Production of Mesenchymal Stem Cells through Stem Cell Reprogramming

 ,
,  ,
, 
Abstract
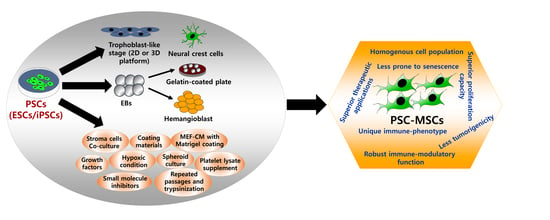
:
1. Introduction
2. Overview of MSCs
2.1. MSC Sources
2.2. MSC Characterization
3. Derivation of MSCs from PSCs: Methods and Applications
3.1. MSCs Derived from ESCs
3.1.1. Basic Methods
3.1.2. MSCs Derived via Repeated Passages Using Trypsinization with MSC Culture Medium
3.1.3. Hemangioblast-Based Methods
3.1.4. Defined Culture-Based Methods
3.1.5. MSCs Derived via Neural Crest Cells
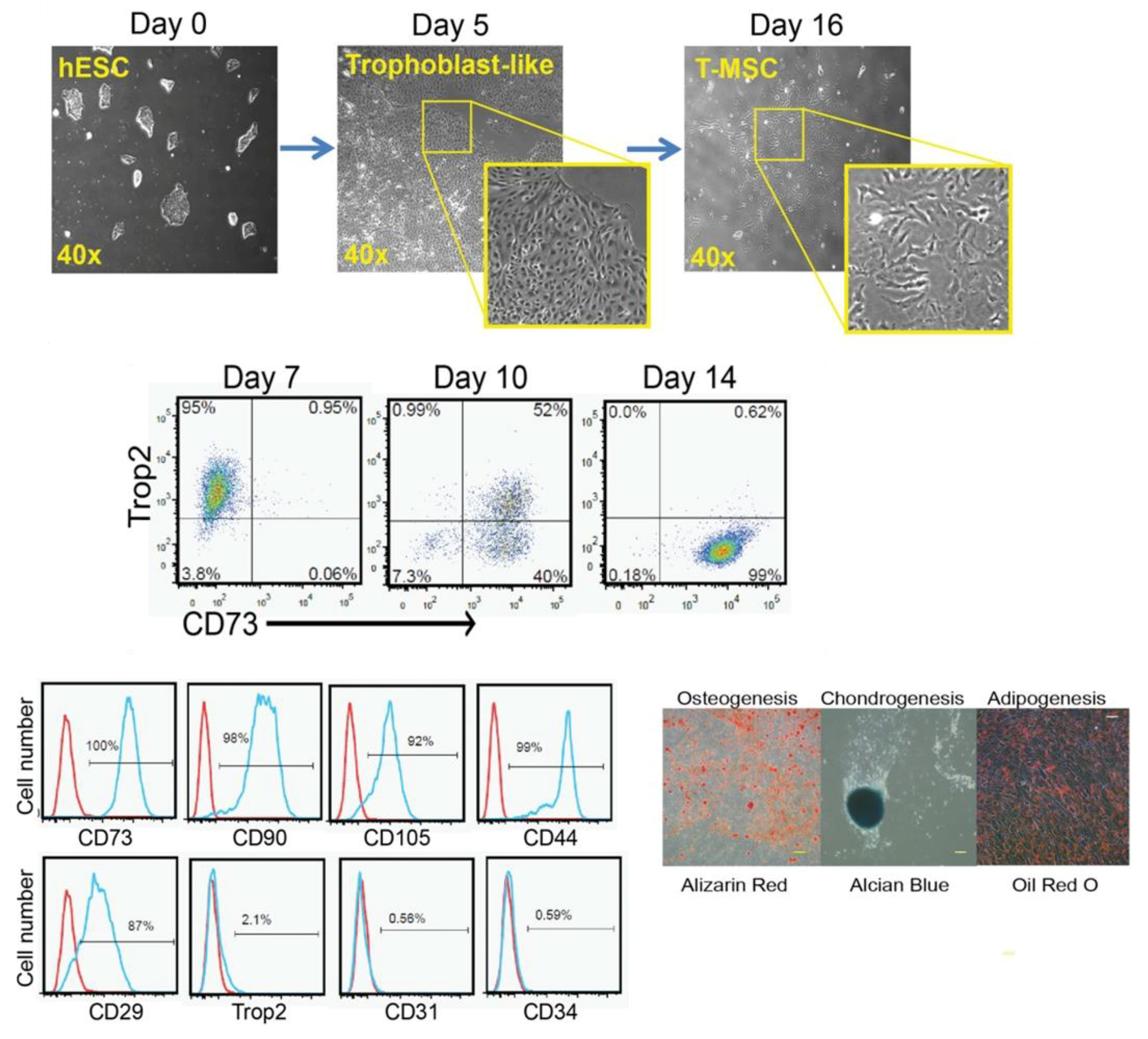
3.1.6. MSCs Derived via the Trophoblast-Like Stage
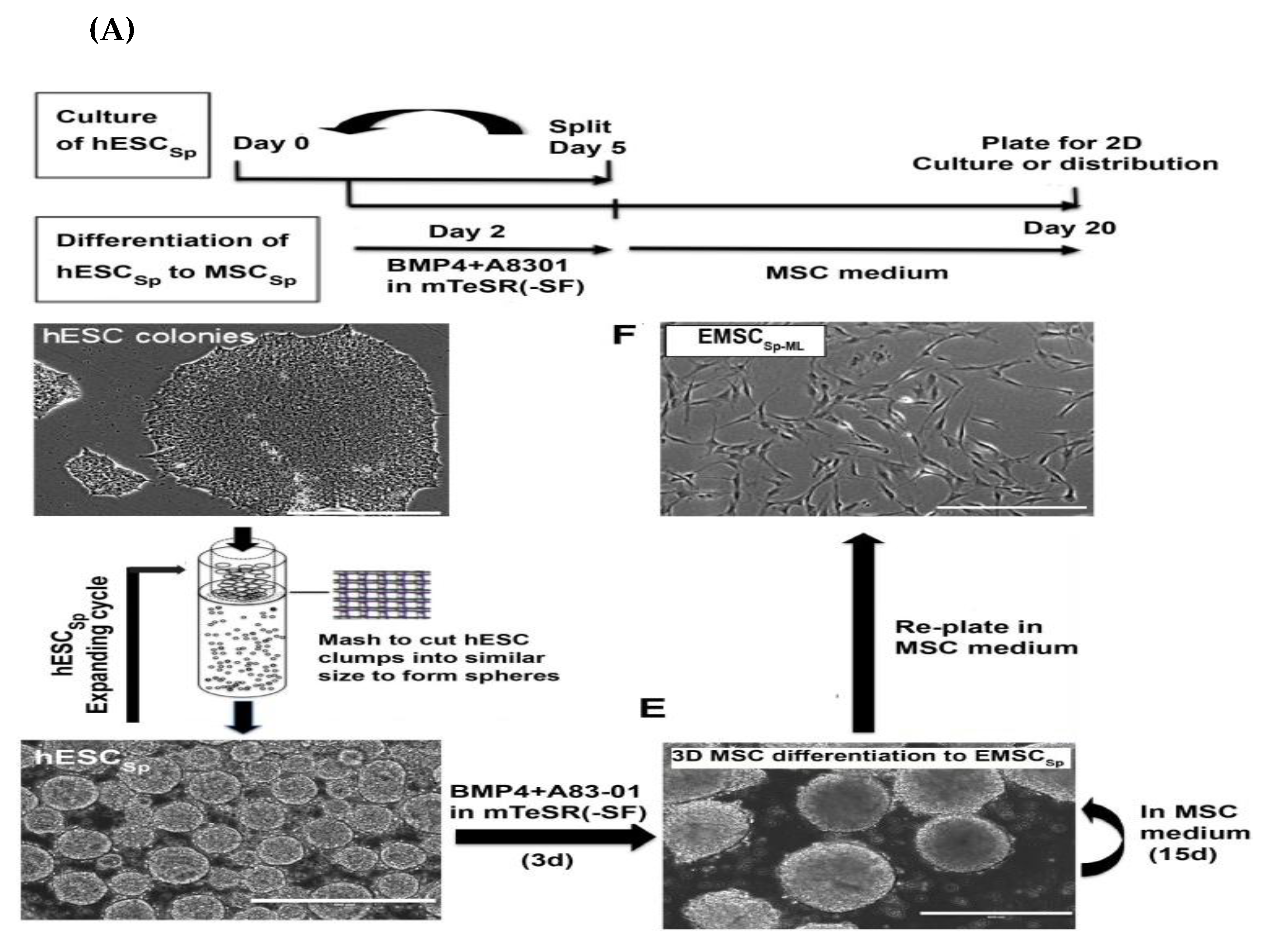
3.1.7. MSCs Derived via Spheroids Culture
3.1.8. MSCs Derived Using Small Molecule Inhibitors and Growth Factors
3.1.9. Therapeutic Applications of ESC-MSCs in Disease Models
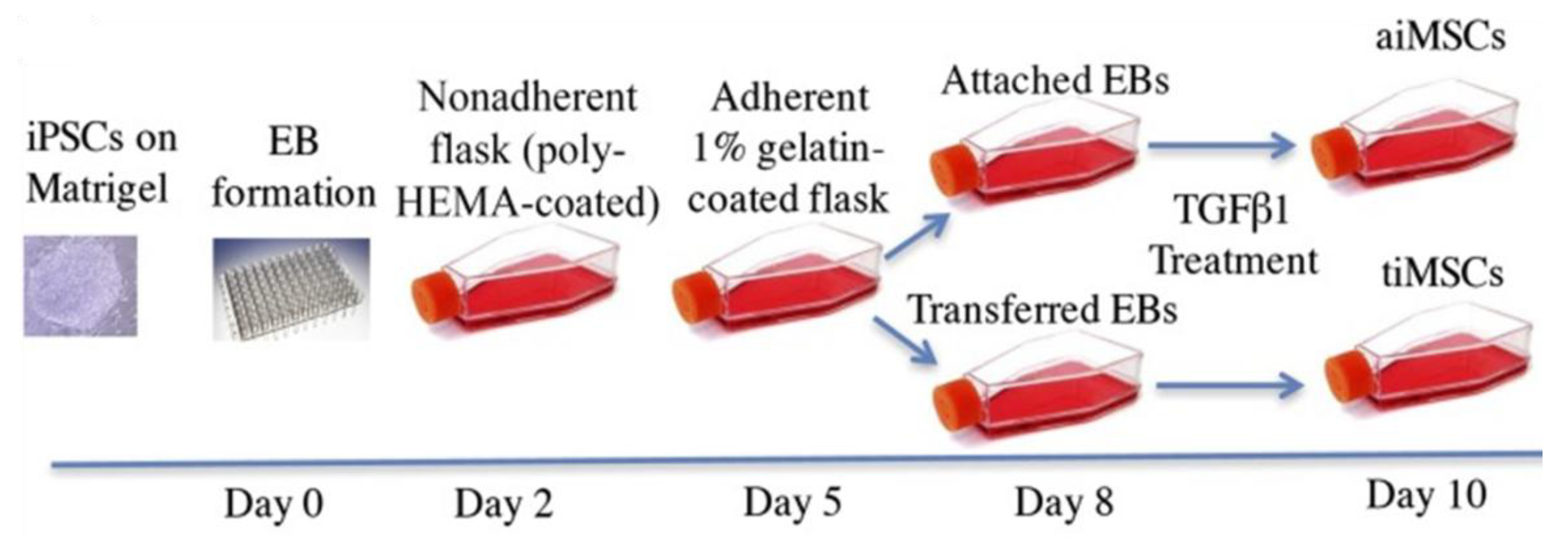
3.2. MSCs Derived from iPSCs
3.2.1. Derivation of iPSC-MSCs via Various Culture Components and Growth Factors
3.2.2. Derivation of iPSC-MSCs via Coating Materials and Small Molecule Inhibitors
3.2.3. Derivation of iPSC-MSCs via Ectopic Expression of MSC-Related Genes
3.2.4. Derivation of iPSC-MSCs via NCCs
3.2.5. iPSC-MSCs with Immunomodulatory and Anti-Inflammatory Functions
3.2.6. iPSC-MSCs for Bone Regeneration
3.2.7. iPSC-MSCs for Diabetes Therapy
3.2.8. Other Therapeutic Efficacies of iPSC-MSCs
4. Two-Edged Sword: Properties of PSC-MSCs and Future Prospects
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD-MSCs | Adipose-Derived MSCs |
| ALP | Alkaline Phosphatase |
| BEAS-2B | Bronchial Epithelial Cells |
| BM-MSCs | Bone Marrow-Derived MSCs |
| bFGF | Basic Fibroblast Growth Factor |
| CD | Cluster of Differentiation |
| CFU-F | Colony Forming Unit Fibroblasts |
| CM | Conditioned Medium |
| CNS | Central Nervous System |
| CPC | Calcium Phosphate Cement |
| DBM | Demineralized Bone Matrix |
| DCs | Dendritic Cells |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| DOX | Doxorubicin |
| ECM | Extracellular Matrix |
| EAE | Experimental Autoimmune Encephalitis |
| EBs | Embryonic Bodies |
| EMT | Epithelial-to-Mesenchymal Transition |
| FACS | Fluorescence-Activated Cell Sorting |
| FBS | Fetal Bovine Serum |
| GDF | Growth/Differentiation Factor |
| hESC-MSCSP | Spheroidal hESC-MSC |
| HLA | Human Leukocyte Antigen |
| IDO1 | Indoleamine 2,3 Dioxygenase |
| IFNγ | Interferon γ |
| IKK | IκB Kinase |
| i.m. | Intramuscular |
| ISCT | International Society for Cellular Therapy |
| i.v. | Intravenous |
| KO-SR | Knockout Serum Replacement |
| LEF | Lymphoid Enhancer-Binding Factor |
| LNGFR | Low-Affinity Nerve Growth Factor Receptor |
| MCT | Monocrotaline |
| MEF | Mouse Embryonic Fibroblast |
| MIF | Macrophage Inhibitory Factor |
| MPs | Mesenchymal Progenitors |
| MSCs | Mesenchymal Stem Cells |
| NCC | Neural Crest Cells |
| NCLCs | NC-Like Cells |
| NF-κB | Nuclear Factor Kappa B |
| NK cells | Natural Killer Cells |
| OA | Osteoarthritis |
| PAH | Pulmonary Arterial Hypertension |
| PDGFRa | Platelet-Derived Growth Factor Receptor Alpha |
| PEGD | PEG-Diacrylate |
| PL | Platelet Lysate |
| PMEDSAH | Poly [2-(methacryloyloxy) Ethyl Dimethyl-(3-Sulfopropyl) Ammonium Hydroxide |
| PBMCs | Peripheral Blood Mononuclear Cells |
| ROCK | Rho-Associated, Coiled-Coil Containing Protein Kinase |
| ROS | Reactive Oxygen Species |
| SSEA-4 | Stage-Specific Embryonic Antigen-4 |
| s.c. | Subcutaneous |
| TGF-β1 | Transforming Growth Factor-Beta 1 |
| THY-1 | Thymocyte Antigen-1 |
| T-MSC | Trophoblast-derived Mesenchymal Stem Cells |
| TNFα | Tumor Necrosis Factor-Alpha |
| TSG | Tumor Necrosis Factor Alpha-Stimulated Gene |
| VW | Vascular Wall |
| VEGF | Vascular Endothelial Growth Factor |
References
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.-W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.; Weißhardt, P.; Kleff, V.; Jastrow, H.; Jakob, H.G.; Ergün, S. Vascular wall-resident cd44+ multipotent stem cells give rise to pericytes and smooth muscle cells and contribute to new vessel maturation. PLoS ONE 2011, 6, e20540. [Google Scholar] [CrossRef] [PubMed]
- Tirino, V.; Paino, F.; d’Aquino, R.; Desiderio, V.; De Rosa, A.; Papaccio, G. Methods for the identification, characterization and banking of human dpscs: Current strategies and perspectives. Stem Cell Rev. Rep. 2011, 7, 608–615. [Google Scholar] [CrossRef]
- Wexler, S.A.; Donaldson, C.; Denning-Kendall, P.; Rice, C.; Bradley, B.; Hows, J.M. Adult bone marrow is a rich source of human mesenchymal ‘stem’cells but umbilical cord and mobilized adult blood are not. Br. J. Haematol. 2003, 121, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Abdal Dayem, A.; Lee, S.; Cho, S.-G. The impact of metallic nanoparticles on stem cell proliferation and differentiation. Nanomaterials 2018, 8, 761. [Google Scholar] [CrossRef]
- THOMAS, E.D.; ASHLEY, C.A.; LOCHTE, H.L.; JARETZKI, A.; SAHLER, O.D.; FERREBEE, J.W. Homografts of bone marrow in dogs after lethal total-body radiation. Blood 1959, 14, 720–736. [Google Scholar]
- Friedenstein, A.; Piatetzky-Shapiro, I.; Petrakova, K. Osteogenesis in transplants of bone marrow cells. Development 1966, 16, 381–390. [Google Scholar]
- Kristjánsson, B.; Honsawek, S. Current perspectives in mesenchymal stem cell therapies for osteoarthritis. Stem Cells Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef]
- Komatsu, K.; Honmou, O.; Suzuki, J.; Houkin, K.; Hamada, H.; Kocsis, J.D. Therapeutic time window of mesenchymal stem cells derived from bone marrow after cerebral ischemia. Brain Res. 2010, 1334, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zheng, S.; Zhou, C.; Wang, J.; Wang, T. Repair mechanisms of bone marrow mesenchymal stem cells in myocardial infarction. J. Cell. Mol. Med. 2011, 15, 1032–1043. [Google Scholar] [CrossRef]
- Otto, W.R.; Wright, N.A. Mesenchymal stem cells: From experiment to clinic. Fibrogenesis Tissue Repair 2011, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.R.; Pollock, K.; Hubel, A.; McKenna, D. Mesenchymal stem or stromal cells: A review of clinical applications and manufacturing practices. Transfusion 2014, 54, 1418–1437. [Google Scholar] [CrossRef]
- Conese, M.; Carbone, A.; Castellani, S.; Di Gioia, S. Paracrine effects and heterogeneity of marrow-derived stem/progenitor cells: Relevance for the treatment of respiratory diseases. Cells Tissues Organs 2013, 197, 445–473. [Google Scholar] [CrossRef] [PubMed]
- De Becker, A.; Riet, I.V. Homing and migration of mesenchymal stromal cells: How to improve the efficacy of cell therapy? World J. Stem Cells 2016, 8, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Leibacher, J.; Henschler, R. Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem Cell Res. Ther. 2016, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Miura, Y.; Fujishiro, A.; Shindo, T.; Shimazu, Y.; Hirai, H.; Tahara, H.; Takaori-Kondo, A.; Ichinohe, T.; Maekawa, T. Graft-versus-host disease amelioration by human bone marrow mesenchymal stromal/stem cell-derived extracellular vesicles is associated with peripheral preservation of naive t cell populations. Stem Cells 2018, 36, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Connick, P.; Kolappan, M.; Crawley, C.; Webber, D.J.; Patani, R.; Michell, A.W.; Du, M.-Q.; Luan, S.-L.; Altmann, D.R.; Thompson, A.J.; et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: An open-label phase 2a proof-of-concept study. Lancet Neurol. 2012, 11, 150–156. [Google Scholar] [CrossRef]
- Ho, P.-J.; Yen, M.-L.; Tang, B.-C.; Chen, C.-T.; Yen, B.L. H2o2 accumulation mediates differentiation capacity alteration, but not proliferative decline, in senescent human fetal mesenchymal stem cells. Antioxid. Redox Signal. 2013, 18, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, C.; Rabin, N.; Pizzey, A.; Nathwani, A.; Yong, K. Factors that influence short-term homing of human bone marrow-derived mesenchymal stem cells in a xenogeneic animal model. Haematologica 2008, 93, 1457–1465. [Google Scholar] [CrossRef]
- Mimeault, M.; Batra, S.K. Recent insights into the molecular mechanisms involved in aging and the malignant transformation of adult stem/progenitor cells and their therapeutic implications. Ageing Res. Rev. 2009, 8, 94–112. [Google Scholar] [CrossRef]
- Miura, M.; Miura, Y.; Padilla-Nash, H.M.; Molinolo, A.A.; Fu, B.; Patel, V.; Seo, B.M.; Sonoyama, W.; Zheng, J.J.; Baker, C.C. Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem Cells 2006, 24, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, W.; Ploemacher, R. Primary murine msc show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia 2003, 17, 160. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, J. The mesenchymal stromal cells dilemma—does a negative phase iii trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Kimbrel, E.A.; Kouris, N.A.; Yavanian, G.J.; Chu, J.; Qin, Y.; Chan, A.; Singh, R.P.; McCurdy, D.; Gordon, L.; Levinson, R.D. Mesenchymal stem cell population derived from human pluripotent stem cells displays potent immunomodulatory and therapeutic properties. Stem Cells Dev. 2014, 23, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Tyndall, A. Mesenchymal stem cell treatments in rheumatology—a glass half full? Nat. Rev. Rheumatol. 2014, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Ho, A.D. Mesenchymal stem cell preparations—comparing apples and oranges. Stem Cell Rev. 2007, 3, 239–248. [Google Scholar] [CrossRef]
- Janzen, V.; Forkert, R.; Fleming, H.E.; Saito, Y.; Waring, M.T.; Dombkowski, D.M.; Cheng, T.; DePinho, R.A.; Sharpless, N.E.; Scadden, D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16 ink4a. Nature 2006, 443, 421. [Google Scholar] [CrossRef]
- Stenderup, K.; Justesen, J.; Clausen, C.; Kassem, M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 2003, 33, 919–926. [Google Scholar] [CrossRef]
- Mueller, S.M.; Glowacki, J. Age-related decline in the osteogenic potential of human bone marrow cells cultured in three-dimensional collagen sponges. J. Cell. Biochem. 2001, 82, 583–590. [Google Scholar] [CrossRef]
- Yan, L.; Jiang, B.; Li, E.; Wang, X.; Ling, Q.; Zheng, D.; Park, J.W.; Chen, X.; Cheung, E.; Du, X. Scalable generation of mesenchymal stem cells from human embryonic stem cells in 3d. Int. J. Biol. Sci. 2018, 14, 1196. [Google Scholar] [CrossRef]
- Jiang, B.; Li, Y.; Wang, X.; Li, E.; Murphy, K.; Vaccaro, K.; Li, Y.; Xu, R.H. Mesenchymal stem cells derived from human pluripotent cells, an unlimited and quality-controllable source, for therapeutic applications. Stem Cells 2018. [Google Scholar]
- Friedenstein, A.; Chailakhjan, R.; Lalykina, K. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Prolif. 1970, 3, 393–403. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Petrakova, K.V.; Kurolesova, A.I.; Frolova, G.P. Heterotopic transplants of bone marrow. Transplantation 1968, 6, 230–247. [Google Scholar] [CrossRef]
- Castro-Malaspina, H.; Gay, R.E.; Resnick, G.; Kapoor, N.; Meyers, P.; Chiarieri, D.; Mckenzie, S.; Broxmeyer, H.E.; Moore, M. Characterization of human bone marrow fibroblast colony-forming cells (cfu-f) and their progeny. Blood 1980, 56, 289–301. [Google Scholar]
- Jackson, W.M.; Nesti, L.J.; Tuan, R.S. Potential therapeutic applications of muscle-derived mesenchymal stem and progenitor cells. Expert Opin. Biol. Ther. 2010, 10, 505–517. [Google Scholar] [CrossRef]
- Aust, L.; Devlin, B.; Foster, S.; Halvorsen, Y.; Hicok, K.; Laney, T.d.; Sen, A.; Willingmyre, G.; Gimble, J. Yield of human adipose-derived adult stem cells from liposuction aspirates. Cytotherapy 2004, 6, 7–14. [Google Scholar] [CrossRef]
- Gay, I.C.; Chen, S.; MacDougall, M. Isolation and characterization of multipotent human periodontal ligament stem cells. Orthod. Craniofacial Res. 2007, 10, 149–160. [Google Scholar] [CrossRef]
- Smiler, D.; Soltan, M.; Albitar, M. Toward the identification of mesenchymal stem cells in bone marrow and peripheral blood for bone regeneration. Implant Dent. 2008, 17, 236–247. [Google Scholar] [CrossRef]
- Rotter, N.; Oder, J.; Schlenke, P.; Lindner, U.; Böhrnsen, F.; Kramer, J.; Rohwedel, J.; Huss, R.; Brandau, S.; Wollenberg, B. Isolation and characterization of adult stem cells from human salivary glands. Stem Cells Dev. 2008, 17, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, M.J.; Bonnet, D.; Janes, S.M. Stem cells of the alveolar epithelium. Lancet 2005, 366, 249–260. [Google Scholar] [CrossRef]
- Corrao, S.; La Rocca, G.; Lo Iacono, M.; Corsello, T.; Farina, F.; Anzalone, R. Umbilical cord revisited: From wharton’s jelly myofibroblasts to mesenchymal stem cells. Histol. Histopathol. 2013, 28, 1235–1244. [Google Scholar] [PubMed]
- Shi, S.; Gronthos, S. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J. Bone Miner. Res. 2003, 18, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Kolaparthy, L.K.; Sanivarapu, S.; Moogla, S.; Kutcham, R.S. Adipose tissue-adequate, accessible regenerative material. Int. J. Stem Cells 2015, 8, 121. [Google Scholar] [CrossRef]
- Kwon, A.; Kim, Y.; Kim, M.; Kim, J.; Choi, H.; Jekarl, D.W.; Lee, S.; Kim, J.M.; Shin, J.-C.; Park, I.Y. Tissue-specific differentiation potency of mesenchymal stromal cells from perinatal tissues. Sci. Rep. 2016, 6, 23544. [Google Scholar] [CrossRef]
- Miao, Z.; Jin, J.; Chen, L.; Zhu, J.; Huang, W.; Zhao, J.; Qian, H.; Zhang, X. Isolation of mesenchymal stem cells from human placenta: Comparison with human bone marrow mesenchymal stem cells. Cell Biol. Int. 2006, 30, 681–687. [Google Scholar] [CrossRef]
- La Rocca, G.; Anzalone, R.; Corrao, S.; Magno, F.; Loria, T.; Iacono, M.L.; Di Stefano, A.; Giannuzzi, P.; Marasà, L.; Cappello, F. Isolation and characterization of oct-4+/hla-g+ mesenchymal stem cells from human umbilical cord matrix: Differentiation potential and detection of new markers. Histochem. Cell Biol. 2009, 131, 267–282. [Google Scholar] [CrossRef]
- Kern, S.; Eichler, H.; Stoeve, J.; Klüter, H.; Bieback, K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar] [CrossRef]
- Secco, M.; Zucconi, E.; Vieira, N.M.; Fogaça, L.L.; Cerqueira, A.; Carvalho, M.D.F.; Jazedje, T.; Okamoto, O.K.; Muotri, A.R.; Zatz, M. Mesenchymal stem cells from umbilical cord: Do not discard the cord! Neuromuscular Disorders 2008, 18, 17–18. [Google Scholar] [CrossRef]
- Elahi, K.C.; Klein, G.; Avci-Adali, M.; Sievert, K.D.; MacNeil, S.; Aicher, W.K. Human mesenchymal stromal cells from different sources diverge in their expression of cell surface proteins and display distinct differentiation patterns. Stem Cells Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.E.; Walker, J.T.; Keating, A. Concise review: Wharton’s jelly: The rich, but enigmatic, source of mesenchymal stromal cells. Stem Cells Transl. Med. 2017, 6, 1620–1630. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Mou, X.-Z.; Du, X.-C.; Xiang, C. Comparative analysis of biological characteristics of adult mesenchymal stem cells with different tissue origins. Asian Pac. J. Trop. Med. 2015, 8, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Billing, A.M.; Hamidane, H.B.; Dib, S.S.; Cotton, R.J.; Bhagwat, A.M.; Kumar, P.; Hayat, S.; Yousri, N.A.; Goswami, N.; Suhre, K. Comprehensive transcriptomic and proteomic characterization of human mesenchymal stem cells reveals source specific cellular markers. Sci. Rep. 2016, 6, 21507. [Google Scholar] [CrossRef]
- Trounson, A.; McDonald, C. Stem cell therapies in clinical trials: Progress and challenges. Cell Stem Cell 2015, 17, 11–22. [Google Scholar] [CrossRef]
- Atoui, R.; Chiu, R.C. Concise review: Immunomodulatory properties of mesenchymal stem cells in cellular transplantation: Update, controversies, and unknowns. Stem Cells Transl. Med. 2012, 1, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Steinert, A.F.; Rackwitz, L.; Gilbert, F.; Nöth, U.; Tuan, R.S. Concise review: The clinical application of mesenchymal stem cells for musculoskeletal regeneration: Current status and perspectives. Stem Cells Transl. Med. 2012, 1, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.M.; Fishman, J.E.; Gerstenblith, G.; Velazquez, D.L.D.; Zambrano, J.P.; Suncion, V.Y.; Tracy, M.; Ghersin, E.; Johnston, P.V.; Brinker, J.A. Comparison of allogeneic vs autologous bone marrow–derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: The poseidon randomized trial. JAMA 2012, 308, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Monsarrat, P.; Vergnes, J.-N.; Planat-Bénard, V.; Ravaud, P.; Kémoun, P.; Sensebé, L.; Casteilla, L. An innovative, comprehensive mapping and multiscale analysis of registered trials for stem cell-based regenerative medicine. Stem Cells Transl. Med. 2016, 5, 826–835. [Google Scholar] [CrossRef]
- Kinkaid, H.Y.M.; Huang, X.P.; Li, R.K.; Weisel, R.D. What’s new in cardiac cell therapy? Allogeneic bone marrow stromal cells as “universal donor cells”. J. Card. Surg. 2010, 25, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, E.T.; Gustafson, M.P.; Dudakovic, A.; Riester, S.M.; Garces, C.G.; Paradise, C.R.; Takai, H.; Karperien, M.; Cool, S.; Im Sampen, H.-J. Identification and validation of multiple cell surface markers of clinical-grade adipose-derived mesenchymal stromal cells as novel release criteria for good manufacturing practice-compliant production. Stem Cell Res. Ther. 2016, 7, 107. [Google Scholar] [CrossRef]
- Ong, W.K.; Tan, C.S.; Chan, K.L.; Goesantoso, G.G.; Chan, X.H.D.; Chan, E.; Yin, J.; Yeo, C.R.; Khoo, C.M.; So, J.B.Y. Identification of specific cell-surface markers of adipose-derived stem cells from subcutaneous and visceral fat depots. Stem Cell Rep. 2014, 2, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Vallone, V.F.; Romaniuk, M.A.; Choi, H.; Labovsky, V.; Otaegui, J.; Chasseing, N.A. Mesenchymal stem cells and their use in therapy: What has been achieved? Differentiation 2013, 85, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Segawa, Y.; Muneta, T.; Makino, H.; Nimura, A.; Mochizuki, T.; Ju, Y.J.; Ezura, Y.; Umezawa, A.; Sekiya, I. Mesenchymal stem cells derived from synovium, meniscus, anterior cruciate ligament, and articular chondrocytes share similar gene expression profiles. J. Orthop. Res. 2009, 27, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Mougiakakos, D. Multipotent mesenchymal stromal cells and the innate immune system. Nat. Rev. Immunol. 2012, 12, 383. [Google Scholar] [CrossRef]
- Gang, E.J.; Bosnakovski, D.; Figueiredo, C.A.; Visser, J.W.; Perlingeiro, R.C. Ssea-4 identifies mesenchymal stem cells from bone marrow. Blood 2007, 109, 1743–1751. [Google Scholar] [CrossRef]
- Sobiesiak, M.; Sivasubramaniyan, K.; Hermann, C.; Tan, C.; Örgel, M.; Treml, S.; Cerabona, F.; de Zwart, P.; Ochs, U.; Müller, C.A. The mesenchymal stem cell antigen msca-1 is identical to tissue non-specific alkaline phosphatase. Stem Cells Dev. 2009, 19, 669–677. [Google Scholar] [CrossRef]
- Gronthos, S.; Zannettino, A.C.; Hay, S.J.; Shi, S.; Graves, S.E.; Kortesidis, A.; Simmons, P.J. Molecular and cellular characterisation of highly purified stromal stem cells derived from human bone marrow. J. Cell Sci. 2003, 116, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Gronthos, S.; Simmons, P.J. The growth factor requirements of stro-1-positive human bone marrow stromal precursors under serum-deprived conditions in vitro. Blood 1995, 85, 929–940. [Google Scholar] [PubMed]
- BÜHRING, H.J.; Battula, V.L.; Treml, S.; Schewe, B.; Kanz, L.; Vogel, W. Novel markers for the prospective isolation of human msc. Ann. N. Y. Acad. Sci. 2007, 1106, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Quirici, N.; Soligo, D.; Bossolasco, P.; Servida, F.; Lumini, C.; Deliliers, G.L. Isolation of bone marrow mesenchymal stem cells by anti-nerve growth factor receptor antibodies. Exper. Hematol. 2002, 30, 783–791. [Google Scholar] [CrossRef]
- Gronthos, S.; Graves, S.; Ohta, S.; Simmons, P. The stro-1+ fraction of adult human bone marrow contains the osteogenic precursors. Blood 1994, 84, 4164–4173. [Google Scholar] [CrossRef]
- Gronthos, S.; Zannettino, A.C.; Graves, S.E.; Ohta, S.; Hay, S.J.; Simmons, P.J. Differential cell surface expression of the stro-1 and alkaline phosphatase antigens on discrete developmental stages in primary cultures of human bone cells. J. Bone Miner. Res. 1999, 14, 47–56. [Google Scholar] [CrossRef]
- Bianco, P.; Cao, X.; Frenette, P.S.; Mao, J.J.; Robey, P.G.; Simmons, P.J.; Wang, C.-Y. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat. Med. 2013, 19, 35. [Google Scholar] [CrossRef]
- Jones, E.A.; English, A.; Kinsey, S.E.; Straszynski, L.; Emery, P.; Ponchel, F.; McGonagle, D. Optimization of a flow cytometry-based protocol for detection and phenotypic characterization of multipotent mesenchymal stromal cells from human bone marrow. Cytom. Part B Clin. Cytom. 2006, 70, 391–399. [Google Scholar] [CrossRef]
- Nombela-Arrieta, C.; Ritz, J.; Silberstein, L.E. The elusive nature and function of mesenchymal stem cells. Nat. Rev. Mol. Cell Biol. 2011, 12, 126. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829. [Google Scholar] [CrossRef]
- Samsonraj, R.M.; Rai, B.; Sathiyanathan, P.; Puan, K.J.; Rötzschke, O.; Hui, J.H.; Raghunath, M.; Stanton, L.W.; Nurcombe, V.; Cool, S.M. Establishing criteria for human mesenchymal stem cell potency. Stem Cells 2015, 33, 1878–1891. [Google Scholar] [CrossRef]
- Samsonraj, R.M.; Raghunath, M.; Nurcombe, V.; Hui, J.H.; van Wijnen, A.J.; Cool, S.M. Concise review: Multifaceted characterization of human mesenchymal stem cells for use in regenerative medicine. Stem Cells Transl. Med. 2017, 6, 2173–2185. [Google Scholar] [CrossRef] [PubMed]
- Nandy, S.B.; Mohanty, S.; Singh, M.; Behari, M.; Airan, B. Fibroblast growth factor-2 alone as an efficient inducer for differentiation of human bone marrow mesenchymal stem cells into dopaminergic neurons. J. Biomed. Sci. 2014, 21, 83. [Google Scholar] [CrossRef] [PubMed]
- Armiñán, A.; Gandía, C.; Bartual, M.; García-Verdugo, J.M.; Lledó, E.; Mirabet, V.; Llop, M.; Barea, J.; Montero, J.A.; Sepúlveda, P. Cardiac differentiation is driven by nkx2. 5 and gata4 nuclear translocation in tissue-specific mesenchymal stem cells. Stem Cells Dev. 2009, 18, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Y.; Wu, Y.; Ni, B.; Liang, Z. Sodium butyrate promotes the differentiation of rat bone marrow mesenchymal stem cells to smooth muscle cells through histone acetylation. PLoS ONE 2014, 9, e116183. [Google Scholar] [CrossRef]
- Huang, G.-J.; Gronthos, S.; Shi, S. Mesenchymal stem cells derived from dental tissues vs. Those from other sources: Their biology and role in regenerative medicine. J. Dent. Res. 2009, 88, 792–806. [Google Scholar] [CrossRef]
- Gugutkov, D.; Awaja, F.; Belemezova, K.; Keremidarska, M.; Krasteva, N.; Kyurkchiev, S.; Gallego-Ferrer, G.; Seker, S.; Elçin, A.E.; Elçin, Y.M. Osteogenic differentiation of mesenchymal stem cells using hybrid nanofibers with different configurations and dimensionality. J. Biomed. Mater. Res. Part A 2017, 105, 2065–2074. [Google Scholar] [CrossRef]
- Scott, M.A.; Nguyen, V.T.; Levi, B.; James, A.W. Current methods of adipogenic differentiation of mesenchymal stem cells. Stem Cells Dev. 2011, 20, 1793–1804. [Google Scholar] [CrossRef]
- Xu, C.; Jiang, J.; Sottile, V.; McWhir, J.; Lebkowski, J.; Carpenter, M.K. Immortalized fibroblast-like cells derived from human embryonic stem cells support undifferentiated cell growth. Stem Cells 2004, 22, 972–980. [Google Scholar] [CrossRef]
- Barberi, T.; Willis, L.M.; Socci, N.D.; Studer, L. Derivation of multipotent mesenchymal precursors from human embryonic stem cells. PLoS Med. 2005, 2, e161. [Google Scholar] [CrossRef]
- Olivier, E.N.; Rybicki, A.C.; Bouhassira, E.E. Differentiation of human embryonic stem cells into bipotent mesenchymal stem cells. Stem Cells 2006, 24, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Hematti, P. Simultaneous generation of cd34+ primitive hematopoietic cells and cd73+ mesenchymal stem cells from human embryonic stem cells cocultured with murine op9 stromal cells. Exp. Hematol. 2007, 35, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Hematti, P. Derivation and immunological characterization of mesenchymal stromal cells from human embryonic stem cells. Exp. Hematol. 2008, 36, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Hwang, N.S.; Varghese, S.; Lee, H.J.; Zhang, Z.; Ye, Z.; Bae, J.; Cheng, L.; Elisseeff, J. In vivo commitment and functional tissue regeneration using human embryonic stem cell-derived mesenchymal cells. Proc. Natl. Acad. Sci. USA 2008, 105, 20641–20646. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.E.; Tong, W.; Krebsbach, P.H. The derivation of mesenchymal stem cells from human embryonic stem cells. Cells Tissues Organs 2009, 189, 256–260. [Google Scholar] [CrossRef]
- Yen, M.L.; Hou, C.H.; Peng, K.Y.; Tseng, P.C.; Jiang, S.S.; Shun, C.T.; Chen, Y.C.; Kuo, M.L. Efficient derivation and concise gene expression profiling of human embryonic stem cell-derived mesenchymal progenitors (emps). Cell Transpl. 2011, 20, 1529–1545. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-J.; Feng, Q.; Caballero, S.; Chen, Y.; Moore, M.A.; Grant, M.B.; Lanza, R. Generation of functional hemangioblasts from human embryonic stem cells. Nat. Methods 2007, 4, 501. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-J.; Luo, C.; Holton, K.; Feng, Q.; Ivanova, Y.; Lanza, R. Robust generation of hemangioblastic progenitors from human embryonic stem cells. Regen. Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kimbrel, E.A.; Ijichi, K.; Paul, D.; Lazorchak, A.S.; Chu, J.; Kouris, N.A.; Yavanian, G.J.; Lu, S.-J.; Pachter, J.S. Human esc-derived mscs outperform bone marrow mscs in the treatment of an eae model of multiple sclerosis. Stem Cell Rep. 2014, 3, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Lian, Q.; Lye, E.; Yeo, K.S.; Tan, E.K.W.; Salto-Tellez, M.; Liu, T.M.; Palanisamy, N.; El Oakley, R.M.; Lee, E.H.; Lim, B. Derivation of clinically compliant mscs from cd105+, cd24− differentiated human escs. Stem Cells 2007, 25, 425–436. [Google Scholar] [CrossRef]
- Karlsson, C.; Emanuelsson, K.; Wessberg, F.; Kajic, K.; Axell, M.Z.; Eriksson, P.S.; Lindahl, A.; Hyllner, J.; Strehl, R. Human embryonic stem cell-derived mesenchymal progenitors—potential in regenerative medicine. Stem Cell Res. 2009, 3, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Ellerström, C.; Strehl, R.; Moya, K.; Andersson, K.; Bergh, C.; Lundin, K.; Hyllner, J.; Semb, H. Derivation of a xeno-free human embryonic stem cell line. Stem Cells 2006, 24, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tang, Y.; Lü, S.; Zhou, J.; Du, Z.; Duan, C.; Li, Z.; Wang, C. The tumourigenicity of ips cells and their differentiated derivates. J. Cell. Mol. Med. 2013, 17, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Le Douarin, N.M.; Dupin, E. Multipotentiality of the neural crest. Curr. Opin. Genet. Dev. 2003, 13, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Fukuta, M.; Nakai, Y.; Kirino, K.; Nakagawa, M.; Sekiguchi, K.; Nagata, S.; Matsumoto, Y.; Yamamoto, T.; Umeda, K.; Heike, T. Derivation of mesenchymal stromal cells from pluripotent stem cells through a neural crest lineage using small molecule compounds with defined media. PLoS ONE 2014, 9, e112291. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.-H.; Chen, X.; Li, D.S.; Li, R.; Addicks, G.C.; Glennon, C.; Zwaka, T.P.; Thomson, J.A. Bmp4 initiates human embryonic stem cell differentiation to trophoblast. Nat. Biotechnol. 2002, 20, 1261. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lazorchak, A.S.; Song, L.; Li, E.; Zhang, Z.; Jiang, B.; Xu, R.H. Immune modulatory mesenchymal stem cells derived from human embryonic stem cells through a trophoblast-like stage. Stem Cells 2016, 34, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Z.; Jiang, B.; Yan, L.; Park, J.W.; Xu, R.H. Generation of mesenchymal stem cells from human embryonic stem cells in a complete serum-free condition. Int. J. Biol. Sci. 2018, 14, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Otsuji, T.G.; Bin, J.; Yoshimura, A.; Tomura, M.; Tateyama, D.; Minami, I.; Yoshikawa, Y.; Aiba, K.; Heuser, J.E.; Nishino, T. A 3d sphere culture system containing functional polymers for large-scale human pluripotent stem cell production. Stem Cell Rep. 2014, 2, 734–745. [Google Scholar] [CrossRef]
- Steiner, D.; Khaner, H.; Cohen, M.; Even-Ram, S.; Gil, Y.; Itsykson, P.; Turetsky, T.; Idelson, M.; Aizenman, E.; Ram, R. Derivation, propagation and controlled differentiation of human embryonic stem cells in suspension. Nat. Biotechnol. 2010, 28, 361. [Google Scholar] [CrossRef]
- Jiang, B.; Yan, L.; Miao, Z.; Li, E.; Wong, K.H.; Xu, R.-H. Spheroidal formation preserves human stem cells for prolonged time under ambient conditions for facile storage and transportation. Biomaterials 2017, 133, 275–286. [Google Scholar] [CrossRef]
- Jiang, B.; Xiang, Z.; Ai, Z.; Wang, H.; Li, Y.; Ji, W.; Li, T. Generation of cardiac spheres from primate pluripotent stem cells in a small molecule-based 3d system. Biomaterials 2015, 65, 103–114. [Google Scholar] [CrossRef]
- Mahmood, A.; Harkness, L.; Schrøder, H.D.; Abdallah, B.M.; Kassem, M. Enhanced differentiation of human embryonic stem cells to mesenchymal progenitors by inhibition of tgf-β/activin/nodal signaling using sb-431542. J. Bone Miner. Res. 2010, 25, 1216–1233. [Google Scholar] [CrossRef]
- Sánchez, L.; Gutierrez-Aranda, I.; Ligero, G.; Rubio, R.; Muñoz-López, M.; García-Pérez, J.L.; Ramos, V.; Real, P.J.; Bueno, C.; Rodríguez, R. Enrichment of human esc-derived multipotent mesenchymal stem cells with immunosuppressive and anti-inflammatory properties capable to protect against experimental inflammatory bowel disease. Stem Cells 2011, 29, 251–262. [Google Scholar] [CrossRef]
- Deng, P.; Zhou, C.; Alvarez, R.; Hong, C.; Wang, C.-Y. Inhibition of ikk/nf-κb signaling enhances differentiation of mesenchymal stromal cells from human embryonic stem cells. Stem Cell Reports 2016, 6, 456–465. [Google Scholar] [CrossRef]
- Zhang, Y.; Liao, S.; Yang, M.; Liang, X.; Poon, M.-W.; Wong, C.-Y.; Wang, J.; Zhou, Z.; Cheong, S.-K.; Lee, C.-N. Improved cell survival and paracrine capacity of human embryonic stem cell-derived mesenchymal stem cells promote therapeutic potential for pulmonary arterial hypertension. Cell Transplant. 2012, 21, 2225–2239. [Google Scholar] [CrossRef]
- Hawkins, K.E.; Corcelli, M.; Dowding, K.; Ranzoni, A.M.; Vlahova, F.; Hau, K.L.; Hunjan, A.; Peebles, D.; Gressens, P.; Hagberg, H. Embryonic stem cell-derived mesenchymal stem cells (mscs) have a superior neuroprotective capacity over fetal mscs in the hypoxic-ischemic mouse brain. Stem Cells Transl. Med. 2018, 7, 439–449. [Google Scholar] [CrossRef]
- Chen, Y.S.; Pelekanos, R.A.; Ellis, R.L.; Horne, R.; Wolvetang, E.J.; Fisk, N.M. Small molecule mesengenic induction of human induced pluripotent stem cells to generate mesenchymal stem/stromal cells. Stem Cells Transl. Med. 2012, 1, 83–95. [Google Scholar] [CrossRef]
- Gonzalo-Gil, E.; Pérez-Lorenzo, M.J.; Galindo, M.; de la Guardia, R.D.; López-Millán, B.; Bueno, C.; Menéndez, P.; Pablos, J.L.; Criado, G. Human embryonic stem cell-derived mesenchymal stromal cells ameliorate collagen-induced arthritis by inducing host-derived indoleamine 2, 3 dioxygenase. Arthritis Res. Ther. 2016, 18, 77. [Google Scholar] [CrossRef]
- Hajizadeh-Saffar, E.; Tahamtani, Y.; Aghdami, N.; Azadmanesh, K.; Habibi-Anbouhi, M.; Heremans, Y.; De Leu, N.; Heimberg, H.; Ravassard, P.; Shokrgozar, M. Inducible vegf expression by human embryonic stem cell-derived mesenchymal stromal cells reduces the minimal islet mass required to reverse diabetes. Sci. Rep. 2015, 5, 9322. [Google Scholar] [CrossRef]
- Yan, L.; Jiang, B.; Niu, Y.; Wang, H.; Li, E.; Yan, Y.; Sun, H.; Duan, Y.; Chang, S.; Chen, G.; et al. Intrathecal delivery of human esc-derived mesenchymal stem cell spheres promotes recovery of a primate multiple sclerosis model. Cell Death Discov. 2018, 4, 018–0091. [Google Scholar] [CrossRef]
- Robinton, D.A.; Daley, G.Q. The promise of induced pluripotent stem cells in research and therapy. Nature 2012, 481, 295. [Google Scholar] [CrossRef] [PubMed]
- Lian, Q.; Zhang, Y.; Zhang, J.; Zhang, H.K.; Wu, X.; Zhang, Y.; Lam, F.F.-Y.; Kang, S.; Xia, J.C.; Lai, W.-H. Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation 2010, 121, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Lian, Q.; Zhang, Y.; Liang, X.; Gao, F.; Tse, H.-F. Directed differentiation of human-induced pluripotent stem cells to mesenchymal stem cells. In Mesenchymal Stem Cells; Springer: Berlin/Heidelberg, Germany, 2016; pp. 289–298. [Google Scholar]
- Zhang, J.; Chan, Y.-C.; Ho, J.C.-Y.; Siu, C.-W.; Lian, Q.; Tse, H.-F. Regulation of cell proliferation of human induced pluripotent stem cell-derived mesenchymal stem cells via ether-a-go-go 1 (heag1) potassium channel. Am. J. Physiol.-Cell Physiol. 2012, 303, C115–C125. [Google Scholar] [CrossRef]
- Zou, L.; Luo, Y.; Chen, M.; Wang, G.; Ding, M.; Petersen, C.C.; Kang, R.; Dagnaes-Hansen, F.; Zeng, Y.; Lv, N.; et al. A simple method for deriving functional mscs and applied for osteogenesis in 3d scaffolds. Sci. Rep. 2013, 3, 2243. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Choo, A.; Lim, S.K. Derivation and characterization of human esc-derived mesenchymal stem cells. In Mesenchymal Stem Cell Assays and Applications; Springer: Berlin/Heidelberg, Germany, 2011; pp. 141–150. [Google Scholar]
- Han, J.; Mistriotis, P.; Lei, P.; Wang, D.; Liu, S.; Andreadis, S.T. Nanog reverses the effects of organismal aging on mesenchymal stem cell proliferation and myogenic differentiation potential. Stem Cells 2012, 30, 2746–2759. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Aprecio, R.M.; Zhou, X.; Wang, Q.; Zhang, W.; Ding, Y.; Li, Y. Therapeutic effect of tsg-6 engineered ipsc-derived mscs on experimental periodontitis in rats: A pilot study. PLoS ONE 2014, 9, e100285. [Google Scholar] [CrossRef] [PubMed]
- Luzzani, C.; Neiman, G.; Garate, X.; Questa, M.; Solari, C.; Espinosa, D.F.; García, M.; Errecalde, A.L.; Guberman, A.; Scassa, M.E. A therapy-grade protocol for differentiation of pluripotent stem cells into mesenchymal stem cells using platelet lysate as supplement. Stem Cell Res. Ther. 2015, 6, 6. [Google Scholar] [CrossRef]
- Medici, D.; Nawshad, A. Type i collagen promotes epithelial–mesenchymal transition through ilk-dependent activation of nf-κb and lef-1. Matrix Biol. 2010, 29, 161–165. [Google Scholar] [CrossRef]
- Liu, Y.; Goldberg, A.J.; Dennis, J.E.; Gronowicz, G.A.; Kuhn, L.T. One-step derivation of mesenchymal stem cell (msc)-like cells from human pluripotent stem cells on a fibrillar collagen coating. PLoS ONE 2012, 7, e33225. [Google Scholar] [CrossRef]
- Villa-Diaz, L.; Brown, S.; Liu, Y.; Ross, A.; Lahann, J.; Parent, J.; Krebsbach, P. Derivation of mesenchymal stem cells from human induced pluripotent stem cells cultured on synthetic substrates. Stem Cells 2012, 30, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Villa-Diaz, L.G.; Nandivada, H.; Ding, J.; Nogueira-de-Souza, N.C.; Krebsbach, P.H.; O’shea, K.S.; Lahann, J.; Smith, G.D. Synthetic polymer coatings for long-term growth of human embryonic stem cells. Nat. Biotechnol. 2010, 28, 581. [Google Scholar] [CrossRef] [PubMed]
- Nandivada, H.; Villa-Diaz, L.G.; O’shea, K.S.; Smith, G.D.; Krebsbach, P.H.; Lahann, J. Fabrication of synthetic polymer coatings and their use in feeder-free culture of human embryonic stem cells. Nat. Protoc. 2011, 6, 1037. [Google Scholar] [CrossRef]
- Wei, H.; Tan, G.; Manasi; Qiu, S.; Kong, G.; Yong, P.; Koh, C.; Ooi, T.H.; Lim, S.Y.; Wong, P.; et al. One-step derivation of cardiomyocytes and mesenchymal stem cells from human pluripotent stem cells. Stem Cell Res. 2012, 9, 87–100. [Google Scholar] [CrossRef]
- Tang, M.; Chen, W.; Liu, J.; Weir, M.D.; Cheng, L.; Xu, H.H. Human induced pluripotent stem cell-derived mesenchymal stem cell seeding on calcium phosphate scaffold for bone regeneration. Tissue Eng. Part A 2014, 20, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Hynes, K.; Menicanin, D.; Mrozik, K.; Gronthos, S.; Bartold, P.M. Generation of functional mesenchymal stem cells from different induced pluripotent stem cell lines. Stem Cells Dev. 2013, 23, 1084–1096. [Google Scholar] [CrossRef]
- Sheyn, D.; Ben-David, S.; Shapiro, G.; De Mel, S.; Bez, M.; Ornelas, L.; Sahabian, A.; Sareen, D.; Da, X.; Pelled, G. Human induced pluripotent stem cells differentiate into functional mesenchymal stem cells and repair bone defects. Stem Cells Transl. Med. 2016, 5, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Steens, J.; Zuk, M.; Benchellal, M.; Bornemann, L.; Teichweyde, N.; Hess, J.; Unger, K.; Görgens, A.; Klump, H.; Klein, D. In vitro generation of vascular wall-resident multipotent stem cells of mesenchymal nature from murine induced pluripotent stem cells. Stem Cell Rep. 2017, 8, 919–932. [Google Scholar] [CrossRef]
- Ouchi, T.; Morikawa, S.; Shibata, S.; Fukuda, K.; Okuno, H.; Fujimura, T.; Kuroda, T.; Ohyama, M.; Akamatsu, W.; Nakagawa, T.; et al. Lngfr(+)thy-1(+) human pluripotent stem cell-derived neural crest-like cells have the potential to develop into mesenchymal stem cells. Differentiation 2016, 92, 270–280. [Google Scholar] [CrossRef]
- Bajpai, R.; Chen, D.A.; Rada-Iglesias, A.; Zhang, J.; Xiong, Y.; Helms, J.; Chang, C.-P.; Zhao, Y.; Swigut, T.; Wysocka, J. Chd7 cooperates with pbaf to control multipotent neural crest formation. Nature 2010, 463, 958. [Google Scholar] [CrossRef]
- Mabuchi, Y.; Morikawa, S.; Harada, S.; Niibe, K.; Suzuki, S.; Renault-Mihara, F.; Houlihan, D.D.; Akazawa, C.; Okano, H.; Matsuzaki, Y. Lngfr+ thy-1+ vcam-1hi+ cells reveal functionally distinct subpopulations in mesenchymal stem cells. Stem Cell Rep. 2013, 1, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Viejo, M.; Menendez-Menendez, Y.; Blanco-Gelaz, M.; Ferrero-Gutierrez, A.; Fernandez-Rodriguez, M.; Gala, J.; Otero-Hernandez, J. Quantifying mesenchymal stem cells in the mononuclear cell fraction of bone marrow samples obtained for cell therapy. In Transplantation Proceedings; Elsevier: Amsterdam, The Netherlands, 2013; pp. 434–439. [Google Scholar]
- Kimura, H.; Ouchi, T.; Shibata, S.; Amemiya, T.; Nagoshi, N.; Nakagawa, T.; Matsumoto, M.; Okano, H.; Nakamura, M.; Sato, K. Stem cells purified from human induced pluripotent stem cell-derived neural crest-like cells promote peripheral nerve regeneration. Sci. Rep. 2018, 8, 018–27952. [Google Scholar] [CrossRef]
- Giuliani, M.; Oudrhiri, N.; Noman, Z.M.; Vernochet, A.; Chouaib, S.; Azzarone, B.; Durrbach, A.; Bennaceur-Griscelli, A. Human mesenchymal stem cells derived from induced pluripotent stem cells downregulate nk cell cytolytic machinery. Blood 2011, 118, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Frobel, J.; Hemeda, H.; Lenz, M.; Abagnale, G.; Joussen, S.; Denecke, B.; Šarić, T.; Zenke, M.; Wagner, W. Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Rep. 2014, 3, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Shao, K.; Koch, C.; Gupta, M.K.; Lin, Q.; Lenz, M.; Laufs, S.; Denecke, B.; Schmidt, M.; Linke, M.; Hennies, H.C. Induced pluripotent mesenchymal stromal cell clones retain donor-derived differences in DNA methylation profiles. Mol. Ther. 2013, 21, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Gregory, C.A.; Lee, R.H.; Reger, R.L.; Qin, L.; Hai, B.; Park, M.S.; Yoon, N.; Clough, B.; McNeill, E. Mscs derived from ipscs with a modified protocol are tumor-tropic but have much less potential to promote tumors than bone marrow mscs. Proc. Natl. Acad. Sci. USA 2015, 112, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.Q.; Zhang, Y.; Li, X.; Deng, M.X.; Gao, W.X.; Yao, Y.; Chiu, S.M.; Liang, X.; Gao, F.; Chan, C.W. Insensitivity of human ips cells-derived mesenchymal stem cells to interferon-γ-induced hla expression potentiates repair efficiency of hind limb ischemia in immune humanized nod scid gamma mice. Stem Cells 2015, 33, 3452–3467. [Google Scholar] [CrossRef]
- Gao, W.-X.; Sun, Y.-Q.; Shi, J.; Li, C.-L.; Fang, S.-B.; Wang, D.; Deng, X.-Q.; Wen, W.; Fu, Q.-L. Effects of mesenchymal stem cells from human induced pluripotent stem cells on differentiation, maturation, and function of dendritic cells. Stem Cell Res. Ther. 2017, 8, 48. [Google Scholar] [CrossRef]
- Xue, Y.; Cai, X.; Wang, L.; Liao, B.; Zhang, H.; Shan, Y.; Chen, Q.; Zhou, T.; Li, X.; Hou, J. Generating a non-integrating human induced pluripotent stem cell bank from urine-derived cells. PLoS ONE 2013, 8, e70573. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Huang, W.; Su, H.; Xue, Y.; Su, Z.; Liao, B.; Wang, H.; Bao, X.; Qin, D. Generation of integration-free neural progenitor cells from cells in human urine. Nat. Methods 2013, 10, 84. [Google Scholar] [CrossRef]
- Wang, D.; Sun, Y.-Q.; Gao, W.-X.; Fan, X.-L.; Shi, J.-B.; Fu, Q.-L. An in vitro and in vivo study of the effect of dexamethasone on immunoinhibitory function of induced pluripotent stem cell-derived mesenchymal stem cells. Cell Transplant. 2018, 27, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gan, Y.; Li, W.; Su, J.; Zhang, Y.; Huang, Y.; Roberts, A.; Han, Y.; Li, J.; Wang, Y. The interaction between mesenchymal stem cells and steroids during inflammation. Cell Death Dis. 2014, 5, e1009. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, S.; Tuan, R.S. Functional comparison of human-induced pluripotent stem cell-derived mesenchymal cells and bone marrow-derived mesenchymal stromal cells from the same donor. Stem Cells Dev. 2014, 23, 1594–1610. [Google Scholar] [CrossRef] [PubMed]
- Teramura, T.; Onodera, Y.; Mihara, T.; Hosoi, Y.; Hamanishi, C.; Fukuda, K. Induction of mesenchymal progenitor cells with chondrogenic property from mouse-induced pluripotent stem cells. Cell. Reprogramming 2010, 12, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Marolt, D.; Campos, I.M.; Bhumiratana, S.; Koren, A.; Petridis, P.; Zhang, G.; Spitalnik, P.F.; Grayson, W.L.; Vunjak-Novakovic, G. Engineering bone tissue from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 8705–8709. [Google Scholar] [CrossRef]
- Lin, H.; Yang, G.; Tan, J.; Tuan, R.S. Influence of decellularized matrix derived from human mesenchymal stem cells on their proliferation, migration and multi-lineage differentiation potential. Biomaterials 2012, 33, 4480–4489. [Google Scholar] [CrossRef]
- Boyd, N.L.; Robbins, K.R.; Dhara, S.K.; West, F.D.; Stice, S.L. Human embryonic stem cell–derived mesoderm-like epithelium transitions to mesenchymal progenitor cells. Tissue Eng. Part A 2009, 15, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Himeno, T.; Kamiya, H.; Naruse, K.; Cheng, Z.; Ito, S.; Kondo, M.; Okawa, T.; Fujiya, A.; Kato, J.; Suzuki, H. Mesenchymal stem cell-like cells derived from mouse induced pluripotent stem cells ameliorate diabetic polyneuropathy in mice. BioMed Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.-P.; Liu, X.-C.; Ma, P.-F.; Gao, C.; Li, J.-L.; Lin, Y.-Y.; Shao, W.; Han, S.; Zhao, B.; Wang, L.-M. Ipsc-mscs combined with low-dose rapamycin induced islet allograft tolerance through suppressing th1 and enhancing regulatory t-cell differentiation. Stem Cells Dev. 2015, 24, 1793–1804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, X.; Liao, S.; Wang, W.; Wang, J.; Li, X.; Ding, Y.; Liang, Y.; Gao, F.; Yang, M.; et al. Potent paracrine effects of human induced pluripotent stem cell-derived mesenchymal stem cells attenuate doxorubicin-induced cardiomyopathy. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Sze, S.K.; de Kleijn, D.P.; Lai, R.C.; Tan, E.K.W.; Zhao, H.; Yeo, K.S.; Low, T.Y.; Lian, Q.; Lee, C.N.; Mitchell, W. Elucidating the secretion proteome of human embryonic stem cell-derived mesenchymal stem cells. Mol. Cell. Proteom. 2007, 6, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Chow, Y.; Sun, S.; Zeng, Q.; Li, H.; Shi, J.; Sun, Y.; Wen, W.; Tse, H.; Lian, Q. Mesenchymal stem cells derived from human induced pluripotent stem cells modulate t-cell phenotypes in allergic rhinitis. Allergy 2012, 67, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Hynes, K.; Bright, R.; Marino, V.; Ng, J.; Verma, P.; Gronthos, S.; Bartold, P. Potential of ipsc-derived mesenchymal stromal cells for treating periodontal disease. Stem Cells Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Hynes, K.; Menicanin, D.; Gronthos, S.; Bartold, M.P. Differentiation of ipsc to mesenchymal stem-like cells and their characterization. In Induced Pluripotent Stem (ips) Cells; Springer: Berlin/Heidelberg, Germany, 2014; pp. 353–374. [Google Scholar]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.; Tse, H.F.; Mak, J.C.; Lian, Q. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am. J. Respir. Cell. Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Ikeya, M. Generation and applications of induced pluripotent stem cell-derived mesenchymal stem cells. Stem Cells Int. 2018, 2018, 9601623. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, Y.; Zhao, B.; Niu, X.; Hu, B.; Li, Q.; Zhang, J.; Ding, J.; Chen, Y.; Wang, Y. Comparison of exosomes secreted by induced pluripotent stem cell-derived mesenchymal stem cells and synovial membrane-derived mesenchymal stem cells for the treatment of osteoarthritis. Stem Cell Res. Ther. 2017, 8, 64. [Google Scholar] [CrossRef]
- Hu, G.-w.; Li, Q.; Niu, X.; Hu, B.; Liu, J.; Zhou, S.-m.; Guo, S.-c.; Lang, H.-l.; Zhang, C.-q.; Wang, Y.; et al. Exosomes secreted by human-induced pluripotent stem cell-derived mesenchymal stem cells attenuate limb ischemia by promoting angiogenesis in mice. Stem Cell Res. Ther. 2015, 6, 10. [Google Scholar] [CrossRef]
- Zhang, J.; Guan, J.; Niu, X.; Hu, G.; Guo, S.; Li, Q.; Xie, Z.; Zhang, C.; Wang, Y. Exosomes released from human induced pluripotent stem cells-derived mscs facilitate cutaneous wound healing by promoting collagen synthesis and angiogenesis. J. Transl. Med. 2015, 13, 49. [Google Scholar] [CrossRef]
- Le Blanc, K.; Tammik, C.; Rosendahl, K.; Zetterberg, E.; Ringdén, O. Hla expression and immunologic propertiesof differentiated and undifferentiated mesenchymal stem cells. Exp. Hematol. 2003, 31, 890–896. [Google Scholar] [CrossRef]
- El Haddad, N.; Heathcote, D.; Moore, R.; Yang, S.; Azzi, J.; Mfarrej, B.; Atkinson, M.; Sayegh, M.H.; Lee, J.-S.; Ashton-Rickardt, P.G.; et al. Mesenchymal stem cells express serine protease inhibitor to evade the host immune response. Blood 2011, 117, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Kim, J.; Cho, J.H.; Chung, H.M.; Chae, J.I. Comparative analysis of human mesenchymal stem cells derived from bone marrow, placenta, and adipose tissue as sources of cell therapy. J. Cell. Biochem. 2016, 117, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Steens, J.; Klein, D. Current strategies to generate human mesenchymal stem cells in vitro. Stem Cells Int. 2018, 2018. [Google Scholar] [CrossRef]
- Ribeiro, A.; Laranjeira, P.; Mendes, S.; Velada, I.; Leite, C.; Andrade, P.; Santos, F.; Henriques, A.; Grãos, M.; Cardoso, C.M. Mesenchymal stem cells from umbilical cord matrix, adipose tissue and bone marrow exhibit different capability to suppress peripheral blood b, natural killer and t cells. Stem Cell Res. Ther. 2013, 4, 125. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.J.; Gopalakrishnan, D.; Shankar, S.R.; Vasandan, A.B. Pro-inflammatory cytokines, ifnγ and tnfα, influence immune properties of human bone marrow and wharton jelly mesenchymal stem cells differentially. PLoS ONE 2010, 5, e9016. [Google Scholar] [CrossRef]
- Montesinos, J.; Flores-Figueroa, E.; Castillo-Medina, S.; Flores-Guzman, P.; Hernandez-Estevez, E.; Fajardo-Orduna, G.; Orozco, S.; Mayani, H. Human mesenchymal stromal cells from adult and neonatal sources: Comparative analysis of their morphology, immunophenotype, differentiation patterns and neural protein expression. Cytotherapy 2009, 11, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Wegmeyer, H.; Bröske, A.-M.; Leddin, M.; Kuentzer, K.; Nisslbeck, A.K.; Hupfeld, J.; Wiechmann, K.; Kuhlen, J.; von Schwerin, C.; Stein, C. Mesenchymal stromal cell characteristics vary depending on their origin. Stem Cells Dev. 2013, 22, 2606–2618. [Google Scholar] [CrossRef]
- Gjorgieva, D.; Zaidman, N.; Bosnakovski, D. Mesenchymal stem cells for anti-cancer drug delivery. Recent Pat. Anti-Cancer Drug Discov. 2013, 8, 310–318. [Google Scholar] [CrossRef]
- Kidd, S.; Spaeth, E.; Dembinski, J.L.; Dietrich, M.; Watson, K.; Klopp, A.; Battula, V.L.; Weil, M.; Andreeff, M.; Marini, F.C. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009, 27, 2614–2623. [Google Scholar] [CrossRef]
- Vogel, G. Ready or not? Human es cells head toward the clinic. Science 2005. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zhou, Y.; Tan, S.; Zhou, G.; Aagaard, L.; Xie, L.; Bünger, C.; Bolund, L.; Luo, Y. Mesenchymal stem cells derived from human induced pluripotent stem cells retain adequate osteogenicity and chondrogenicity but less adipogenicity. Stem Cell Res. Ther. 2015, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Moslem, M.; Eberle, I.; Weber, I.; Henschler, R.; Cantz, T. Mesenchymal stem/stromal cells derived from induced pluripotent stem cells support cd34pos hematopoietic stem cell propagation and suppress inflammatory reaction. Stem Cells Int. 2015. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
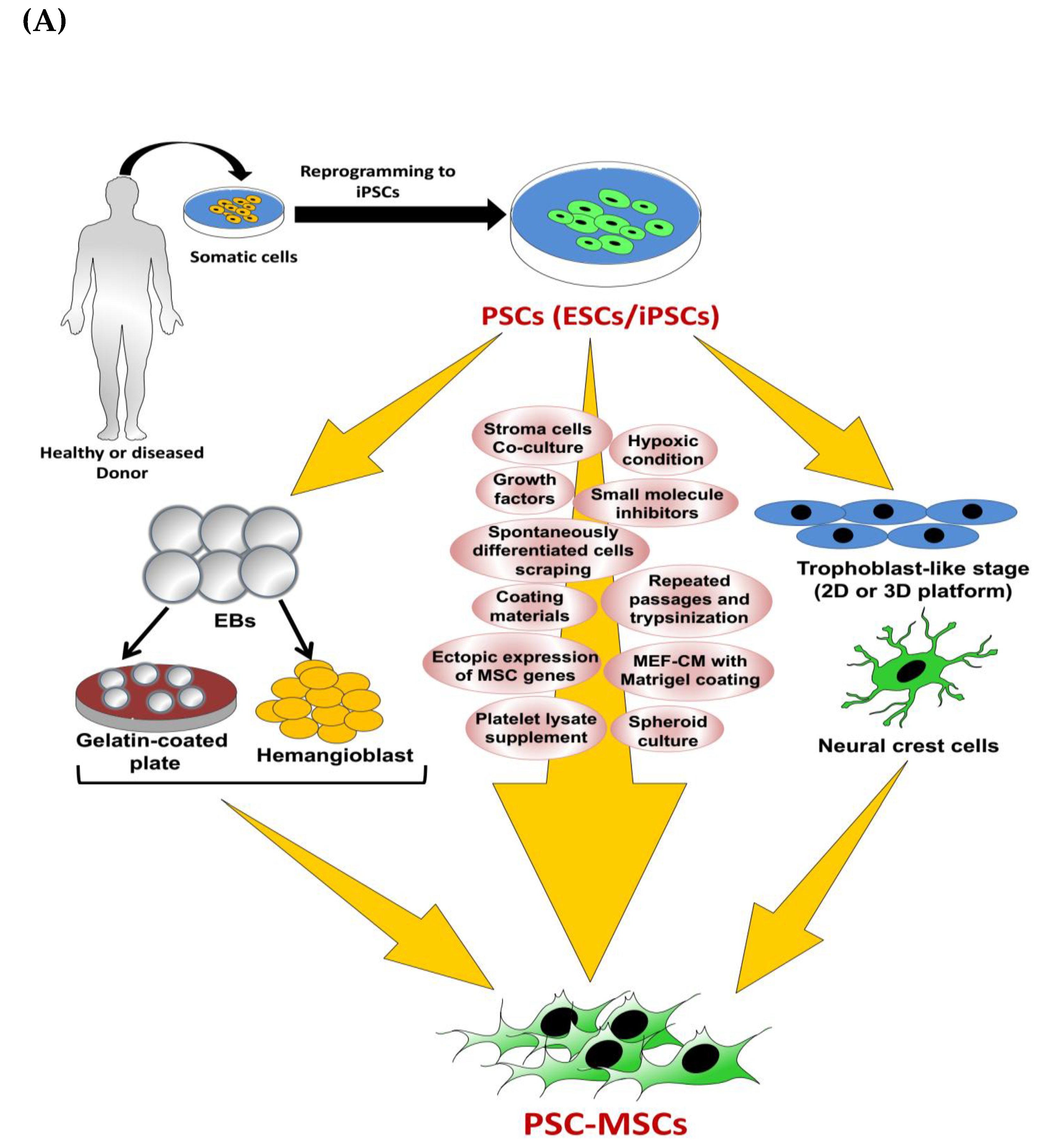{kind=link}
{kind=link}
{kind=link}
| PSC Lines | Derivation Method | Characteristic Features | Therapeutic Efficacy |
|---|---|---|---|
| ESC-MSC | Co-culture of hESCs with murine OP9 stromal cells (Barberi et al., 2005 [89]) | -Spindle-like morphology. -Positively expressed CD105, STRO-1, CD106, CD29, CD44, CD54, CD166, vimentin, and alpha smooth muscle actin and negatively expressed CD34, CD45, and CD14. -Late detection of CD73+ cells (at day 40). -Tri-lineage differentiation. | -n.d. * |
| Co-culture method of Barberi et al., but with irradiated murine OP9 stromal cells (Trivedi et al., 2007 [91]). | -Positive for CD73, CD29, CD44, CD54, CD90 and, CD105. -Negative for CD34 and CD45. -Early detection of CD73+ cells that followed by the emergence of CD34+ cells (within first 2 weeks). - Tri-lineage differentiation. | -n.d. * | |
| Culture on Matrigel plate with MEF-CM+bFGF (Trivedi et al., 2008 [92]) | -Positive expression for CD29, CD44, CD54, CD73, 90, and CD105. -Negative expression for CD34, CD45, and CD31 -High expression of HLA class I, but no expression of HLA class II. - Tri-lineage differentiation. | -Inhibited the proliferation of responder T-lymphocytes [92]. -Co-transplantation of hESC-MSCs (conditionally overexpressed with VEGF) with islet resulted in resulted in 50% reduction of the required islet mass in diabetic mice through promoting islet revascularization [119]. | |
| EBs’ formation with gelatin coating and mechanical scraping (Hwang et al., 2008 [93]) | -Fibroblast-like morphology. -Positive expression for CD13, CD14, CD29, CD44, CD73, CD90, CD105, CD146, CD166, STRO-1, and PDGFR-α. -Negative expression for CD34, CD45, CD117, and CD133. -Tri-lineage differentiation. | -New cartilage formation (rich in ECM) upon transplantation into the athymic mice for 12 weeks. -Cartilage defects complete recovery after the transplantation in the articular cartilage defects in the femoral condyle of athymic nude rats. | |
| EBs formation with gelatin coating + bFGF (Brown et al., 2009 [94]) | -Similar characteristics to hBM-MSCs with a higher proliferative capacity. -Positive for CD73 and STRO-1, and lacked the expression of CD45. -Osteogenic and adipogenic differentiations. | -In vitro generation of osteoprogenitor cells after the transduction with bone-associated lentiviral Col2.3-GFP. | |
| Repeated passage with trypsinization with MSC culture medium (Yen et al., 2011 [95]) | -Similar characteristics to hBM-MSCs. -Positively expressed CD29, CD44, CD73, CD90, and CD105, whereas negatively expressed CD14, CD34, and CD45. -Weak expression of SSEA-4 as well as CD9 and no expression of TRA-1-60 or TRA-1-81. -No teratoma formation when injected in immune-compromised mice. -Tri-lineage differentiation. | -Highly expressed geneses associated with transcriptional and proliferative processes (Transcriptome profiling analysis) | |
| Hemangioblast: Bi-potential progenitors derived from EBs (using cytokine-rich media) (Kimbrel et al., 2014 [26]) | -Similar characteristics to BM-MSCs with a higher proliferative capacity and smaller size. -Higher expression of CD10 and CD24 than that of adult BM-MSCs. -Tri-lineage differentiation. | -Suppression of dendritic cell-associated high production of IL-12p70 and the high level of CD83 [26]. -Therapeutic activity against autoimmune disorder mouse models, such as EAE and lupus nephritis and uveitis [26]. -Showed a superior activity than that of BM-MSCs in against EAE mouse model and the neuronal demyelination that attributed to the low expression of IL-6 [98]. | |
| Defined culture condition-based method with PDGF-AB, and bFGF (Lian et al., 2007 [99]) | -Similar characteristics to BM-MSCs. -Reduced expression of pluripotency-related genes (HESX1, POUFL5, SOX-2, UTF-1, and ZFP42) and decreased protein level of OCT4 and SOX2. -Positive expression for CD29, CD44, CD49a, CD105, and CD166. -Negative expression for CD34 and CD45. -No teratoma formation 4 months post-transplantation. -Tri-lineage differentiation. | -Efficient therapeutic activity against MCT-induced PAH mouse model [115]. | |
| Defined culture condition-based method (using xeno-free hESCs and culture conditions) (Karlsson et al., 2009 [100]) | -Fibroblast-like morphology. -Loss of expression of Oct-4, Nanog, TRA 1-60, TRA 1-81, SSEA-3, and SSEA-4. -Loss of expression of the endoderm- and neuroectoderm-related markers. -Positive for CD105, CD166, CD10, and CD13, whereas negative for CD133 and CD117. -Tri-lineage differentiation. | -Transplantation into SCID mice resulted in formation of well-defined tissues of MSC origin without teratoma formation. | |
| Trophoblast-like stage (With BMP4 and activin-like receptor kinases inhibitor) (Wang et al., 2016 [106]) | -Downregulation of trophoblast-related genes (from day 11 to day 16). -Positive for CD73, CD90, CD105, CD29, and CD44. -Negative for Trop2, CD31, and CD34. -Tri-lineage differentiation. | -In vitro and in vivo immunomodulatory activity. | |
| 3D platform (formation of trophoblast-like stage in spheroid) (Yan et al., 2018 [32]) | On day 10: Positive expression for trophoblast- and MSC-related markers. -On day 20: No detection of trophoblast-associated markers and upregulation of MSC-related markers. -Positive for CD90, CD105, and CD44. -Negative for apoptotic markers. -Tri-lineage differentiation. | -Potentially adhered and differentiated into bone and cartilage in DBM scaffold [32]. -Potent in vitro immunomodulatory activity [32]. -Robust therapeutic activity in mouse model of inflammatory colitis [32]. -Recovery of multiple sclerosis using EAE model in cynomolgus monkeys through the i.t. injection [120]. | |
| Small molecule inhibitors (TGF-β/activin/nodal signaling pathway inhibitor, SB-431542 (Mahmood et al., 2010 [112]) | -Positive for CD44, CD73, CD146, and CD166. -Downregulation of myogenesis-related genes. | -In vitro and in vivo tri-lineage differentiation capacities. | |
| Small molecule inhibitors (SMAD-2/3 signaling pathway inhibitor) (Sanchez et al., 2011 [113]) | -Positive expression for CD73 and CD90, whereas negative for CD34 expression. -Low expression of the pluripotency markers (Oct4 and Tra-1-60). -Tri-lineage differentiation. -Higher proliferation capacity than BM-MSCs. | -Potent in vivo anti-inflammatory and immunosuppressive activities in a mouse model of experimental colitis. -Alleviation of collagen-mediated arthritis in mice through the upregulation of the expression of IDO1 [118]. | |
| Small molecule inhibitors (IKK/NF-κB signaling inhibitor) (Deng et al., 2016 [114]) | -Loss pluripotency markers expression level. -Decrease in ALP activity. -Positive expression for CD51 and CD90, whereas negative for CD34 and CD45. | -In vitro and in vivo bone formation. | |
| Small molecule inhibitors (TGF-β pathway inhibitor, SB431542) (Chen et al., 2012 [117]) | -MSC-like morphology. -Positive for the expression of CD105, CD73, and CD90, whereas lacked the expression of CD45 and CD14. -Marked reduction in the expression level of the pluripotency-associated markers. | -Potent neuroprotective effect in a hypoxic-ischemic mouse brain model and better than that shown by fetal MSCs [116]. | |
| iPSC-MSC | Defined culture conditions with growth factors bFGF, EGF, and PDGF-AB (Lian et al., 2010 [122]) | -Similar to BM-MSCs with higher proliferation capacity. -Positive for CD44, CD49a, CD73, CD105, and CD166. -Negative for CD34, CD45, and CD133. -Tri-lineage differentiation. | -Therapeutic activity against severe hind-limb ischemia mouse model [122]. -Increased hEAG1 potassium channel encoded by KCNH1 [124]. -Robust immunomodulatory function through marked reduction of phytohaemagglutinin-induced lymphocyte proliferation as well as decreased the proliferation of CD3-positive T cells [164]. -Alleviation of cigarette smoke-related pulmonary damage in rat model via the mitochondrial transfer mechanism [167]. -In vitro and in vivo attenuation of DOX-mediated cardiomyopathy via reduction of ROS generation [162]. -Suppression the early stage differentiation of CD14+ monocytes to DCs and blocked DC-mediated T cell activation [150]. -Combination with the low concentration of rapamycin markedly increased the survival rate of the islet allograft in the diabetic mice [161]. |
| Repeated passage with trypsinization with MSC culture medium (Zou et al., 2013 [125]) | -Fibroblast-like morphology. -Positive expression for CD90, CD73, and CD105. -Loss of the expression of pluripotency markers (OCT3/4 and TRA-1-81), whereas still positive for Nanog -Tri-lineage differentiation. | -In vitro osteogenic differentiation. | |
| Hypoxic condition with growth factor (Yang et al., 2014 [128]) | -Similar to rat BM-MSCs. -Positively expressed CD29 and CD90, whereas negatively expressed CD34 and CD45. | -In vivo anti-inflammatory activity using a rat model of experimental periodontitis. | |
| Using PL supplement (Luzzani et al., 2015 [129]) | -Share characteristics with the UC-MSC. -Positive for CD90, CD73, CD105, CD166, and CD271. -Tri-lineage differentiation. | -In vitro immunomodulating activity through the suppression of concanavalin-A-induced lymphocyte proliferation. | |
| Biomimetic, fibrillar, type I collagen coatings (Liu et al., 2012 [131] | -Positive expression for CD90, CD105, CD166, CD73, and CD146, whereas negative for CD34 and CD45. -Tri-lineage differentiation. -Spindle-shaped morphology. | -n.d. * | |
| PMEDSAH coating (Villa Diaz et al., 2012 [132]) | -Positively expressed CD44, CD73, CD105, and CD166, whereas lacked the expression of CD34 and CD45. -Tri-lineage differentiation. | -In vivo bone regenerative capacity in calvaria defects mouse model. | |
| EB formation with gelatin coating (Tang et al., 2014 [136]) | -Positive expression of MSC markers. -Lacked the expression of the hematopoietic markers and the pluripotency markers. -Tri-lineage differentiation. | -Efficient in vitro osteogenic differentiation in CPC scaffold. | |
| Small molecule inhibitors (TGF-β pathway inhibitor, SB431542) (Chen et al., 2012 [117]) | -MSC-like morphology. -Decrease in the pluripotency markers. -Tri-lineage differentiation. -No in vivo teratoma formation. -High expression of vimentin and N-cadherin. | -Potent neuroprotective effect in a hypoxic-ischemic mouse brain model and better than that shown by fetal MSCs [116]. | |
| Gelatin coating (Hyunes et al., 2013 [137]) | -Fibroblastic-like morphology. -Positive expression for CD73, CD90, CD105, CD146, and CD166. -Lacked the expression of the pluripotency markers. -Negative expression for CD14, CD34, and CD45. -Tri-lineage differentiation. | -In vitro immunomodulating activity through the suppression of concanavalin-A-induced mouse splenocyte proliferation [165]. -Anti-inflammatory activity in mouse model implanted with P. gingivalis containing sponge [165]. | |
| EB formation with poly-hydroxyethyl methacrylate and gelatin coatings (Sheyn et al., 2016 [138]) | -Similar characteristics to BM-MSCs. -Tri-lineage differentiation. | -In vivo bone formation. | |
| Using NCCs (Ouchi et al., 2016 [140]) | -Spindle-like morphology. - High expression of LNGFR and THY-1. -Differentiation into neural crest-related cells. | -High proliferative capacity upon transplantation into chicken embryo and can migrate to the sclerotome region [140]. -Recovery of the peripheral nerve injury in NOD/SCID mice with sciatic nerve injury [144]. | |
| MSC culture medium supplemented with bFGF (Giuliani et al., 2011 [145]) | -Spindle-shaped morphology. -High expression of CD90, CD105, CD146, CD54, and CD73. -Lacked the expression of CD45), HLA class II (HLA-DR), and costimulatory molecules. -No expression of the pluripotency factors | -Potent superior immunomodulatory activity than of BM-MSCs and after various passages. -Decreased NK proliferation and its cytolytic property. | |
| Redifferentiation of iPSC reprogrammed from the reprogramming of BM-MSCs (Frobel et al., 2014 [146]) | -MSC-like morphology. -Tri-lineage differentiation. -Positive expression for CD29, CD73, CD90, and CD105 (less expression), whereas negative expression for CD14, CD31, CD34, and CD45. | -Immunomodulatory function, but lower than the original MSCs. | |
| Small molecule inhibitors (SMAD-2/3 inhibitor, SB-431542)(Zhao et al., 2015 [148]) | -Spindle-like morphology. -Positive expression for CD73, CD105, CD166, CD44 and CD90), whereas negative for HLA-DR, CD11b, CD24, CD34, and CD45. -High expression of mesodermal markers CD140A/PDGFRα -Significant decrease in the expression of the pluripotency factors and the neuroectodermal factors. | -Potent in vivo tumor homing activity similar to that of BM-MSCs, whereas with lower pro-tumor activity than BM-MSCs and thus avoiding tumor progression. | |
| Commercially purchased iPSC-MSCs derived from fetal and adult BM (Sun et al., 2015 [149]) | -Positive expression for CD44, CD105, CD90, and CD73, whereas lacked the expression of CD45, CD14, CD34, CD3, and CD56. -High expression level of of HLA class I. | -Superior effect in the attenuation of the in vivo inflammation in induced hind limb ischemia mouse model than that of BM-MSCs. | |
| EB formation method (With MSCs differentiation medium + all trans retinoic acid and the continuous passage for 4 months (Himeno et al., 2013 [160]) | -Positively expressed CD105, CD140a, Sca-1, and CD44. -Negatively expressed CD34, TER119, CD31, CD45, and CD11b. -Tri-lineage differentiation. | -In vivo attenuation of diabetes-related polyneuropathy in streptozotocin-diabetic mice. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdal Dayem, A.; Lee, S.B.; Kim, K.; Lim, K.M.; Jeon, T.-i.; Seok, J.; Cho, S.-G. Production of Mesenchymal Stem Cells through Stem Cell Reprogramming. Int. J. Mol. Sci. 2019, 20, 1922. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081922
Abdal Dayem A, Lee SB, Kim K, Lim KM, Jeon T-i, Seok J, Cho S-G. Production of Mesenchymal Stem Cells through Stem Cell Reprogramming. International Journal of Molecular Sciences. 2019; 20(8):1922. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081922
Chicago/Turabian StyleAbdal Dayem, Ahmed, Soo Bin Lee, Kyeongseok Kim, Kyung Min Lim, Tak-il Jeon, Jaekwon Seok, and Ssang-Goo Cho. 2019. "Production of Mesenchymal Stem Cells through Stem Cell Reprogramming" International Journal of Molecular Sciences 20, no. 8: 1922. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081922