Tuning the Interactions in Multiresponsive Complex Coacervate-Based Underwater Adhesives

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Material Analysis
2.1.1. Injectability
2.1.2. Liquid-to-Solid Transition
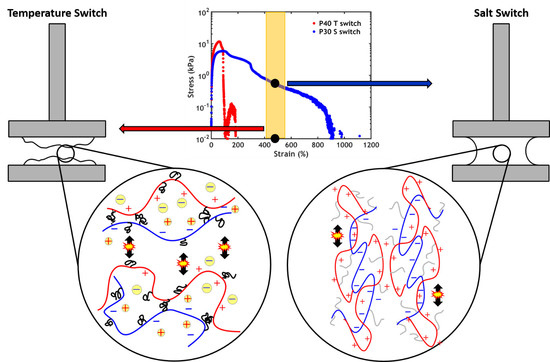
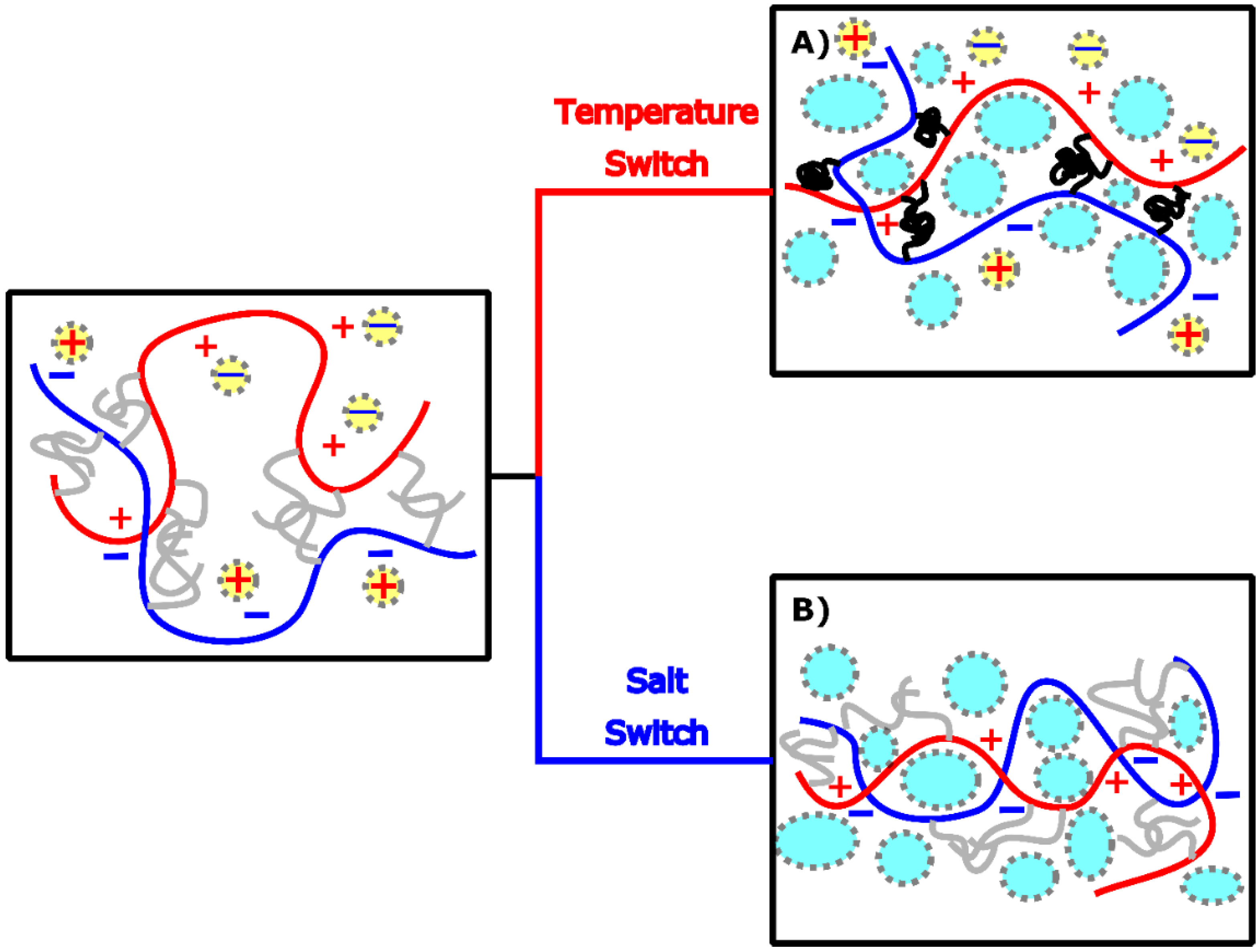
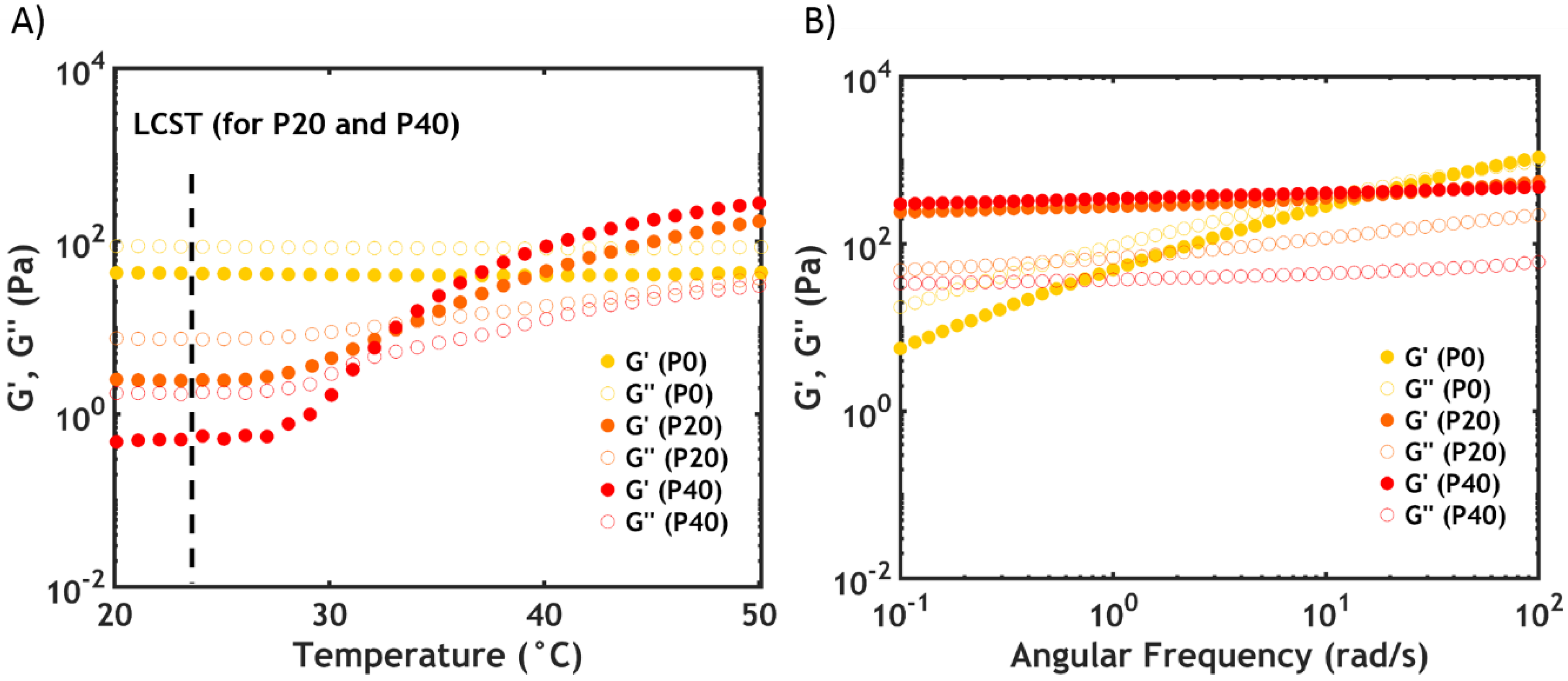
2.2. Temperature Switch
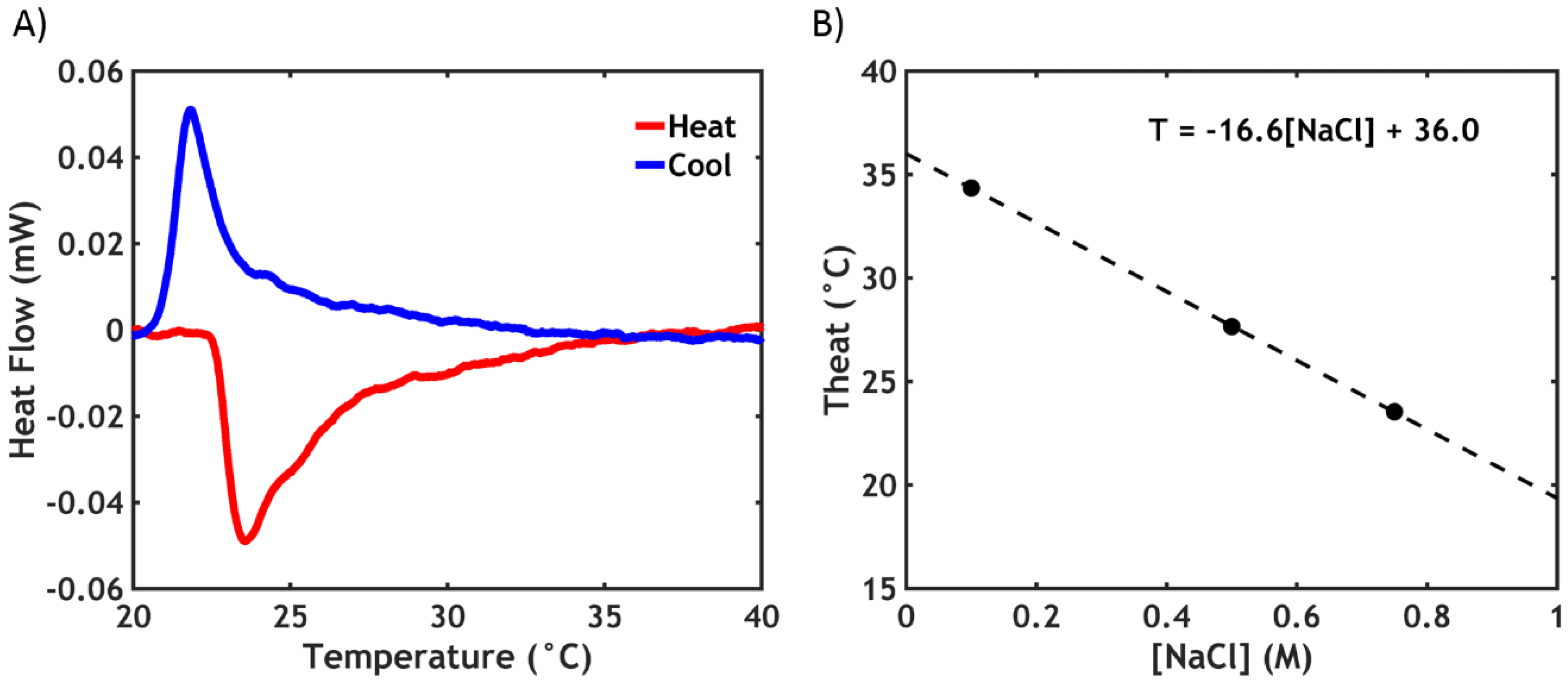
2.2.1. Differential Scanning Calorimetry (DSC)
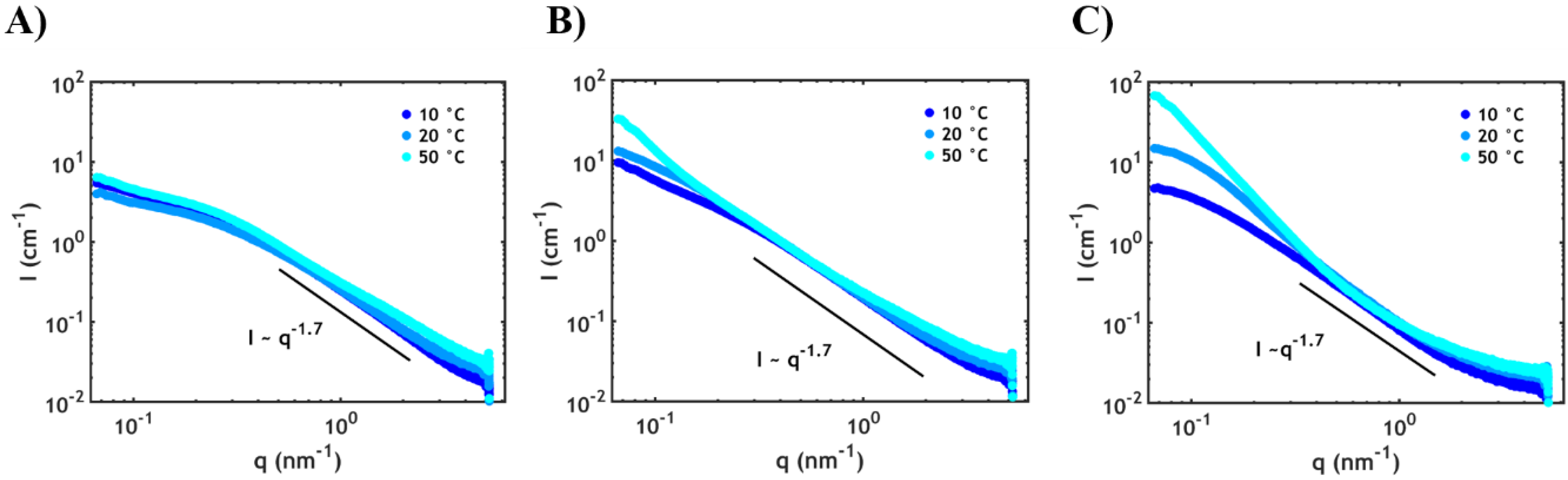
2.2.2. Small Angle X-ray Scattering (SAXS)
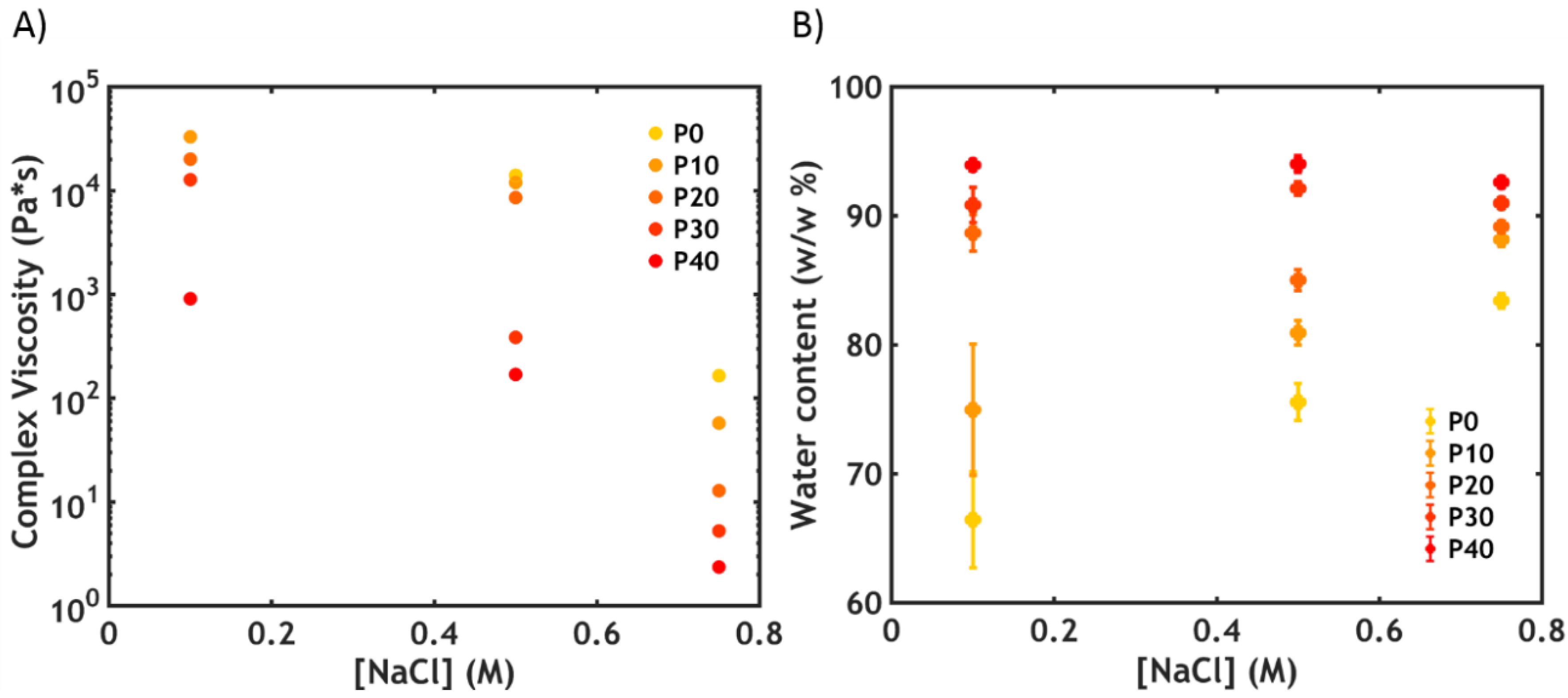
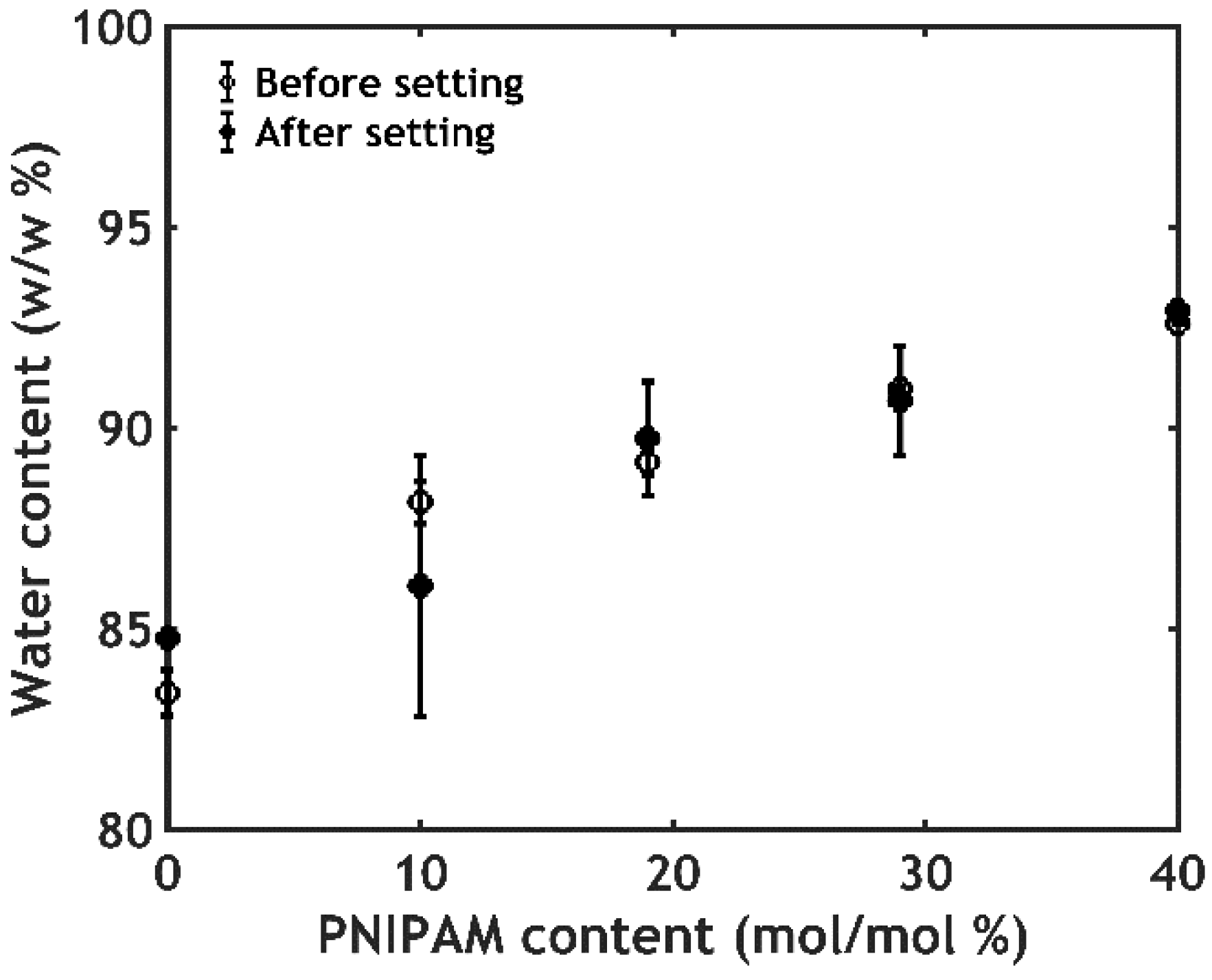
2.2.3. Water Content Analysis
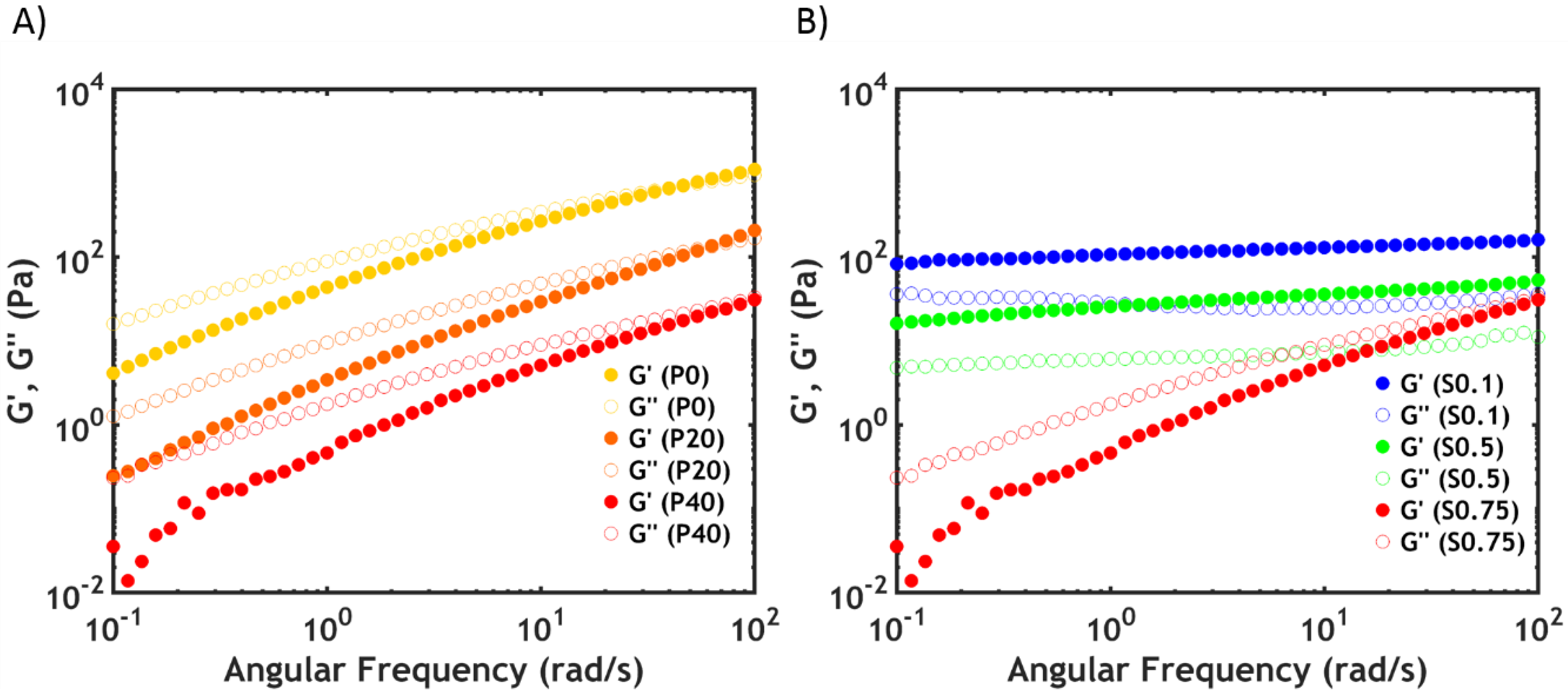
2.2.4. Linear Rheology
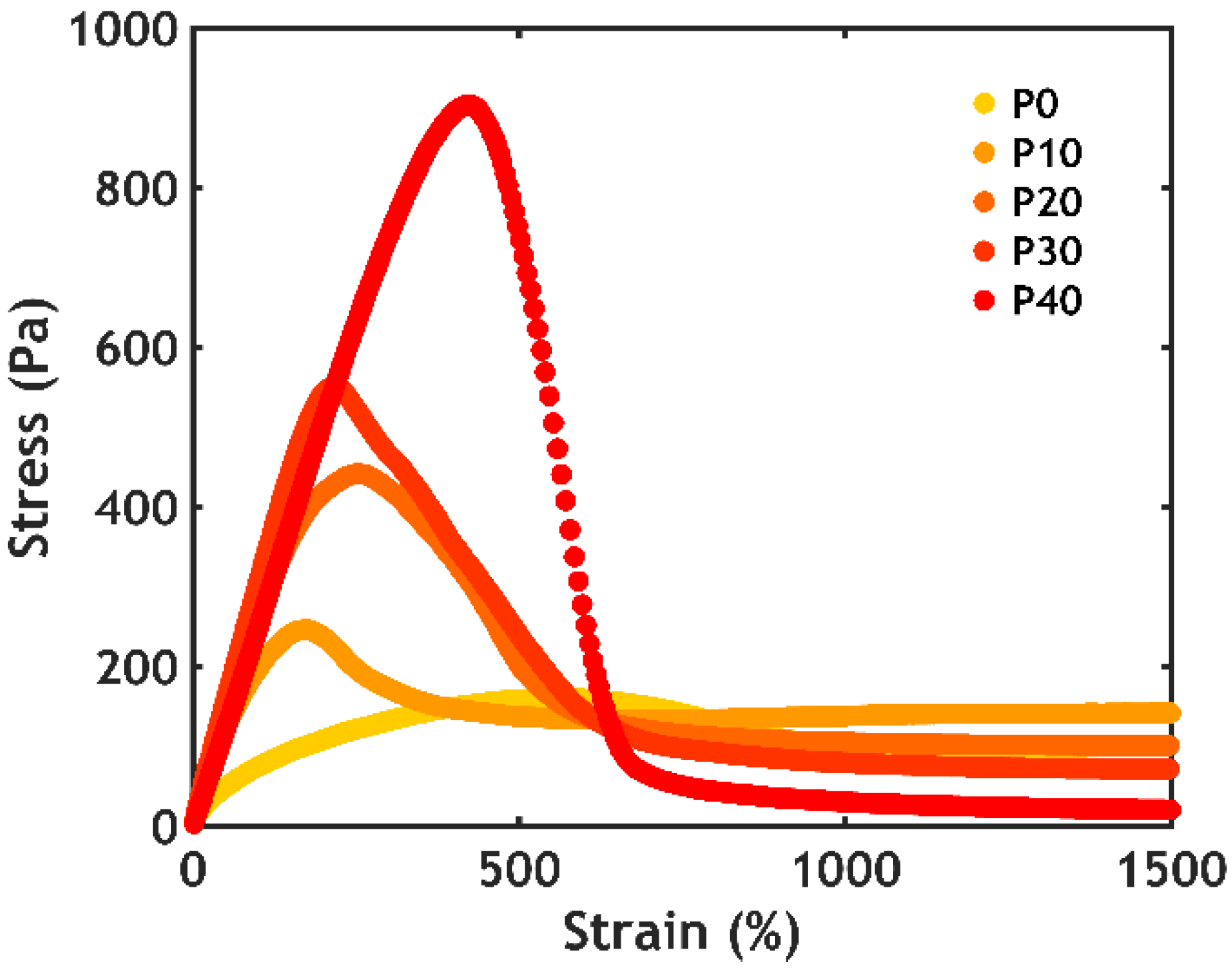
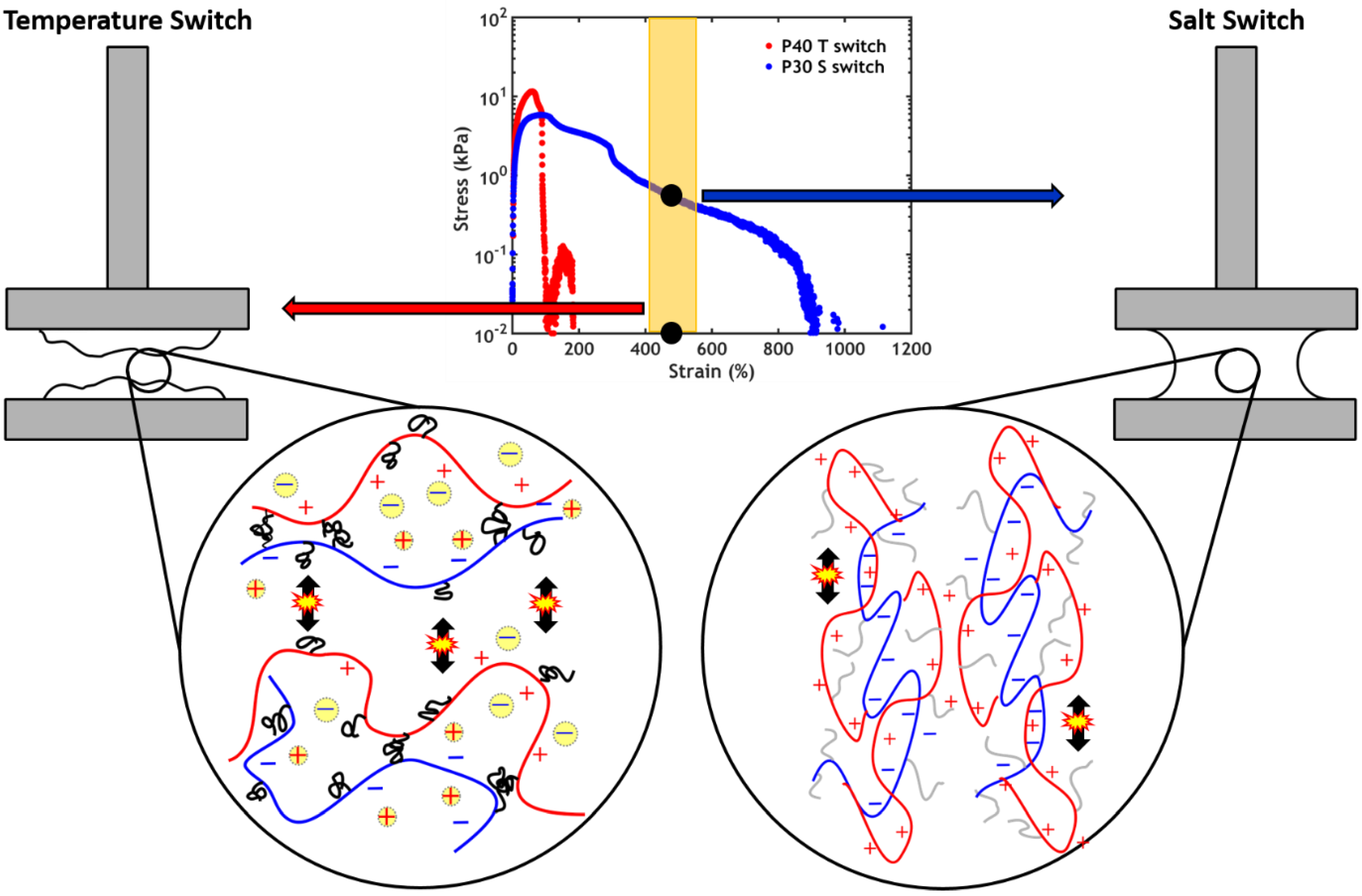
2.2.5. Non-linear Rheology
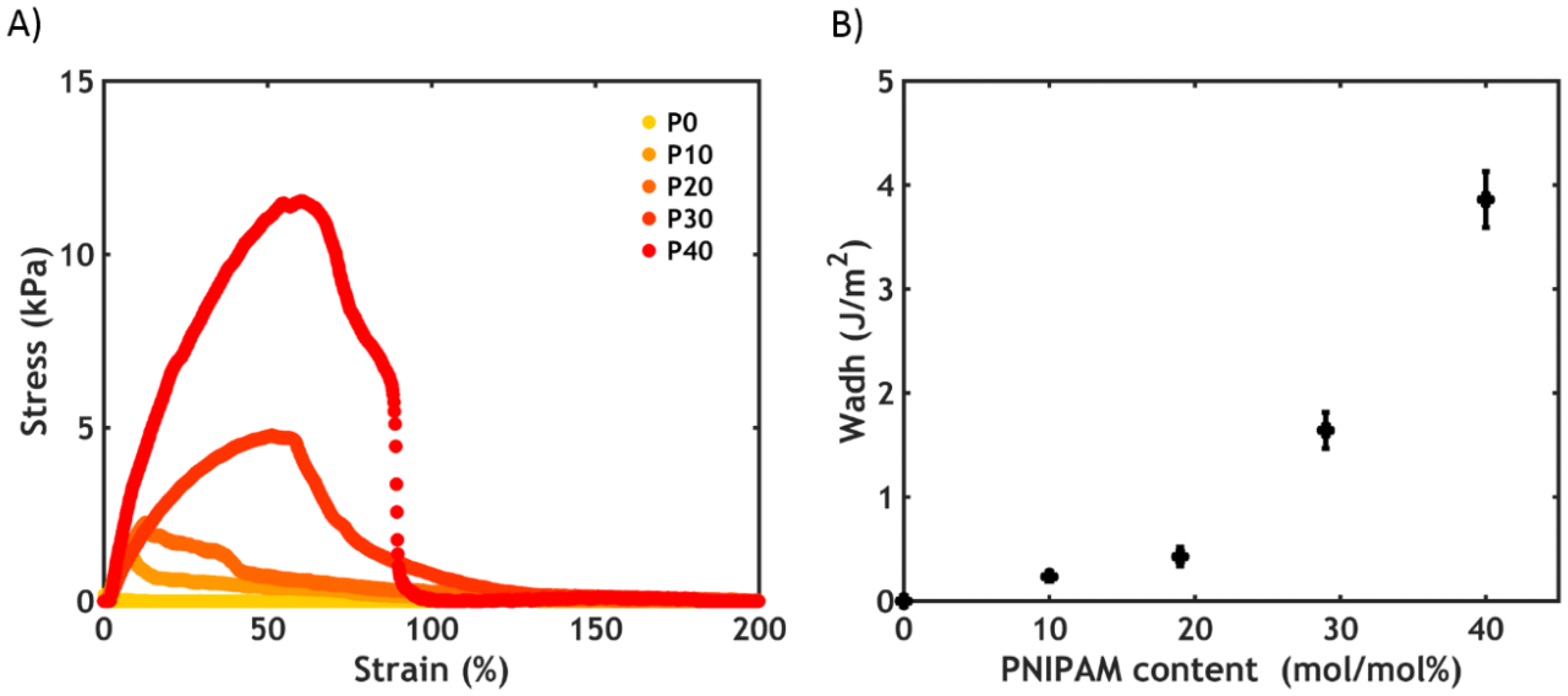
2.2.6. Underwater Adhesion
2.3. Salt Switch
2.3.1. Water Content Analysis
2.3.2. Linear Rheology
2.3.3. Underwater Adhesion
3. Materials and Methods
3.1. Materials
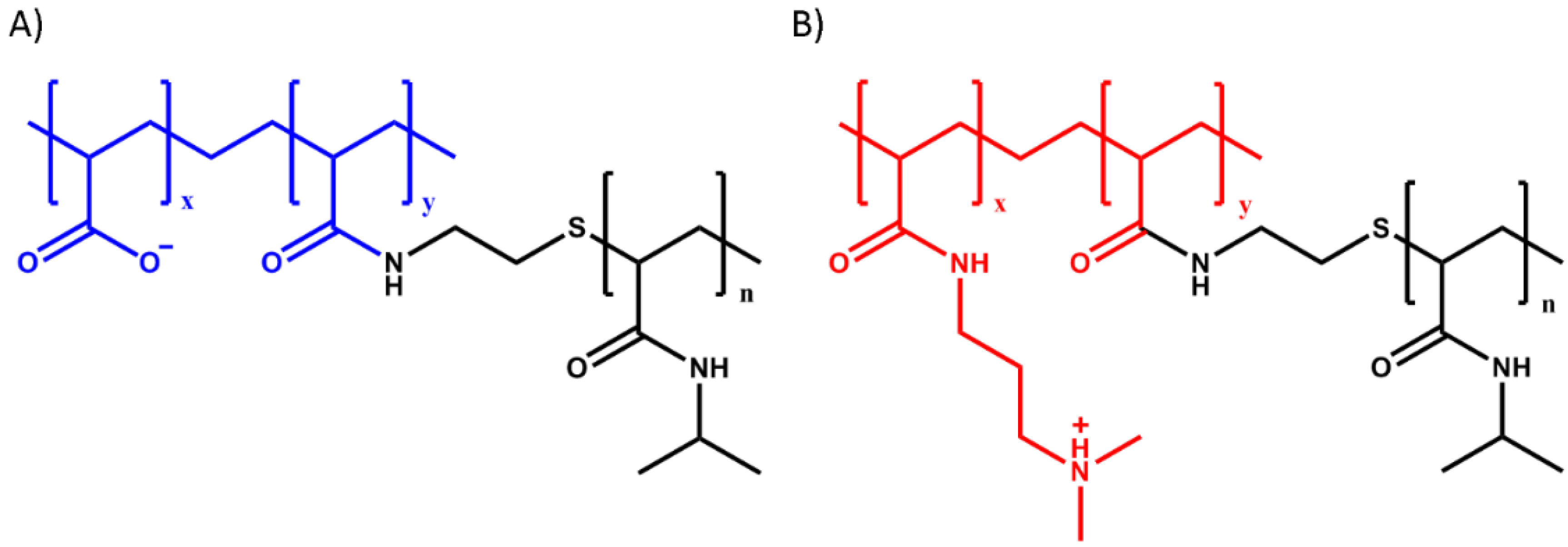
3.2. Polymer Synthesis
3.3. Complex Coacervation
3.4. Optical Microscopy
3.5. Water Content Analysis
3.6. Rheology
3.6.1. Linear Rheology
3.6.2. Non-Linear Rheology
3.7. Differential Scanning Calorimetry (DSC)
3.8. Small Angle X-ray Scattering (SAXS)
3.9. Underwater Adhesion
3.10. Poly(acrylic acid) PAA Hydrogel Thin-Film Synthesis
4. Conclusions
- A high salt concentration, close to the CSC, is necessary to allow injectability of the adhesive;
- The addition of PNIPAM allows the activation of the setting process via a temperature and/or a ionic strength gradient, resulting in a better performance when compared to PNIPAM-free complex coacervates;
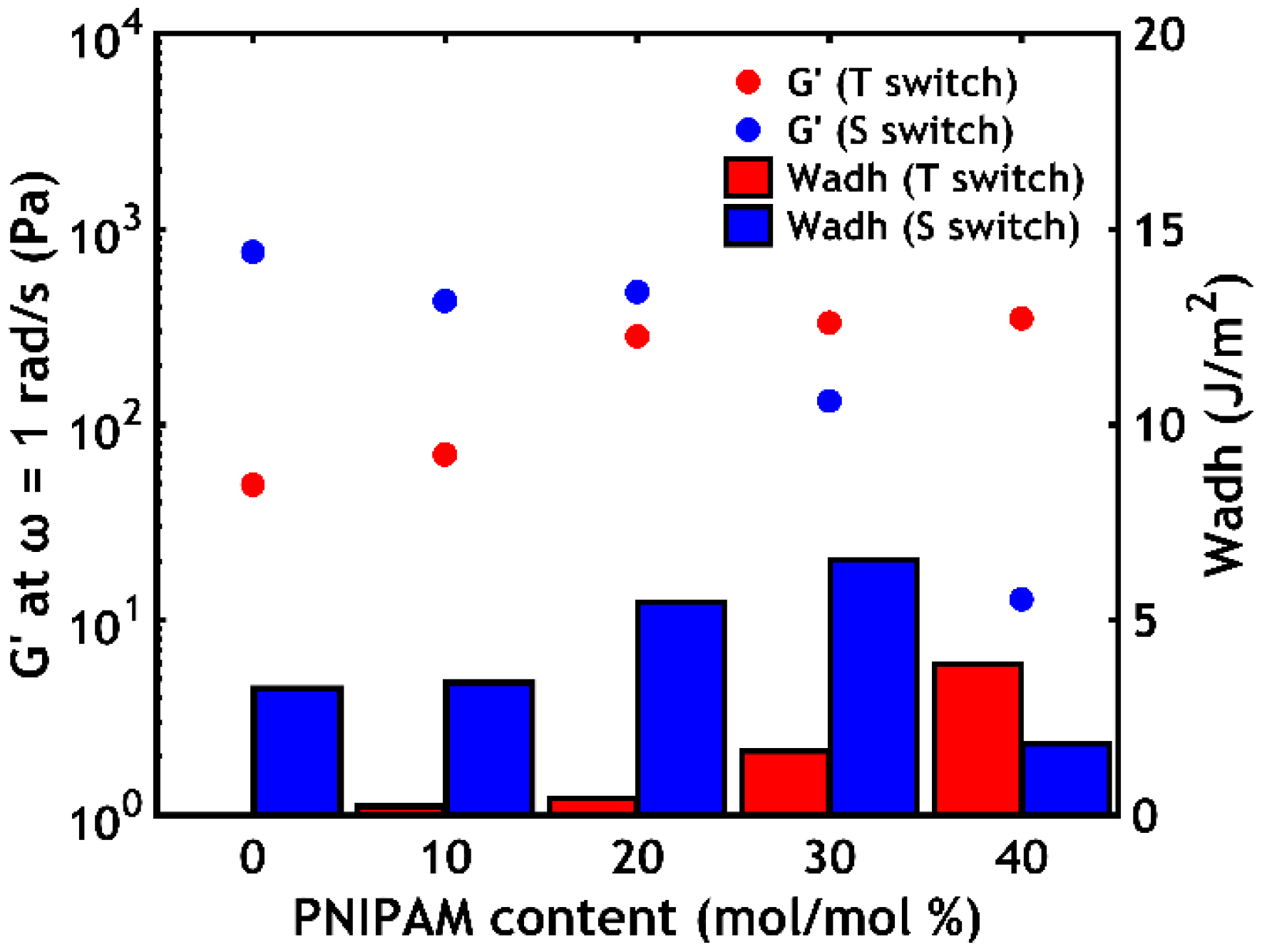
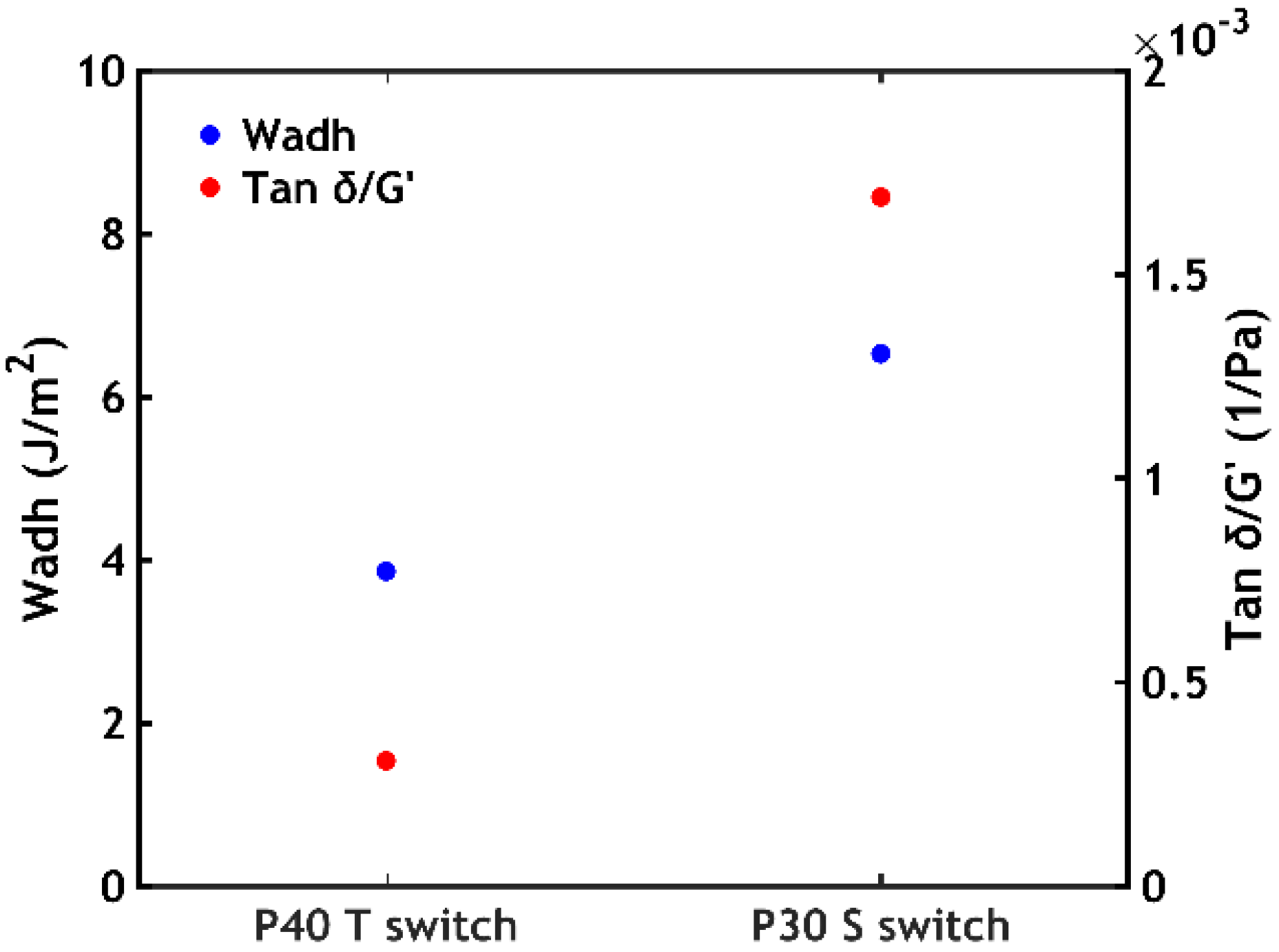
- When performing a temperature switch, a PNIPAM content of 40% leads to the highest work of adhesion (Wadh = 3.9 J/m2);
- When performing a salt switch, a PNIPAM content of 30% leads to the highest work of adhesion (Wadh = 6.5 J/m2).
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bouten, P.J.M.; Zonjee, M.; Bender, J.; Yauw, S.T.K.; Van Goor, H.; Van Hest, J.C.M.; Hoogenboom, R. The chemistry of tissue adhesive materials. Prog. Polym. Sci. 2014, 39, 1375–1405. [Google Scholar] [CrossRef]
- Ferzli, G.S.; Frezza, E.E.; Pecoraro, A.M., Jr.; Ahern, K.D. Prospective randomized study of stapled versus unstapled mesh in a laparoscopic preperitoneal inguinal hernia repair 1. J. Am. Coll. Surg. 1999, 188, 461–465. [Google Scholar] [CrossRef]
- Lang, N.; Pereira, M.J.; Lee, Y.; Friehs, I.; Vasilyev, N.V.; Feins, E.N.; Ablasser, K.; O’Cearbhaill, E.D.; Xu, C.; Fabozzo, A.; et al. A blood-resistant surgical glue for minimally invasive repair of vessels and heart defects. Sci. Transl. Med. 2014, 6, 218ra6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waite, J.H. Nature’s underwater adhesive specialist. Int. J. Adhes. Adhes. 1987, 7, 9–14. [Google Scholar] [CrossRef]
- Waite, J.H.; Andersen, N.H.; Jewhurst, S.; Sun, C. Mussel adhesion: Finding the tricks worth mimicking. J. Adhes. 2005, 81, 297–317. [Google Scholar] [CrossRef]
- Walker, G. The histology, histochemistry and ultrastructure of the cement apparatus of three adult sessile barnacles, Elminius modestus, Balanus balanoides and Balanus hameri. Mar. Biol. 1970, 7, 239–248. [Google Scholar] [CrossRef]
- Stewart, R.J.; Weaver, J.C.; Morse, D.E.; Waite, J.H. The tube cement of Phragmatopoma californica: A solid foam. J. Exp. Biol. 2004, 207, 4727–4734. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.J.; Wang, C.S.; Song, I.T.; Jones, J.P. The role of coacervation and phase transitions in the sandcastle worm adhesive system. Adv. Colloid Interface Sci. 2017, 239, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Gucht, J.V.D.; Spruijt, E.; Lemmers, M.; Cohen Stuart, M.A. Polyelectrolyte complexes: Bulk phases and colloidal systems. J. Colloid Interface Sci. 2011, 361, 407–422. [Google Scholar] [CrossRef]
- Jones, J.P.; Sima, M.; O’Hara, R.G.; Stewart, R.J. Water-borne endovascular embolics inspired by the undersea adhesive of marine sandcastle worms. Adv. Healthc. Mater. 2016, 5, 795–801. [Google Scholar] [CrossRef]
- Stewart, R.J.; Wang, C.S.; Shao, H. Complex coacervates as a foundation for synthetic underwater adhesives. Adv. Colloid Interface Sci. 2011, 167, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spruijt, E.; Sprakel, J.; Cohen Stuart, M.A.; Van Der Gucht, J. Interfacial tension between a complex coacervate phase and its coexisting aqueous phase. Soft Matter 2010, 6, 172–178. [Google Scholar] [CrossRef]
- Creton, C. Pressure-sensitive adhesives: An introductory course. MRS Bull. 2003, 28, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Hofman, A.H.; Van Hees, I.A.; Yang, J.; Kamperman, M. Bioinspired underwater adhesives by using the supramolecular toolbox. Adv. Mater. 2018, 30, 1704640. [Google Scholar] [CrossRef]
- Ahn, B.K.; Das, S.; Linstadt, R.; Kaufman, Y.; Martinez-Rodriguez, N.R.; Mirshafian, R.; Kesselman, E.; Talmon, Y.; Lipshutz, B.H.; Israelachvili, J.N.; et al. High-performance mussel-inspired adhesives of reduced complexity. Nat. Commun. 2015, 6, 8663. [Google Scholar] [CrossRef]
- Kaur, S.; Weerasekare, G.M.; Stewart, R.J. Multiphase adhesive coacervates inspired by the sandcastle worm. ACS Appl. Mater. Interfaces 2011, 3, 941–944. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Stewart, R.J. Biomimetic underwater adhesives with environmentally triggered setting mechanisms. Adv. Mater. 2010, 22, 729–733. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.; Das, S.; Zalicki, P.J.; Mirshafian, R.; Eisenbach, C.D.; Israelachvili, J.N.; Waite, J.H.; Ahn, B.K. Microphase behavior and enhanced wet-cohesion of synthetic copolyampholytes inspired by a mussel foot protein. J. Am. Chem. Soc. 2015, 137, 9214–9217. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, P.G.; Lapitsky, Y. Ionically Cross-linked poly(allylamine) as a stimulus-responsive underwater adhesive: Ionic strength and pH effects. Langmuir 2015, 31, 1564–1574. [Google Scholar] [CrossRef]
- Kim, S.; Yoo, H.Y.; Huang, J.; Lee, Y.; Park, S.; Park, Y.; Jin, S.; Jung, Y.M.; Zeng, H.; Hwang, D.S.; et al. Salt triggers the simple coacervation of an underwater adhesive when cations meet aromatic π electrons in seawater. ACS Nano 2017, 11, 6764–6772. [Google Scholar] [CrossRef]
- Zhao, Q.; Lee, D.W.; Ahn, B.K.; Seo, S.; Kaufman, Y.; Israelachvili, J.N.; Waite, J.H. Underwater contact adhesion and microarchitecture in polyelectrolyte complexes actuated by solvent exchange. Nat. Mater. 2016, 15, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dompé, M.; Cedano-Serrano, F.J.; Heckert, O.; Van Den Heuvel, N.; Van Der Gucht, J.; Tran, Y.; Hourdet, D.; Creton, C.; Kamperman, M. Thermoresponsive complex coacervate-based underwater adhesive. Adv. Mater. 2019, 31, 1808179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dompé, M.; Cedano-Serrano, F.J.; Vahdati, M.; Westerveld, L.V.; Hourdet, D.; Creton, C.; Kodger, T.; Kamperman, M. Underwater Adhesion of Multi-Responsive Complex Coacervates. Adv. Mater. Interfaces. (in press).
- Heskins, M.; Guillet, J.E. Solution properties of poly(N-isopropylacrylamide). J. Macromol. Sci. Part A Chem. 1968, 2, 1441–1455. [Google Scholar] [CrossRef]
- Sun, T.L.; Kurokawa, T.; Kuroda, S.; Ihsan, A.B.; Akasaki, T.; Sato, K.; Haque, M.A.; Nakajima, T.; Gong, J.P. Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nat. Mater. 2013, 12, 932–937. [Google Scholar] [CrossRef] [Green Version]
- Feldstein, M.M. Molecular nature of pressure-sensitive adhesion. In Fundamentals of Pressure Sensitivity, 1st ed.; Benedek, I., Feldstein, M.M., Eds.; Taylor & Francis Group, LLC: Boca Raton, FL, USA, 2008; pp. 241–284. [Google Scholar]
- Asplund, P.; Blomstedt, P.; Bergenheim, A.T. Percutaneous balloon compression vs percutaneous retrogasserian glycerol rhizotomy for the primary treatment of trigeminal neuralgia. Neurosurgery 2016, 78, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Spruijt, E.; Westphal, A.H.; Borst, J.W.; Cohen Stuart, M.A.; Van Der Gucht, J. Binodal compositions of polyelectrolyte complexes. Macromolecules 2010, 43, 6476–6484. [Google Scholar] [CrossRef]
- Zhang, Y.; Furyk, S.; Bergbreiter, D.E.; Cremer, P.S. Specific ion effects on the water solubility of macromolecules: PNIPAM and the hofmeister series. J. Am. Chem. Soc. 2005, 127, 14505–14510. [Google Scholar] [CrossRef]
- Spruijt, E.; Cohen Stuart, M.A.; Van Der Gucht, J. Linear viscoelasticity of polyelectrolyte complex coacervates. Macromolecules 2013, 46, 1633–1641. [Google Scholar] [CrossRef]
- Guo, H.; Brûlet, A.; Rajamohanan, P.R.; Marcellan, A.; Sanson, N.; Hourdet, D. Influence of topology of LCST-based graft copolymers on responsive assembling in aqueous media. Polymer 2015, 60, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Hourdet, D.; L’Alloret, F.; Audebert, R. Reversible thermothickening of aqueous polymer solutions. Polymer 1994, 35, 2624–2630. [Google Scholar] [CrossRef]
- Siband, E.; Tran, Y.; Hourdet, D. Thermoresponsive interpolyelectrolyte complexation: Application to macromolecular assemblies. Macromolecules 2011, 44, 8185–8194. [Google Scholar] [CrossRef]
- Petit, L.; Bouteiller, L.; Brûlet, A.; Lafuma, F.; Hourdet, D. Responsive hybrid self-assemblies in aqueous media. Langmuir 2007, 23, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Schild, H.G.; Tirrell, D.A. Microcalorimetric detection of lower critical solution temperatures in aqueous polymer solutions. J. Phys. Chem. 1990, 94, 4352–4356. [Google Scholar] [CrossRef]
- Marciel, A.B.; Srivastava, S.; Tirrell, M.V. Structure and rheology of polyelectrolyte complex coacervates. Soft Matter 2018, 14, 2454–2464. [Google Scholar] [CrossRef]
- Barrett, D.G.; Bushnell, G.G.; Messersmith, P.B. Mechanically robust, negative-swelling, mussel-inspired tissue adhesives. Adv. Healthc. Mater. 2013, 2, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Liu, K.; Wang, M.; Wang, K.; Fang, L.; Chen, H.; Zhou, J.; Lu, X. Mussel-inspired adhesive and conductive hydrogel with long-lasting moisture and extreme temperature tolerance. Adv. Funct. Mater. 2018, 28, 1704195. [Google Scholar] [CrossRef]
- Kaneko, Y.; Yoshida, R.; Sakai, K.; Sakurai, Y.; Okano, T. Temperature-responsive shrinking kinetics of poly (N-isopropylacrylamide) copolymer gels with hydrophilic and hydrophobic comonomers. J. Membr. Sci. 1995, 101, 13–22. [Google Scholar] [CrossRef]
- Porcel, C.H.; Schlenoff, J.B. Compact polyelectrolyte complexes: “Saloplastic” candidates for biomaterials. Biomacromolecules 2009, 10, 2968–2975. [Google Scholar] [CrossRef] [Green Version]
- Courtois, J.; Baroudi, I.; Nouvel, N.; Degrandi, E.; Pensec, S.; Ducouret, G.; Chanéac, C.; Bouteiller, L.; Creton, C. Supramolecular soft adhesive materials. Adv. Funct. Mater. 2010, 20, 1803–1811. [Google Scholar] [CrossRef]
- Skrzeszewska, P.J.; Sprakel, J.; De Wolf, F.A.; Fokkink, R.; Cohen Stuart, M.A.; Van Der Gucht, J. Fracture and self-healing in a well-defined self-assembled polymer network. Macromolecules 2010, 43, 3542–3548. [Google Scholar] [CrossRef]
- Chung, H.; Glass, P.; Pothen, J.M.; Sitti, M.; Washburn, N.R. Enhanced adhesion of dopamine methacrylamide elastomers via viscoelasticity tuning. Biomacromolecules 2011, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Guvendiren, M.; Messersmith, P.B.; Shull, K.R. Self-assembly and adhesion of DOPA-modified methacrylic triblock hydrogels. Biomacromolecules 2008, 9, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariri, H.H.; Lehaf, A.M.; Schlenoff, J.B. Mechanical properties of osmotically stressed polyelectrolyte complexes and multilayers: Water as a plasticizer. Macromolecules 2012, 45, 9364–9372. [Google Scholar] [CrossRef]
- Deplace, F.; Carelli, C.; Mariot, S.; Retsos, H.; Chateauminois, A.; Ouzineb, K.; Creton, C. Fine tuning the adhesive properties of a soft nanostructured adhesive with rheological measurements. J. Adhes. 2009, 85, 18–54. [Google Scholar] [CrossRef]
- Guo, H.; Sanson, N.; Marcellan, A.; Hourdet, D. Thermoresponsive toughening in LCST-type hydrogels: Comparison between semi-interpenetrated and grafted networks. Macromolecules 2016, 49, 9568–9577. [Google Scholar] [CrossRef] [Green Version]
- Durand, A.; Hourdet, D. Synthesis and thermoassociative properties in aqueous solution of graft copolymers containing poly(N-isopropylacrylamide) side chains. Polymer 1999, 40, 4941–4951. [Google Scholar] [CrossRef]
- Petit, L.; Karakasyan, C.; Pantoustier, N.; Hourdet, D. Synthesis of graft polyacrylamide with responsive self-assembling properties in aqueous media. Polymer 2007, 48, 7098–7112. [Google Scholar] [CrossRef]
- Sudre, G.; Olanier, L.; Tran, Y.; Hourdet, D.; Creton, C. Reversible adhesion between a hydrogel and a polymer brush. Soft Matter 2012, 8, 8184–8193. [Google Scholar] [CrossRef] [Green Version]
- Chollet, B.; Li, M.; Martwong, E.; Bresson, B.; Fretigny, C.; Tabeling, P.; Tran, Y. Multiscale surface-attached hydrogel thin films with tailored architecture. ACS Appl. Mater. Interfaces 2016, 8, 11729–11738. [Google Scholar] [CrossRef] [Green Version]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex Coacervate | PNIPAM/Total Polymer Molar Ratio (% mol/mol) | NaCl Concentration (M) |
|---|---|---|
| P0S0.1 | 0 | 0.10 |
| P0S0.5 | 0 | 0.50 |
| P0S0.75 | 0 | 0.75 |
| P10S0.1 | 10 | 0.10 |
| P10S0.5 | 10 | 0.50 |
| P10S0.75 | 10 | 0.75 |
| P20S0.1 | 19 | 0.10 |
| P20S0.5 | 19 | 0.50 |
| P20S0.75 | 19 | 0.75 |
| P30S0.1 | 29 | 0.10 |
| P30S0.5 | 29 | 0.50 |
| P30S0.75 | 29 | 0.75 |
| P40S0.1 | 40 | 0.10 |
| P40S0.5 | 40 | 0.50 |
| P40S0.75 | 40 | 0.75 |
| Complex Coacervate | Theat (°C) | ΔH⦵heat (kJ/mol PNIPAM) | Tcool (°C) | ΔH⦵cool (kJ/mol PNIPAM) |
|---|---|---|---|---|
| P0S0.75 | - | - | - | - |
| P10S0.75 | 23.8 | - | 21.6 | - |
| P20S0.75 | 23.5 | - | 21.3 | - |
| P30S0.75 | 22.7 | 1.8 | 21.8 | 1.4 |
| P40S0.75 | 23.5 | 1.8 | 21.8 | 1.3 |
| P40S0.5 | 27.7 | 1.8 | 26.4 | 1.3 |
| P40S0.1 | 34.4 | 1.9 | 32.4 | 1.8 |
| Polymer | PNIPAM Molar Ratio (mol/mol %) | Mn Graft Copolymer (kg/mol) | PNIPAM Chains Per Backbone | Polydispersity Index (PDI) |
|---|---|---|---|---|
| PAA | 0 | 239 | 0 | 4.3 |
| PAA-g-PNIPAM10 | 11 | 359 | 9 | - |
| PAA-g-PNIPAM10 | 20 | 405 | 17 | - |
| PAA-g-PNIPAM10 | 33 | 499 | 35 | - |
| PAA-g-PNIPAM10 | 46 | 636 | 60 | - |
| PDMAPAA | 0 | 139 | 0 | 4.6 |
| PDMAPAA-g-PNIPA10 | 9 | 104 | 1 | 5.3 |
| PDMAPAA-g-PNIPA20 | 18 | 147 | 3 | 6.4 |
| PDMAPAA-g-PNIPA30 | 26 | 248 | 7 | 4.4 |
| PDMAPAA-g-PNIPA40 | 33 | 244 | 9 | 4.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dompé, M.; Cedano-Serrano, F.J.; Vahdati, M.; Sidoli, U.; Heckert, O.; Synytska, A.; Hourdet, D.; Creton, C.; van der Gucht, J.; Kodger, T.; et al. Tuning the Interactions in Multiresponsive Complex Coacervate-Based Underwater Adhesives. Int. J. Mol. Sci. 2020, 21, 100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010100
Dompé M, Cedano-Serrano FJ, Vahdati M, Sidoli U, Heckert O, Synytska A, Hourdet D, Creton C, van der Gucht J, Kodger T, et al. Tuning the Interactions in Multiresponsive Complex Coacervate-Based Underwater Adhesives. International Journal of Molecular Sciences. 2020; 21(1):100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010100
Chicago/Turabian StyleDompé, Marco, Francisco J. Cedano-Serrano, Mehdi Vahdati, Ugo Sidoli, Olaf Heckert, Alla Synytska, Dominique Hourdet, Costantino Creton, Jasper van der Gucht, Thomas Kodger, and et al. 2020. "Tuning the Interactions in Multiresponsive Complex Coacervate-Based Underwater Adhesives" International Journal of Molecular Sciences 21, no. 1: 100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010100