Structure-Based Approach for the Prediction of Mu-opioid Binding Affinity of Unclassified Designer Fentanyl-Like Molecules



Abstract
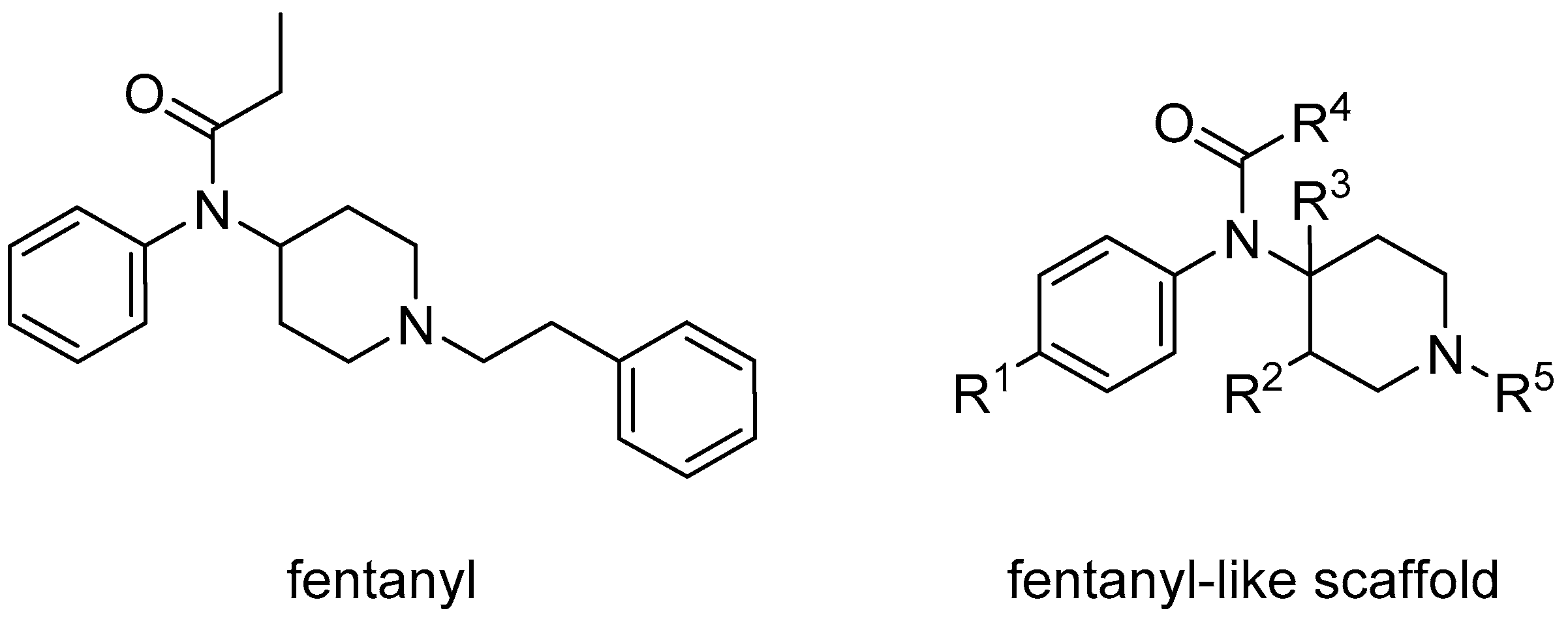
:1. Introduction
2. Results and Discussion
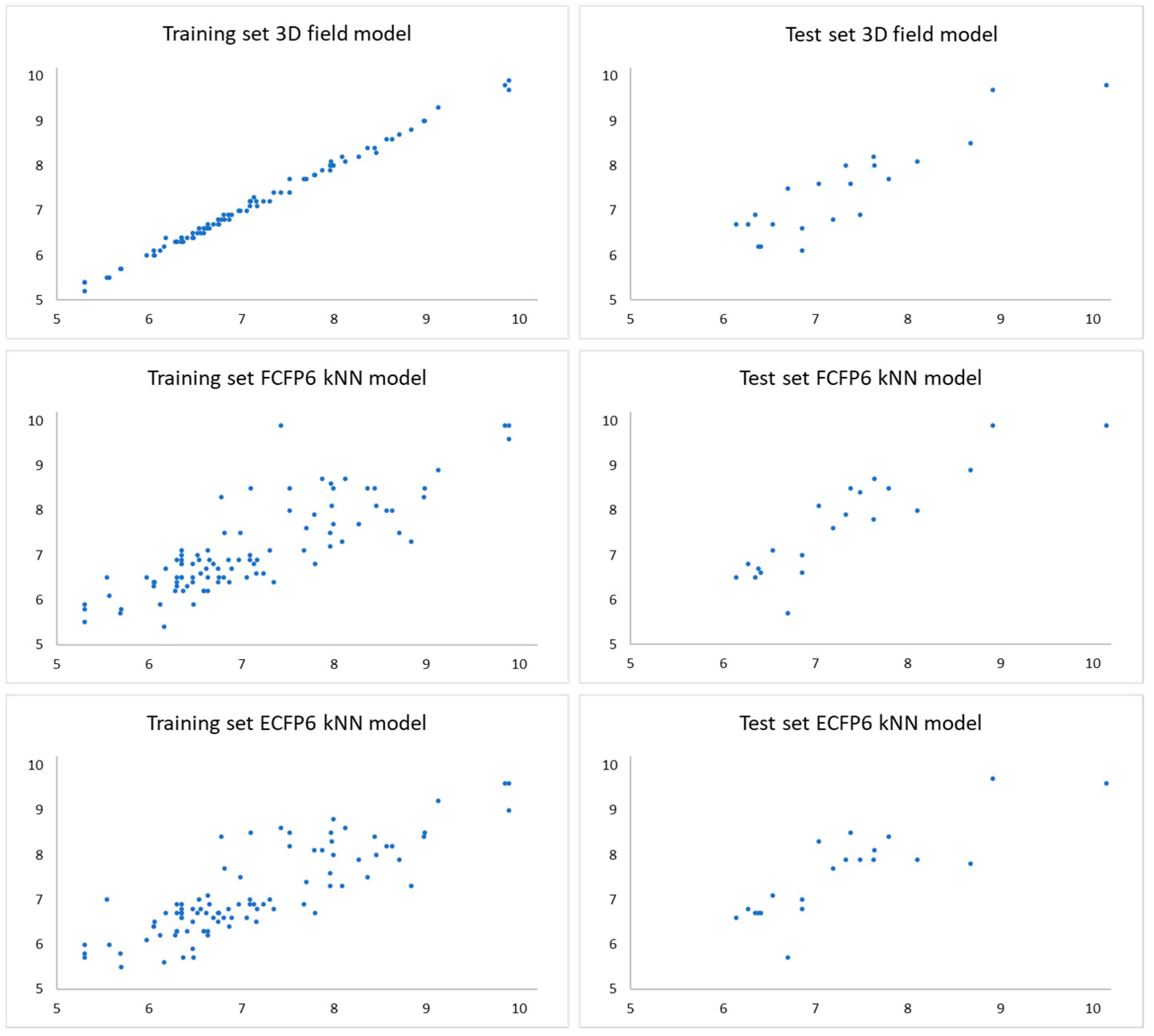
2.1. Statistical Analysis and Results
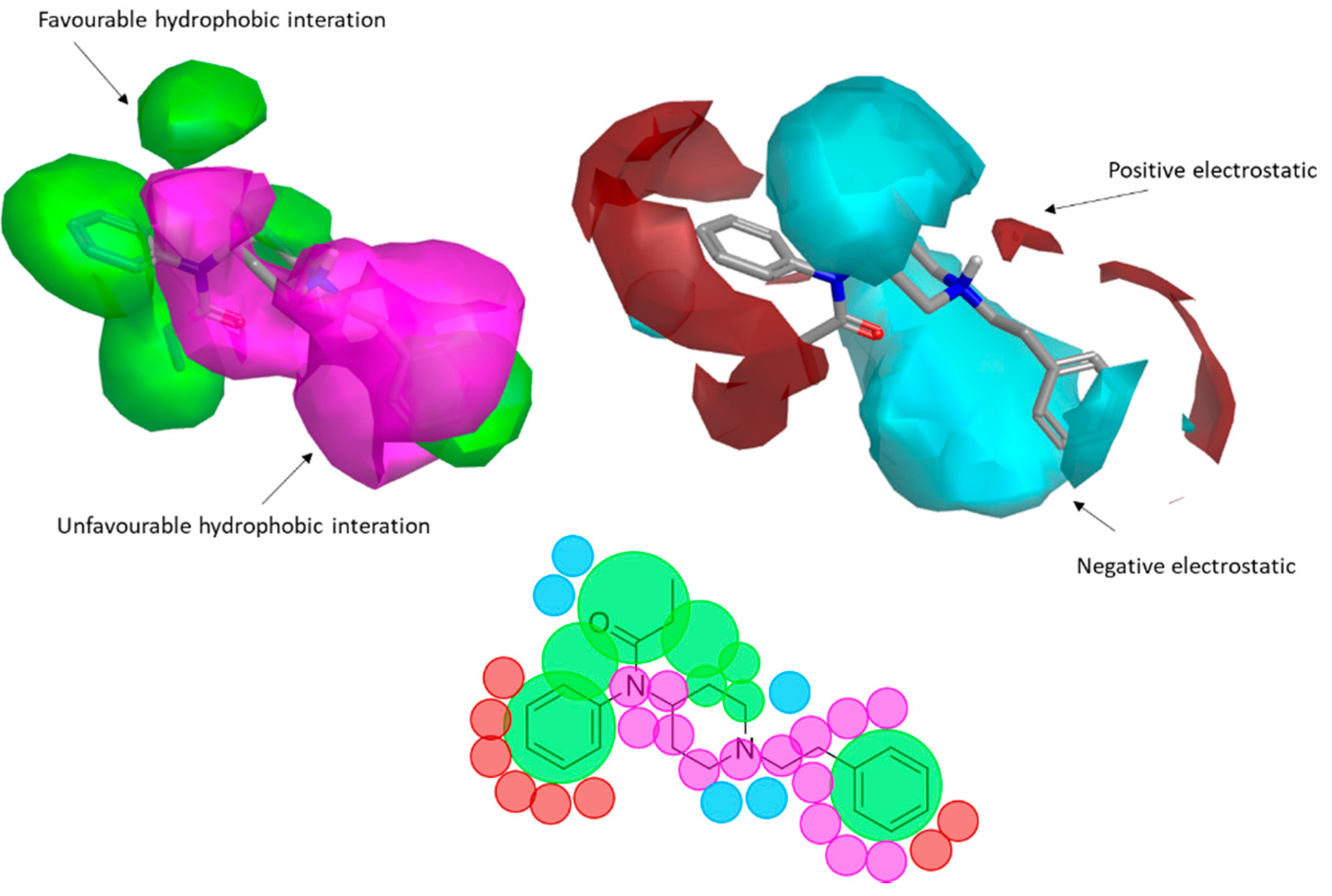
2.2. Activity Cliffs in the Activity Landscape of the QSAR Set
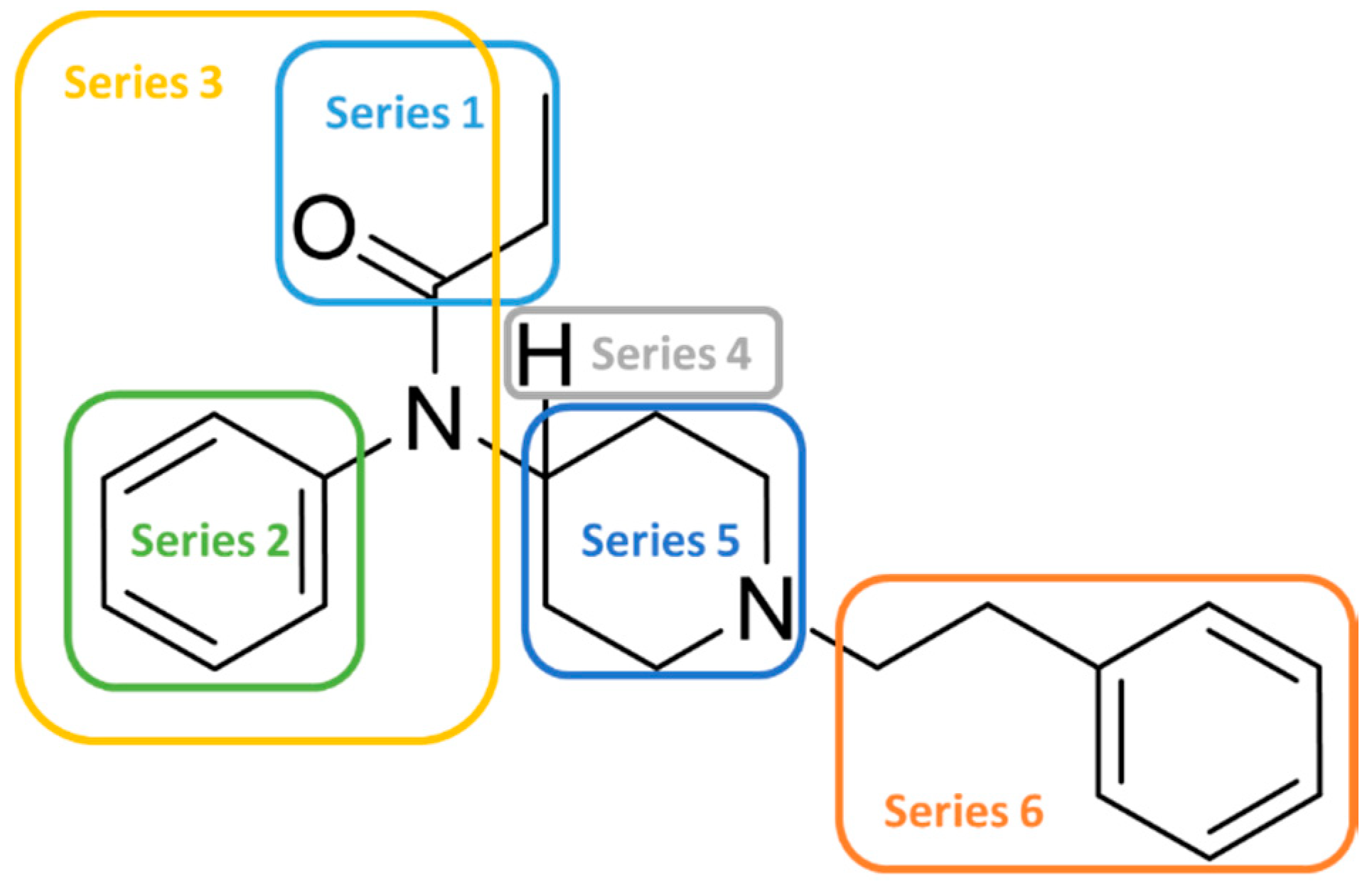
2.3. Enlarging the Activity Landscape of Fentanyl-like Compounds
2.4. Designer Drugs from the Scaffold-Hopping Results
3. Materials and Methods
3.1. Biological Data
3.2. Molecular Modeling
3.3. Compound Alignment for the 3D Model and kNN Models Information
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, S. Historical Review: Opiate Addiction and Opioid Receptors. Cell Transpl. 2018. [Google Scholar] [CrossRef]
- Gracies, J.M.; O’Dell, M.; Vecchio, M.; Hedera, P.; Kocer, S.; Rudzinska-Bar, M.; Rubin, B.; Timerbaeva, S.L.; Lusakowska, A.; Boyer, F.C.; et al. Effects of Repeated Abobotulinumtoxina Injections in Upper Limb Spasticity. Muscle Nerve 2018, 57, 245–254. [Google Scholar] [CrossRef]
- Vecchio, M.; Gracies, J.M.; Panza, F.; Fortunato, F.; Vitaliti, G.; Malaguarnera, G.; Cinone, N.; Beatrice, R.; Ranieri, M.; Santamato, A. Change in Coefficient of Fatigability Following Rapid, Repetitive Movement Training in Post-Stroke Spastic Paresis: A Prospective Open-Label Observational Study. J. Stroke Cerebrovasc. Dis. 2017, 26, 2536–2540. [Google Scholar] [CrossRef]
- Vecchio, M.; Malaguarnera, G.; Giordano, M.; Malaguarnera, M.; Volti, G.L.; Galvano, F.; Drago, F.; Basile, F.; Malaguarnera, M. A musician’s dystonia. Lancet 2012, 379, 2116. [Google Scholar] [CrossRef]
- Lake, S.; Kennedy, M.C. Health outcomes associated with illicit prescription opioid injection: A systematic review. J. Addict. Dis. 2016, 35, 73–91. [Google Scholar] [CrossRef]
- Frank, R.G.; Pollack, H.A. Addressing the Fentanyl Threat to Public Health. N. Engl. J. Med. 2017, 376, 605–607. [Google Scholar] [CrossRef]
- Rudd, R.A.; Seth, P.; David, F.; Scholl, L. Increases in Drug and Opioid-Involved Overdose Deaths—United States, 2010–2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1445–1452. [Google Scholar] [CrossRef]
- Rudd, R.A.; Aleshire, N.; Zibbell, J.E.; Gladden, R.M. Increases in Drug and Opioid Overdose Deaths--United States, 2000–2014. MMWR Morb. Mortal. Wkly. Rep. 2016, 64, 1378–1382. [Google Scholar] [CrossRef]
- Fentanyl Remains the Most Significant Synthetic Opioid Threat and Poses the Greatest Threat to the Opioid User Market in the United States. Report Number: DEA-DCT-DIB-003-18. Available online: https://ndews.umd.edu/sites/ndews.umd.edu/files/fentanyl-remains-most-significant-synthetic-opioid-threat-2018.pdf (accessed on 22 January 2019).
- Fomin, D.; Baranauskaite, V.; Usaviciene, E.; Sumkovskaja, A.; Laima, S.; Jasulaitis, A.; Minkuviene, Z.N.; Chmieliauskas, S.; Stasiuniene, J. Human deaths from drug overdoses with carfentanyl involvement-new rising problem in forensic medicine: A STROBE-compliant retrospective study. Medicine (Baltimore) 2018, 97, e13449. [Google Scholar] [CrossRef]
- Ellis, C.R.; Kruhlak, N.L.; Kim, M.T.; Hawkins, E.G.; Stavitskaya, L. Predicting opioid receptor binding affinity of pharmacologically unclassified designer substances using molecular docking. PLoS ONE 2018, 13, e0197734. [Google Scholar] [CrossRef]
- Floresta, G.; Amata, E.; Dichiara, M.; Marrazzo, A.; Salerno, L.; Romeo, G.; Prezzavento, O.; Pittala, V.; Rescifina, A. Identification of Potentially Potent Heme Oxygenase 1 Inhibitors through 3D-QSAR Coupled to Scaffold-Hopping Analysis. ChemMedChem 2018. [Google Scholar] [CrossRef]
- Floresta, G.; Rescifina, A.; Marrazzo, A.; Dichiara, M.; Pistara, V.; Pittala, V.; Prezzavento, O.; Amata, E. Hyphenated 3D-QSAR statistical model-scaffold hopping analysis for the identification of potentially potent and selective sigma-2 receptor ligands. Eur. J. Med. Chem. 2017, 139, 884–891. [Google Scholar] [CrossRef]
- Rescifina, A.; Floresta, G.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Nastasi, G.; Dichiara, M.; Amata, E. Sigma-2 receptor ligands QSAR model dataset. Data Brief 2017, 13, 514–535. [Google Scholar] [CrossRef]
- Rescifina, A.; Floresta, G.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Nastasi, G.; Dichiara, M.; Amata, E. Development of a Sigma-2 Receptor affinity filter through a Monte Carlo based QSAR analysis. Eur. J. Pharm. Sci. 2017, 106, 94–101. [Google Scholar] [CrossRef]
- Floresta, G.; Cilibrizzi, A.; Abbate, V.; Spampinato, A.; Zagni, C.; Rescifina, A. FABP4 inhibitors 3D-QSAR model and isosteric replacement of BMS309403 datasets. Data Brief 2019, 22, 471–483. [Google Scholar] [CrossRef]
- Floresta, G.; Cilibrizzi, A.; Abbate, V.; Spampinato, A.; Zagni, C.; Rescifina, A. 3D-QSAR assisted identification of FABP4 inhibitors: An effective scaffold hopping analysis/QSAR evaluation. Bioorg. Chem. 2019, 84, 276–284. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- De Jong, S. SIMPLS: An alternative approach to partial least squares regression. Chemom. Intell. Lab. Syst. 1993, 18, 251–263. [Google Scholar] [CrossRef]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: a basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Kubinyi, H.; Hamprecht, F.A.; Mietzner, T. Three-dimensional quantitative similarity-activity relationships (3D QSiAR) from SEAL similarity matrices. J. Med. Chem. 1998, 41, 2553–2564. [Google Scholar] [CrossRef]
- Jiang, H.L.; Huang, X.Q.; Rong, S.B.; Luo, X.M.; Chen, J.Z.; Tang, Y.; Chen, K.X.; Zhu, Y.C.; Jin, W.Q.; Chi, Z.Q.; et al. Theoretical studies on opioid receptors and ligands. I. Molecular modeling and QSAR studies on the interaction mechanism of fentanyl analogs binding to μ-opioid receptor. Int. J. Quantum Chem. 2000, 78, 285–293. [Google Scholar] [CrossRef]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef]
- Olesen, P.H. The use of bioisosteric groups in lead optimization. Curr. Opin. Drug Discov. Dev. 2001, 4, 471–478. [Google Scholar]
- Floresta, G.; Pittala, V.; Sorrenti, V.; Romeo, G.; Salerno, L.; Rescifina, A. Development of new HO-1 inhibitors by a thorough scaffold-hopping analysis. Bioorg. Chem. 2018, 81, 334–339. [Google Scholar] [CrossRef]
- Floresta, G.; Apirakkan, O.; Rescifina, A.; Abbate, V. Discovery of High-Affinity Cannabinoid Receptors Ligands through a 3D-QSAR Ushered by Scaffold-Hopping Analysis. Molecules 2018, 23, 2183. [Google Scholar] [CrossRef]
- Clarke, D.L.; Drobatz, K.J.; Korzekwa, C.; Nelson, L.S.; Perrone, J. Trends in Opioid Prescribing and Dispensing by Veterinarians in Pennsylvania. JAMA Netw. Open 2019, 2, e186950. [Google Scholar] [CrossRef]
- Armenian, P.; Vo, K.T.; Barr-Walker, J.; Lynch, K.L. Fentanyl, fentanyl analogs and novel synthetic opioids: A comprehensive review. Neuropharmacology 2018, 134, 121–132. [Google Scholar] [CrossRef]
- Mounteney, J.; Giraudon, I.; Denissov, G.; Griffiths, P. Fentanyls: Are we missing the signs? Highly potent and on the rise in Europe. Int. J. Drug Policy 2015, 26, 626–631. [Google Scholar] [CrossRef]
- Lopez-Lopez, E.; Naveja, J.J.; Medina-Franco, J.L. DataWarrior: An evaluation of the open-source drug discovery tool. Expert Opin. Drug Discov. 2019, 1–7. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef]
- Alemán, C.; Luque, F.J.; Orozco, M. Suitability of the PM3-derived molecular electrostatic potentials. J. Comput. Chem. 1993, 14, 799–808. [Google Scholar] [CrossRef]
- Roy, P.P.; Leonard, J.T.; Roy, K. Exploring the impact of size of training sets for the development of predictive QSAR models. Chemom. Intell. Lab. Syst. 2008, 90, 31–42. [Google Scholar] [CrossRef]
- Choudhari, P.B.; Bhatia, M.S.; Jadhav, S.D. Pharmacophore Identification and QSAR Studies on Substituted Benzoxazinone as Antiplatelet Agents: kNN-MFA Approach. Sci. Pharm. 2012, 80, 283–294. [Google Scholar] [CrossRef]
- Gupta, S.P.; Samanta, S.; Patil, V.M. A 3D-QSAR study on a series of benzimidazole derivatives acting as hepatitis C virus inhibitors: Application of kNN-molecular field analysis. Med. Chem. 2010, 6, 87–90. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | r2 Training Set | q2 Training Set | r2 Test Set | MSE a Training Set | MSE a Test Set | MAE b Training Set | MAE b Test Set | MAPE c Training Set | MAPE c Test Set |
|---|---|---|---|---|---|---|---|---|---|
| 3D-field | 0.99 | 0.68 | 0.77 | 0.005 | 0.22 | 0.05 | 0.41 | 0.75 | 5.75 |
| FCFP6 kNN | 0.68 | 0.65 | 0.59 | 0.35 | 0.40 | 0.44 | 0.53 | 6.25 | 7.26 |
| ECFP6 kNN | 0.71 | 0.70 | 0.61 | 0.31 | 0.39 | 0.44 | 0.54 | 6.26 | 7.39 |
| Entry | Structure and pKi of the Most Active Analogs | Structure and pKi of the Less Active Analogs | Disparity | Δ Activity |
|---|---|---|---|---|
| 1 |  10.15 |  8.92 | −24.6 | −1.23 |
| 2 |  9.89 |  8.92 | −19.4 | −0.97 |
| 3 |  10.15 |  8.68 | −15.4 | −1.47 |
| 4 |  9.85 |  8.92 | −14.2 | −0.93 |
| 5 |  9.89 |  8.68 | −14.1 | −1.21 |
| 6 |  10.15 |  9.13 | −13.8 | −1.02 |
| 7 |  10.15 |  7.87 | −12.8 | −2.28 |
| Series 1 | Series 2 | Series 3 |
 9.0 |  9.0 |  8.9 |
 8.8 |  8.9 |  8.8 |
| Series 4 | Series 5 | Series 6 |
 10.8 |  9.8 |  9.5 |
 10.6 |  9.7 |  9.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Floresta, G.; Rescifina, A.; Abbate, V. Structure-Based Approach for the Prediction of Mu-opioid Binding Affinity of Unclassified Designer Fentanyl-Like Molecules. Int. J. Mol. Sci. 2019, 20, 2311. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092311
Floresta G, Rescifina A, Abbate V. Structure-Based Approach for the Prediction of Mu-opioid Binding Affinity of Unclassified Designer Fentanyl-Like Molecules. International Journal of Molecular Sciences. 2019; 20(9):2311. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092311
Chicago/Turabian StyleFloresta, Giuseppe, Antonio Rescifina, and Vincenzo Abbate. 2019. "Structure-Based Approach for the Prediction of Mu-opioid Binding Affinity of Unclassified Designer Fentanyl-Like Molecules" International Journal of Molecular Sciences 20, no. 9: 2311. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092311