 2009, 10, 58. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Dubin, K.; Callahan, M.; Ren, B.; Khanin, R.; Viale, A.; Ling, L.; No, D.; Gobourne, A.; Littmann, E.; Huttenhower, C.; et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat. Commun. 2016, 7, 10391. [Google Scholar] [CrossRef] [Green Version]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Blacher, E.; Levy, M.; Tatirovsky, E.; Elinav, E. Microbiome-modulated metabolites at the interface of host immunity. J. Immunol. 2017, 198, 572–580. [Google Scholar] [CrossRef]
- Derosa, L.; Routy, B.; Enot, D.; Baciarello, G.; Massard, C.; Loriot, Y.; Fizazi, K.; Escudier, B.J.; Zitvogel, L.; Albiges, L. Impact of antibiotics on outcome in patients with metastatic renal cell carcinoma treated with immune checkpoint inhibitors. J. Clin. Oncol. 2017, 35, 462. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Wargo, J.A.; Gopalakrishnan, V.; Spencer, C.; Karpinets, T.; Reuben, A.; Andrews, M.C.; Tetzlaff, M.T.; Lazar, A.; Hwu, P.; Hwu, W.J.; et al. Association of the diversity and composition of the gut microbiome with responses and survival (PFS) in metastatic melanoma (MM) patients (pts) on anti-PD-1 therapy. J. Clin. Oncol. 2017, 35, 3008. [Google Scholar] [CrossRef]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillère, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Hou, B.; Tang, Y.; Li, W.; Zeng, Q.; Chang, D. Efficiency of CAR-T Therapy for Treatment of Solid Tumor in Clinical Trials: A Meta-Analysis. Dis. Markers 2019, 2019, 3425291. [Google Scholar] [CrossRef]
- Migliorini, D.; Dietrich, P.Y.; Stupp, R.; Linette, G.P.; Posey, A.D., Jr.; June, C.H. CAR T-Cell Therapies in Glioblastoma: A First Look. Clin. Cancer Res. 2018, 24, 535–540. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Orpilla, J.; Moughon, D.; Shin, N.; Sedighim, S.; Yong, W.H.; Li, G.; Cloughesy, T.F.; et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2016, 1, e87059. [Google Scholar] [CrossRef]
- Chuu, C.P.; Hiipakka, R.A.; Fukuchi, J.; Kokontis, J.M.; Liao, S. Androgen causes growth suppression and reversion of androgen independent prostate cancer xenografts to an androgen-stimulated phenotype in athymic mice. Cancer Res. 2005, 65, 2082–2084. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood 2017, 29, 1039–1041. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; De Nizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2.;3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef]
- Soliman, H.H.; Minton, S.E.; Han, H.S.; Ismail-Khan, R.; Neuger, A.; Khambati, F.; Noyes, D.; Lush, R.; Chiappori, A.A.; Roberts, J.D. A phase I study of indoximod in patients with advanced malignancies. Oncotarget 2016, 7, 22928–22938. [Google Scholar] [CrossRef] [Green Version]
- Gangadhar, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Kaufman, D.R.; Zhao, Y.; et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: Updated phase 1 results from ECHO-202/ KEYNOTE-037. Ann. Oncol. 2016, 27. [Google Scholar] [CrossRef]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Xie, J.; Zhou, Z.; Jiao, S.; Li, X. Construction of an anti-programmed death-ligand 1 chimeric antigen receptor and determination of its antitumor function with transduced cells. Oncol Lett. 2018, 16, 157–166. [Google Scholar] [CrossRef]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): Combination or built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Rosenberg, S.A.; Restifo, N.P. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat. Med. 2016, 22, 26–36. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sacchetti, B.; Botticelli, A.; Pierelli, L.; Nuti, M.; Alimandi, M. CAR-T with License to Kill Solid Tumors in Search of a Winning Strategy. Int. J. Mol. Sci. 2019, 20, 1903. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081903
Sacchetti B, Botticelli A, Pierelli L, Nuti M, Alimandi M. CAR-T with License to Kill Solid Tumors in Search of a Winning Strategy. International Journal of Molecular Sciences. 2019; 20(8):1903. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081903
Chicago/Turabian StyleSacchetti, Benedetto, Andrea Botticelli, Luca Pierelli, Marianna Nuti, and Maurizio Alimandi. 2019. "CAR-T with License to Kill Solid Tumors in Search of a Winning Strategy" International Journal of Molecular Sciences 20, no. 8: 1903. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081903