Autophagy is Activated In Vivo during Trimethyltin-Induced Apoptotic Neurodegeneration: A Study in the Rat Hippocampus


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
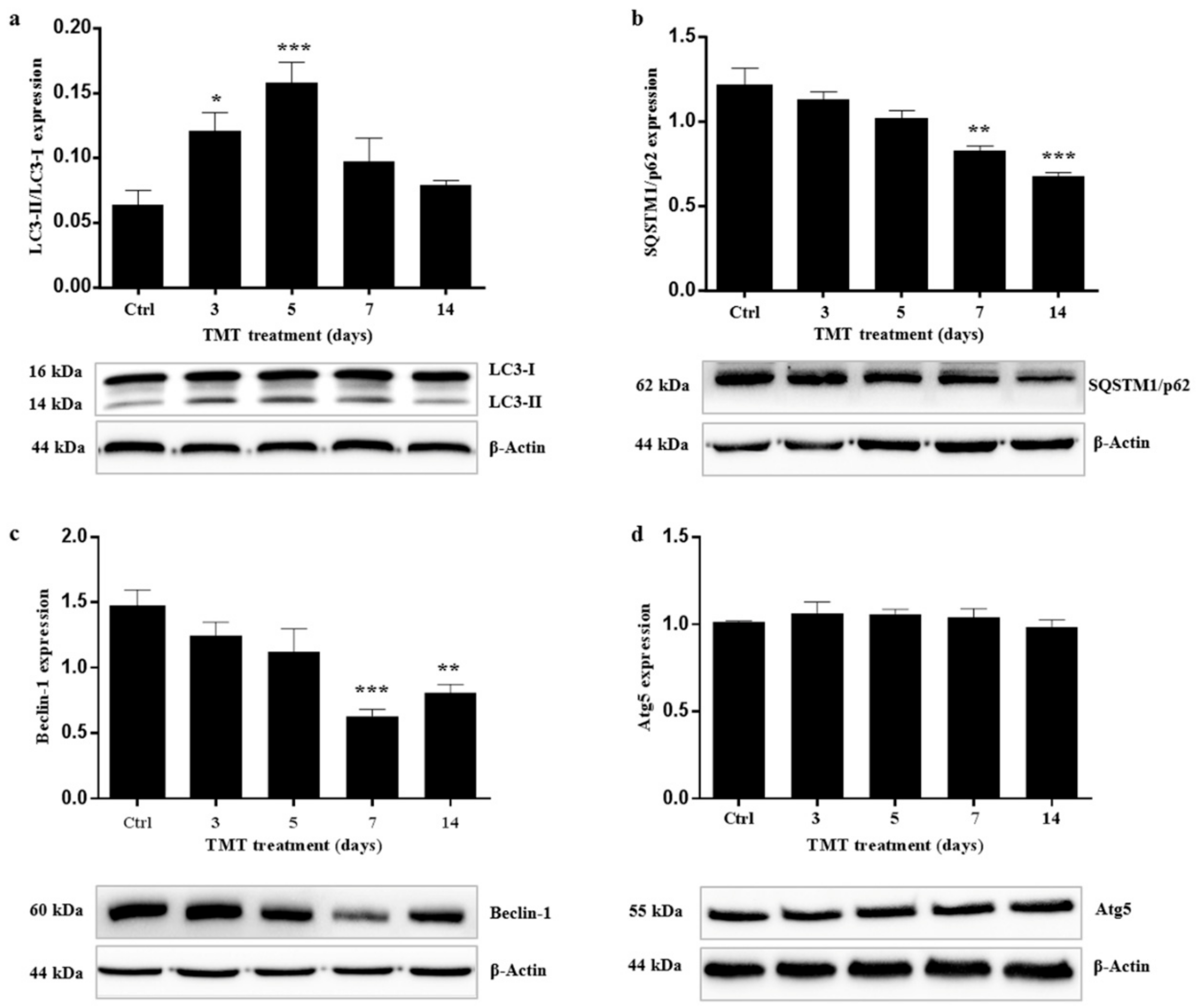
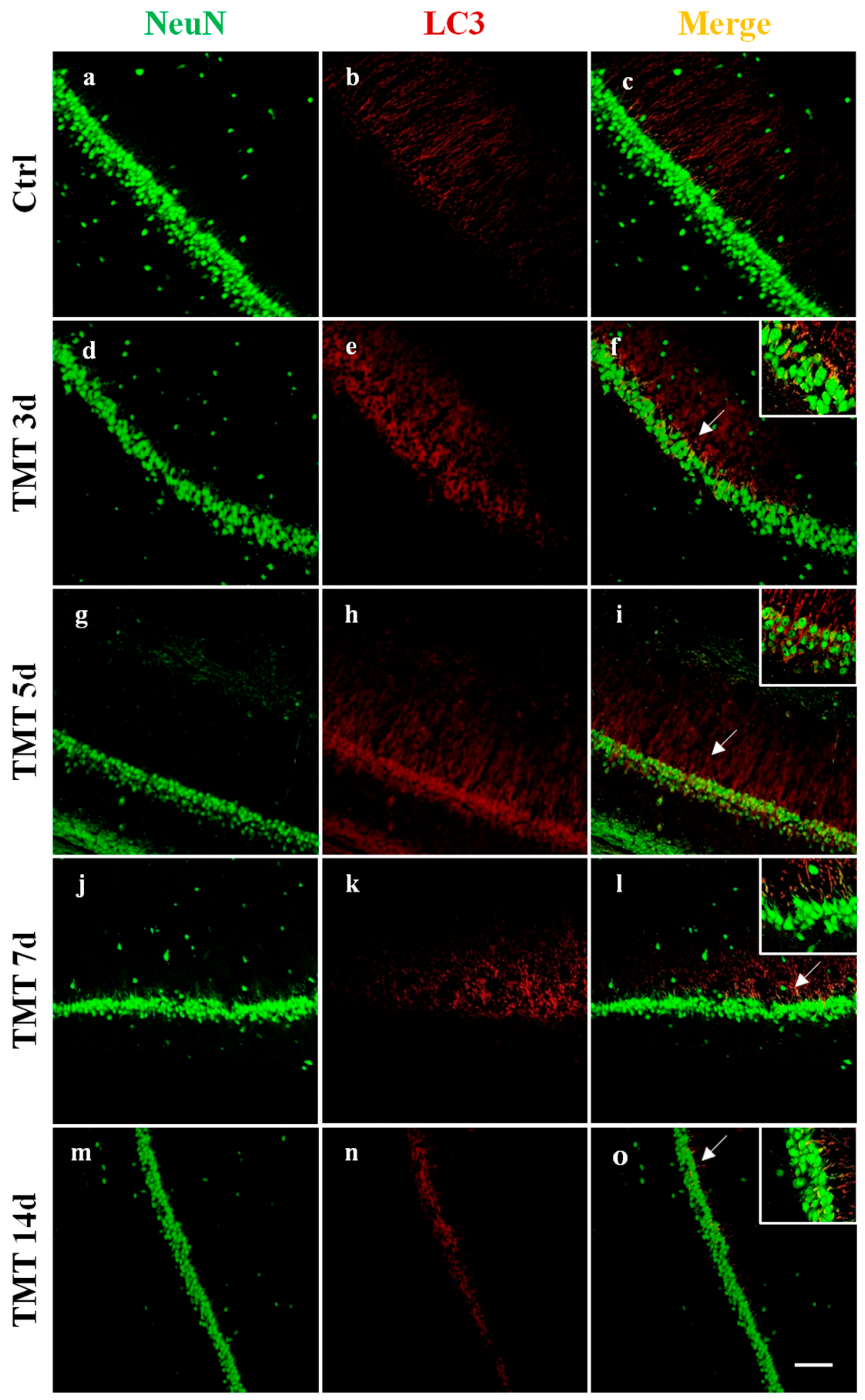
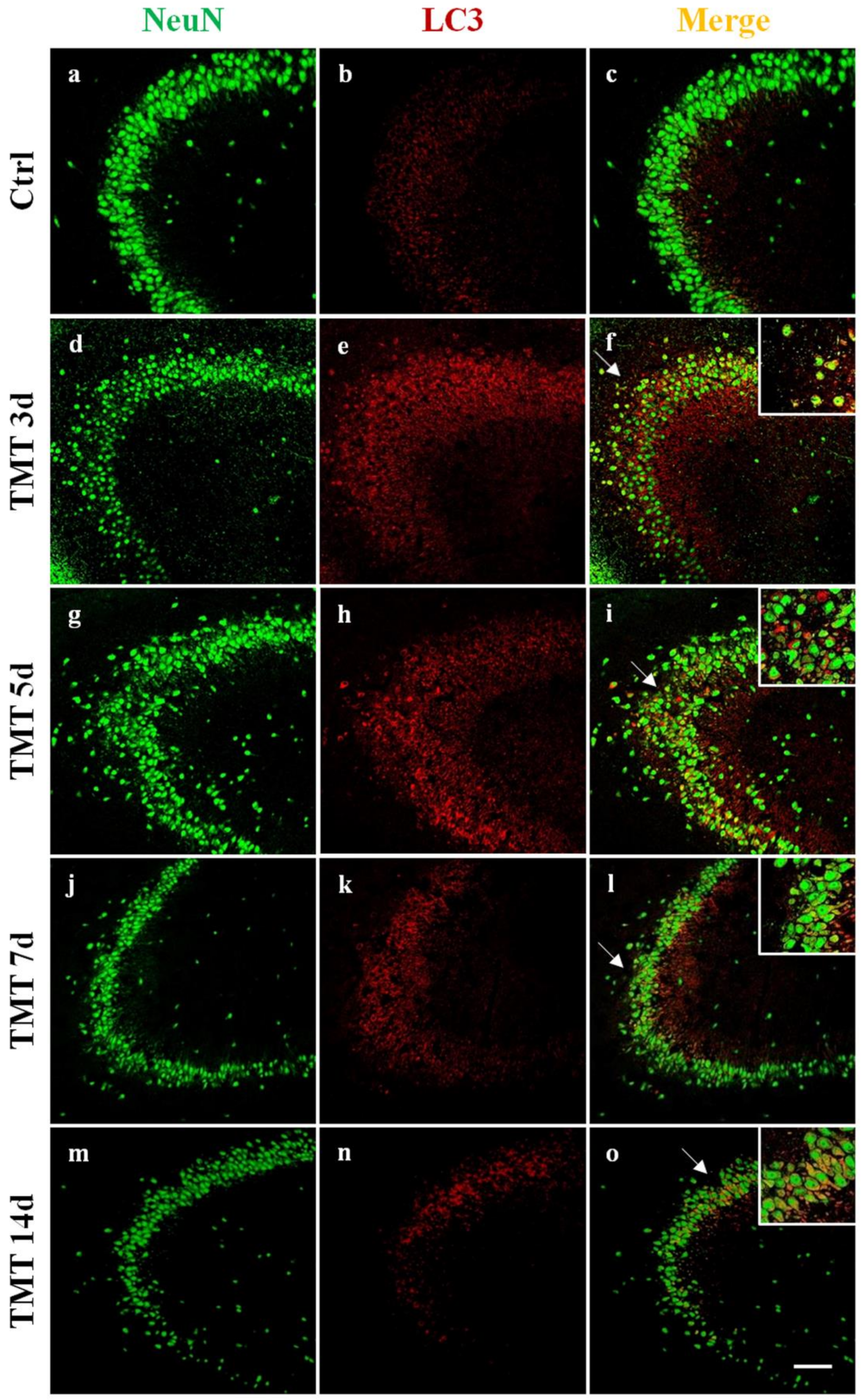
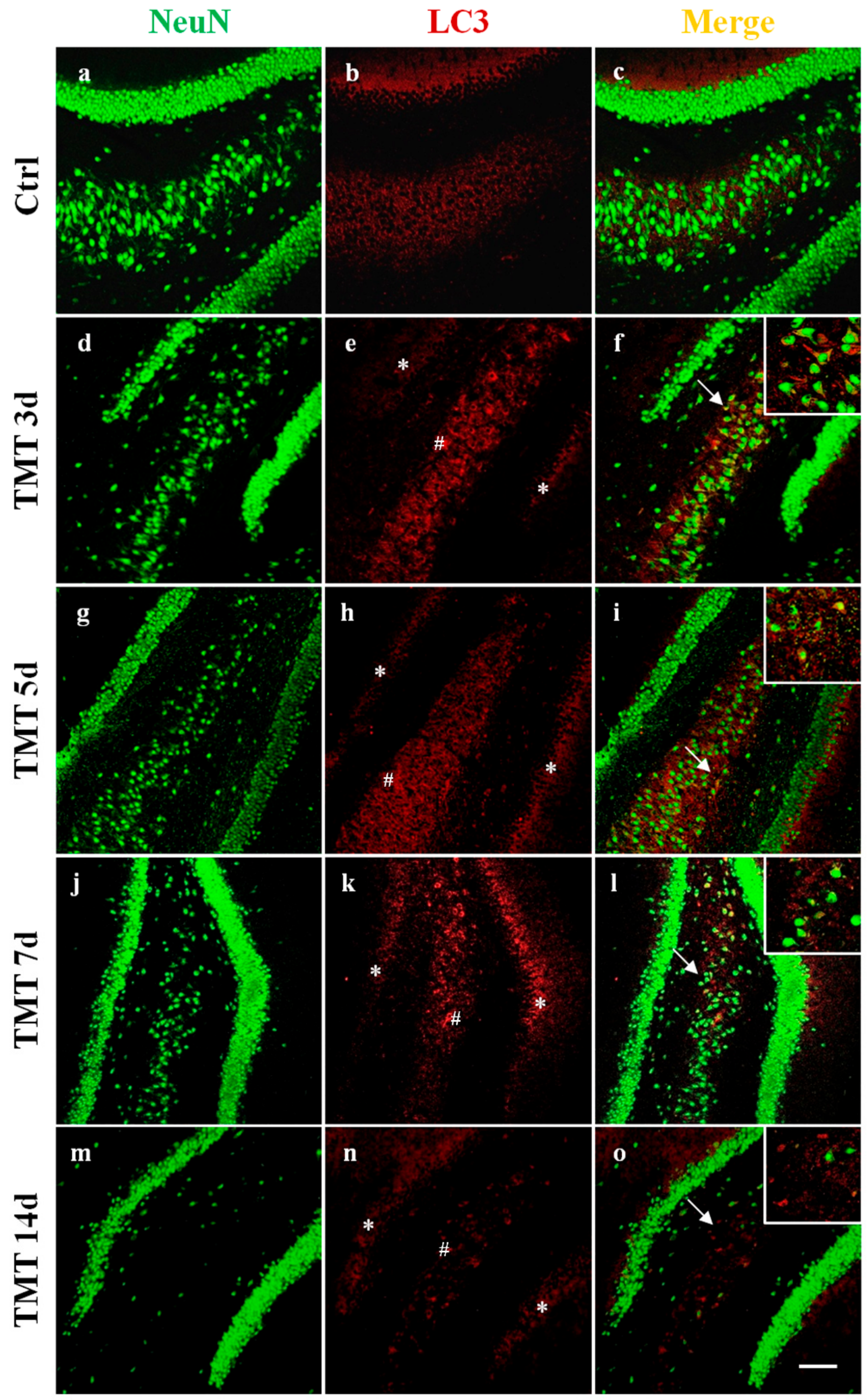
2.1. Autophagy is Activated in the Hippocampus of TMT-Treated Rats
2.2. TMT Reduces Beclin-1 Levels and Has No Effect on Atg5 Expression
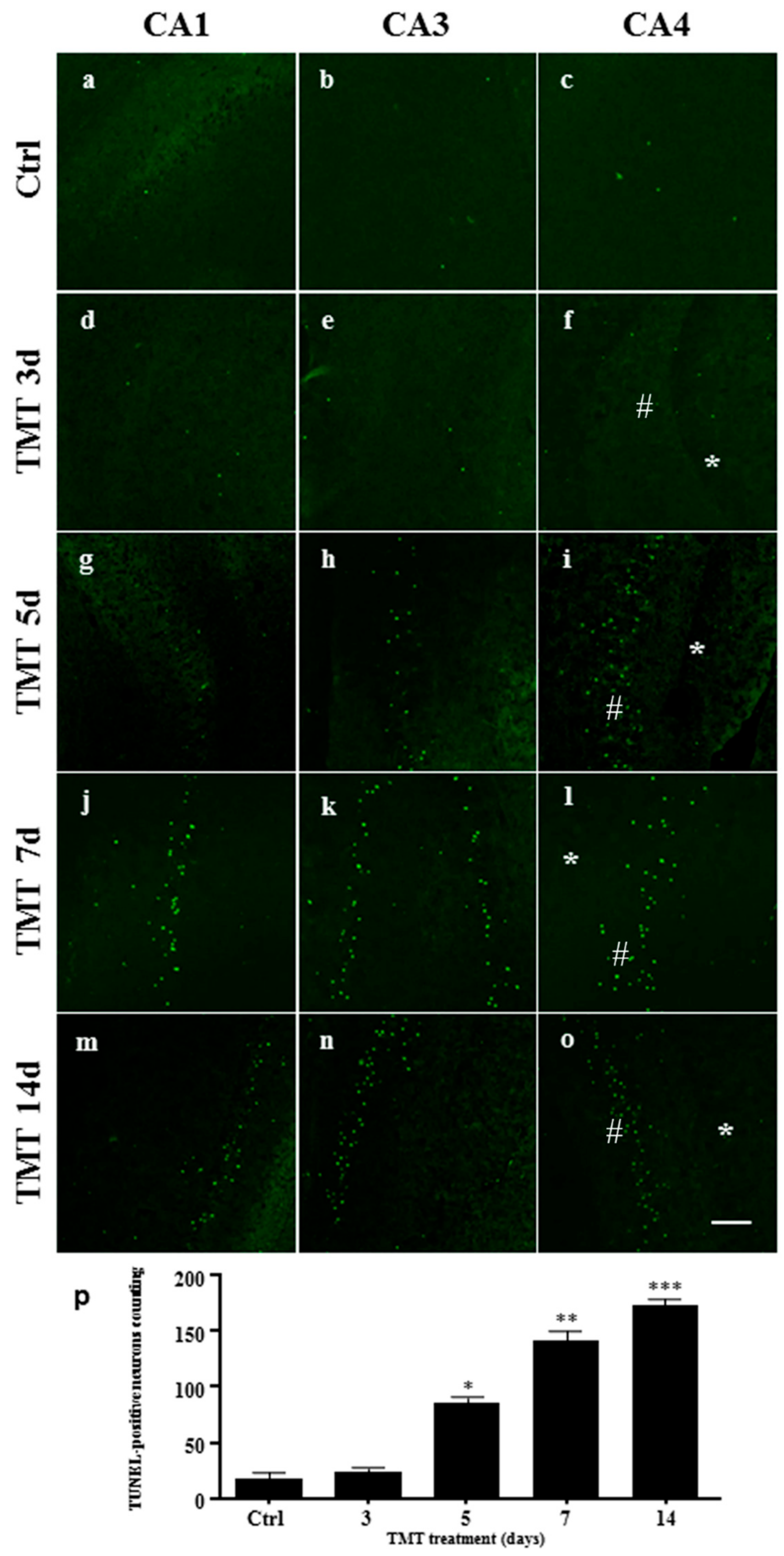
2.3. Apoptotic Cells Increase after TMT-Treatment
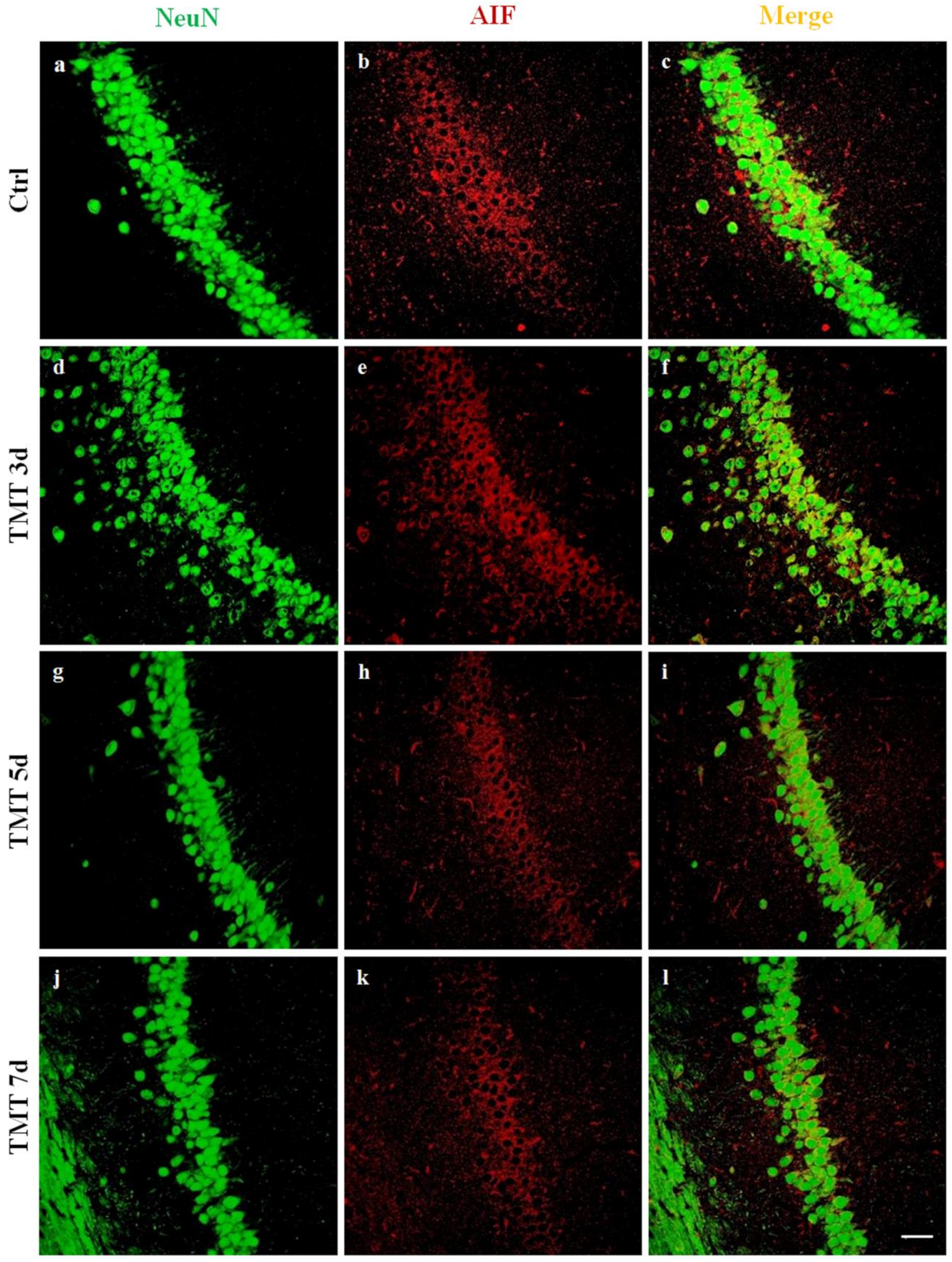
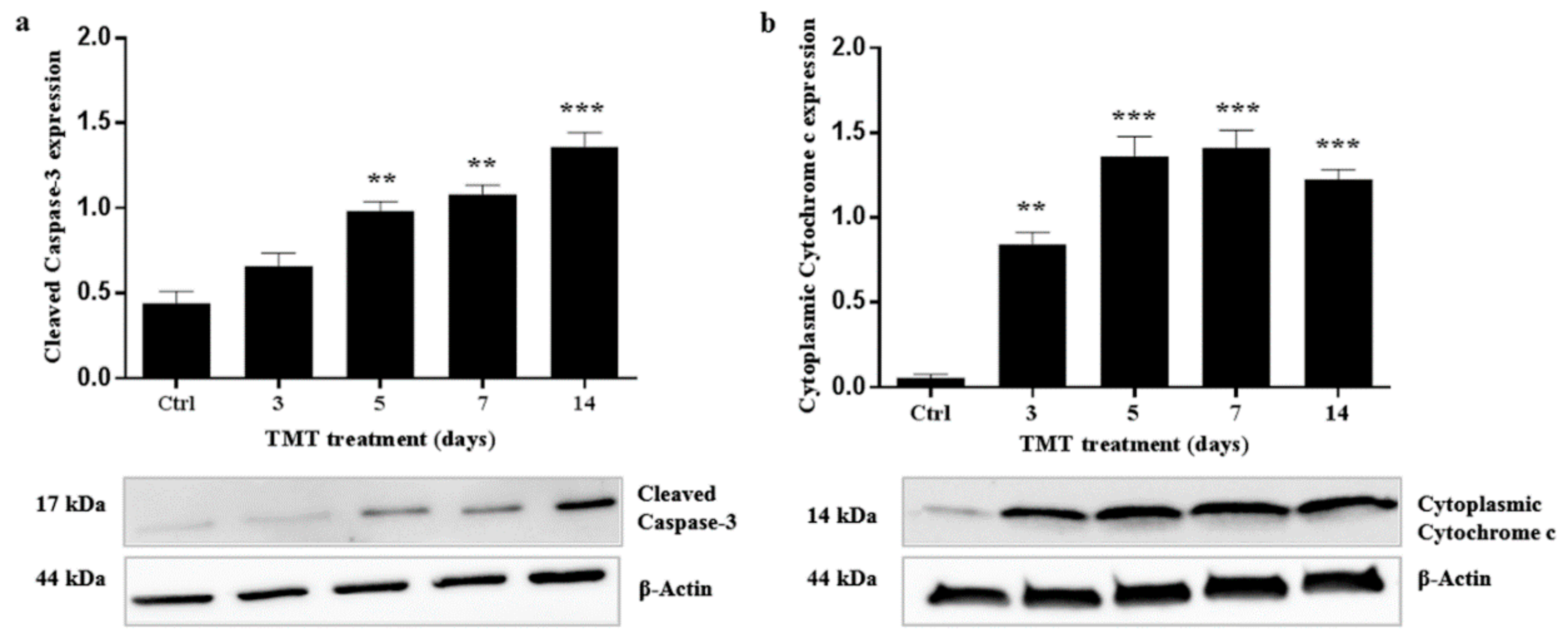
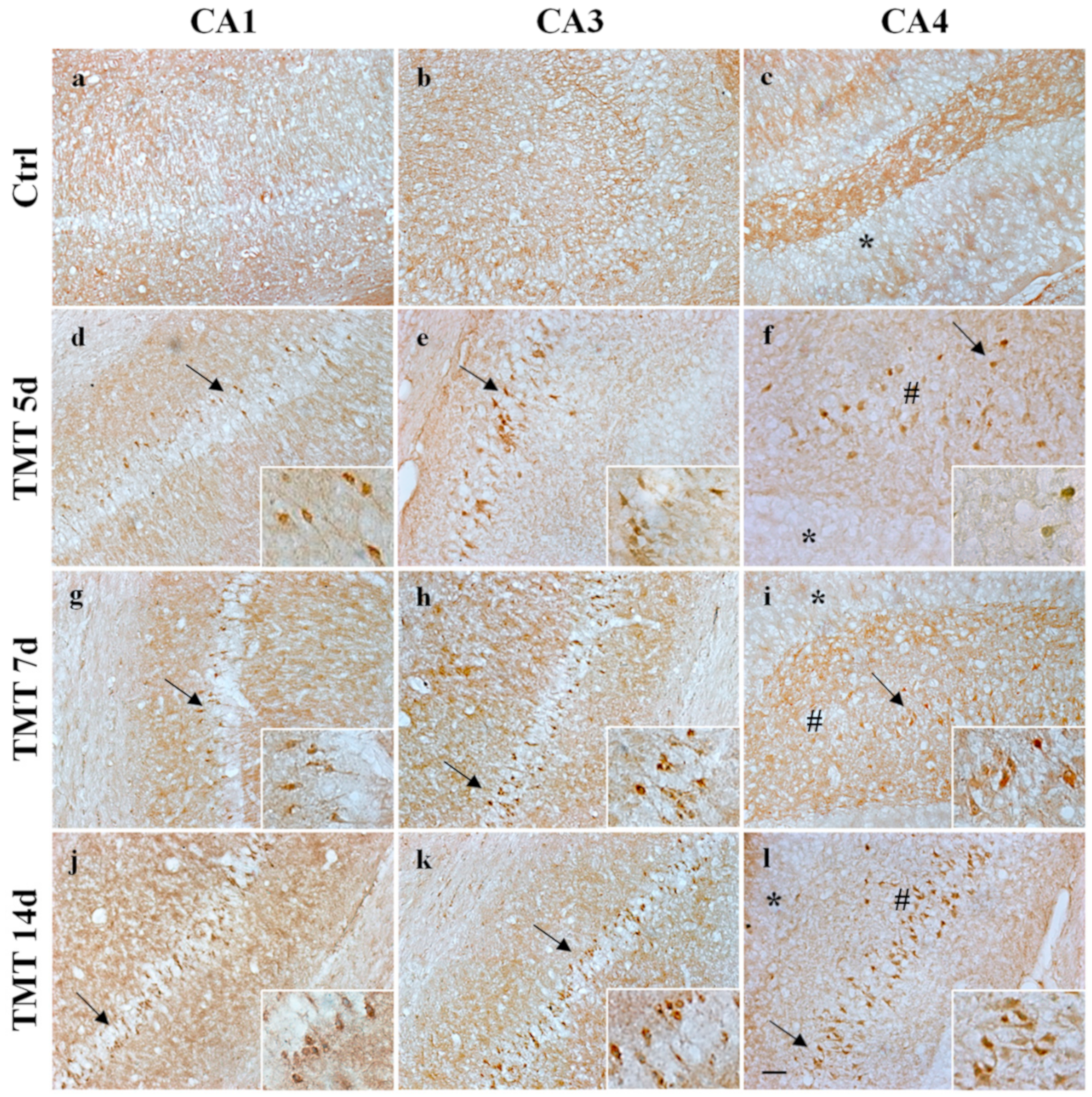
2.4. TMT Did Not Induce AIF Nuclear Translocation but Activated Caspase-3 Expression
2.5. Cytoplasmic Expression of Cytochrome c Increases after TMT-Treatment
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Western Blotting Experiments
4.3. Neuronal Death Assay: TUNEL
4.4. Immunofluorescence Localization of LC3 and AIF
4.5. Immunohistochemistry for Cleaved Caspase-3
4.6. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIF | Apoptosis-inducing factor |
| ATG | Autophagy-related |
| DAB | Diaminobenzidine |
| GFAP | Glial fibrillary astrocytic protein |
| LC3 | Microtubule-associated protein light chain |
| NeuN | Neuronal nuclear antigen |
| PBS | Phosphate buffer saline |
| PFA | Paraformaldehyde |
| PVDF | Polyvinylidene fluoride |
| RT | Room temperature |
| SDS | Sodium dodecyl sulfate |
| TBS-T | Tris buffered saline-tween |
| TMT | Trimethyltin |
| TUNEL | Terminal deoxynucleotidyltransferase dUTP nick end labeling |
References
- Dyer, R.S.; Walsh, T.J.; Wonderlin, W.F.; Bercegeay, M. The trimethyltin syndrome in rats. Neurobehav. Toxicol. Teratol. 1982, 4, 127–133. [Google Scholar] [PubMed]
- Ishida, N.; Akaike, M.; Tsutsumi, S.; Kanai, H.; Masui, A.; Sadamatsu, M.; Kuroda, Y.; Watanabe, Y.; McEwen, B.S.; Kato, N. Trimethyltin syndrome as a hippocampal degeneration model: Temporal changes and neurochemical features of seizure susceptibility and learning impairment. Neuroscience 1997, 81, 1183–1191. [Google Scholar] [CrossRef]
- Geloso, M.C.; Corvino, V.; Michetti, F. Trimethyltin-induced hippocampal degeneration as a tool to investigate neurodegenerative processes. Neurochem. Int. 2011, 58, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, R.; Gangitano, C.; Ceccariglia, S.; Del Fà, A.; Azzena, G.B.; Michetti, F.; Grassi, C. Dysregulation of intracellular calcium homeostasis is responsible for neuronal death in an experimental model of selective hippocampal degeneration induced by trimethyltin. J. Neurochem. 2008, 105, 2109–2121. [Google Scholar] [CrossRef]
- Geloso, M.C.; Vinesi, P.; Michetti, F. Parvalbumin-immunoreactive neurons are not affected by trimethyltin-induced neurodegeneration in the rat hippocampus. Exp. Neurol. 1996, 139, 269–277. [Google Scholar] [CrossRef]
- Geloso, M.C.; Vinesi, P.; Michetti, F. Calretinin-containing neurons in trimethyltin-induced neurodegeneration in the rat hippocampus: An immunocytochemical study. Exp. Neurol. 1997, 146, 67–73. [Google Scholar] [CrossRef]
- Geloso, M.C.; Vinesi, P.; Michetti, F. Neuronal subpopulations of developing rat hippocampus containing different calcium-binding proteins behave distinctively in trimethyltin- induced neurodegeneration. Exp. Neurol. 1998, 154, 645–653. [Google Scholar] [CrossRef]
- Businaro, R.; Corvino, V.; Geloso, M.C.; De Santis, E.; Fumagalli, L.; Michetti, F. De novo expression of calretinin in trimethyltin-induced degeneration of developing rat hippocampus. Mol. Brain Res. 2002, 98, 141–144. [Google Scholar] [CrossRef]
- Gangitano, C.; Falasca, C.; Del Fà, A.; Corvino, V.; Ceccariglia, S.; Zelano, G.; Geloso, M.; Monego, G.; Michetti, F. Hippocampal calretinin-containing neurons cultured in vitro are resistant to trimethyltin-induced neurodegeneration. Calcium Bind. Proteins 2006, 1, 120–124. [Google Scholar]
- Ceccariglia, S.; D’Altocolle, A.; Del Fa’, A.; Pizzolante, F.; Caccia, E.; Michetti, F.; Gangitano, C. Cathepsin D plays a crucial role in the trimethyltin-induced hippocampal neurodegeneration process. Neuroscience 2011, 174, 160–170. [Google Scholar] [CrossRef]
- Fiedorowicz, A.; Figureiel, L.; Kaminska, B.; Zaremba, M.; Wilk, S.; Oderfeld-Nowak, B. Dentate granule neuron apoptosis and glia activation in murine hippocampus induced by trimethyltin exposure. Brain Res. 2001, 912, 116–127. [Google Scholar] [CrossRef]
- Haga, S.; Haga, C.; Aizawa, T.; Ikeda, K. Neuronal degeneration and glial cell-responses following trimethyltin intoxication in the rat. Acta Neuropathol. 2002, 103, 575–582. [Google Scholar] [PubMed]
- Geloso, M.C.; Corvino, V.; Cavallo, V.; Toesca, A.; Guadagni, E.; Passalacqua, R.; Michetti, F. Expression of astrocytic nestin in the rat hippocampus during trimethyltin-induced neurodegeneration. Neurosci. Lett. 2004, 357, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Little, A.R.; Miller, D.B.; Li, S.; Kashon, M.L.; O’Callaghan, J.P. Trimethyltin-induced neurotoxicity: Gene expression pathway analysis, q-RT-PCR and immunoblotting reveal early effects associated with hippocampal damage and gliosis. Neurotoxicol. Teratol. 2012, 34, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Ceccariglia, S.; D’altocolle, A.; Del Fa’, A.; Silvestrini, A.; Barba, M.; Pizzolante, F.; Repele, A.; Michetti, F.; Gangitano, C. Increased expression of Aquaporin 4 in the rat hippocampus and cortex during trimethyltin-induced neurodegeneration. Neuroscience 2014, 274, 273–288. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimorim, T.; Levine, B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 443. [Google Scholar] [CrossRef] [Green Version]
- Kundu, M.; Thompson, C. Autophagy: Basic Principles and Relevance to Disease. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 427–455. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, N.N.; Inagaki, F. Mechanisms of Autophagy. Annu. Rev. Biophys. 2015, 44, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? Rev. Ser. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Sever, O.; Demir, O. Autophagy: Cell death or survive mechanism. J. Oncol. Sci. 2017, 3, 37–44. [Google Scholar] [CrossRef]
- Gabryel, B.; Kost, A.; Kasprowska, D. Neuronal autophagy in cerebral ischemia—A potential target for neuroprotective strategies? Pharmacol. Rep. 2012, 64, 1–15. [Google Scholar] [CrossRef]
- Chen, S.; Atkins, C.M.; Liu, C.L.; Alonso, O.F.; Dietrich, W.D.; Hu, B.R. Alterations in mammalian target of rapamycin signaling pathways after traumatic brain injury. J. Cereb. Blood Flow Metab. 2007, 27, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.L.; Chen, S.; Dietrich, D.; Hu, B.R. Changes in autophagy after traumatic brain injury. J. Cereb. Blood Flow Metab. 2008, 28, 674–683. [Google Scholar] [CrossRef]
- Li, Q.; Han, Y.; Du, J.; Jin, H.; Zhang, J.; Niu, M.; Qin, J. Alterations of apoptosis and autophagy in developing brain of rats with epilepsy: Changes in LC3, P62, Beclin-l and Bcl2 levels. Neurosci. Res. 2018, 130, 47–55. [Google Scholar] [CrossRef]
- Koike, M.; Shibata, M.; Tadakoshi, M.; Gotoh, K.; Komatsu, M.; Waguri, S.; Kawahara, N.; Kuida, K.; Nagata, S.; Kominami, E.; et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am. J. Pathol. 2008, 172, 454–469. [Google Scholar] [CrossRef] [Green Version]
- Ginet, V.; Spiehlmann, A.; Rummel, C.; Rudinskiy, N.; Grishchuk, Y.; Luthi-Carter, R.; Clarke, P.G.H.; Truttmann, A.C.; Puyal, J. Involvement of autophagy in hypoxic excitotokic.neuronal death. Autophagy 2014, 10, 846–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descloux, C.; Ginet, V.; Rummel, C.; Truttmann, A.C.; Puyal, J. Enhanced autophagy contributes to excitotoxic lesions in a rat model of preterm brain injury. Cell Death Dis. 2018, 9, 853. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Gindi, R.; Adame, A.; Wyss-coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of parkinson’s and lewy body disease. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Uchiyama, Y.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PSI Mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Hochfeld, W.E.; Lee, S.; Rubinsztein, D.C. Therapeutic induction of autophagy to modulate neurodegenerative disease progression. Acta Pharmacol. Sin. 2013, 34, 600–604. [Google Scholar] [CrossRef]
- Ruffoli, R.; Bartalucci, A.; Frati, A.; Fornai, F. Ultrastructural studies of ALS mitochondria connect altered function and permeability with defects of mitophagy and mitochondriogenesis. Front. Cell Neurosci. 2015, 9, 341. [Google Scholar] [CrossRef] [Green Version]
- Bouldin, T.W.; Goines, N.D.; Bagnell, R.C.; Krigman, M.R. Pathogenesis of trimethyltin neuronal toxicity. Ultrastructural and cytochemical observations. Am. J. Pathol. 1981, 104, 237–249. [Google Scholar]
- Fabrizi, C.; Somma, F.; Pompili, E.; Biagioni, F.; Lenzi, P.; Fornai, F.; Fumagalli, L. Role of autophagy inhibitors and inducers in modulating the toxicity of trimethyltin in neuronal cell cultures. J. Neural Transm. 2012, 119, 1295–1305. [Google Scholar] [CrossRef]
- Fabrizi, C.; Pompili, E.; De Vito, S.; Somma, F.; Catizone, A.; Ricci, G.; Lenzi, P.; Fornai, F.; Fumagalli, L. Impairment of the autophagic flux in astrocytes intoxicated by trimethyltin. Neurotoxicology 2016, 52, 12–22. [Google Scholar] [CrossRef]
- Kane, M.D.; Yang, C.W.; Gunasekar, P.G.; Isom, G.E. Trimethyltin Stimulates Protein Kinase C Translocation Through Receptor-Mediated Phospholipase C Activation in PC12 Cells. J. Neurochem. 1998, 70, 509–514. [Google Scholar] [CrossRef]
- Gunasekar, P.; Li, L.; Prabhakaran, K.; Eybl, V.; Borowitz, J.; Isom, G. Mechanisms of the apoptotic and necrotic actions of trimethyltin in cerebellar granule cells. Toxicol. Sci. 2001, 64, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geloso, M.C.; Vercelli, A.; Corvino, V.; Repici, M.; Boca, M.; Haglid, K.; Zelano, G.; Michetti, F. Cyclooxygenase-2 and caspase 3 expression in trimethyltin-induced apoptosis in the mouse hippocampus. Exp. Neurol. 2002, 175, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.M.; Barone, S. The neurotoxicant trimethyltin induces apoptosis via caspase activation, p38 protein kinase, and oxidative stress in PC 12 cells. Toxicol. Lett. 2004, 147, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Buck-Koehntop, B.A.; Mascioni, A.; Buffy, J.J.; Veglia, G. Structure, dynamics, and membrane topology of stannin: A mediator of neuronal cell apoptosis induced by trimethyltin chloride. J. Mol. Biol. 2005, 354, 652–665. [Google Scholar] [CrossRef]
- Yuliani, S.; Widyarini, S.; Mustofa; Partadiredja, G. Turmeric extract inhibits apoptosis of hippocampal neurons of trimethyltin-exposed rats. Bratisl. Med. J. 2017, 118, 142–148. [Google Scholar] [CrossRef]
- Gavrieli, Y.; Sherman, Y.; Ben-Sasson, S.A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 1992, 119, 493–501. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011, 134, 258–277. [Google Scholar] [CrossRef]
- Klionsky, D.; Abeliovich, A.; Agostinis, P.; Al, E. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. P62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knævelsrud, H.; Simonsen, A. Figurehting disease by selective autophagy of aggregate-prone proteins. FEBS Lett. 2010, 584, 2635–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Ichimura, Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010, 584, 1374–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between Autophagy Receptors and Ubiquitin-like Proteins Form the Molecular Basis for Selective Autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.B.; Li, S.X.; Chen, X.P.; Yang, L.; Zhang, Y.G.; Liu, R.; Tao, L.Y. Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci. Bull. 2008, 24, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.L.; Li, B.X.; Li, Q.Q.; Chen, X.P.; Sun, Y.X.; Bao, H.J.; Dai, D.K.; Shen, Y.W.; Xu, H.F.; Ni, H.; et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience 2011, 184, 54–63. [Google Scholar] [CrossRef]
- Shi, R.; Weng, J.; Zhao, L.; Li, X.M.; Gao, T.M.; Kong, J. Excessive Autophagy Contributes to Neuron Death in Cerebral Ischemia. CNS Neurosci. Ther. 2012, 18, 250–260. [Google Scholar] [CrossRef]
- Guo, D.; Ma, J.; Yan, L.; Li, T.; Li, Z.; Han, X.; Shui, S. Down-Regulation of Lncrna MALATI Attenuates Neuronal Cell Death Through Suppressing Beclin l-Dependent Autophagy by Regulating Mir-30a in Cerebral Ischemic Stroke. Cell. Physiol. Biochem. 2017, 43, 182–194. [Google Scholar] [CrossRef]
- Grishchuk, Y.; Ginet, V.; Truttmann, A.C.; Clarke, P.G.H.; Puyal, J. Beclin I-independent autophagy contributes to apoptosis in cortical neurons. Autophagy 2011, 7, 1115–1131. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Jacob, A.; Freiberger, S.; Hentschel, K.; Stierhof, Y.D.; Codogno, P.; Proikas-Cezanne, T. Resveratrol-mediated autophagy requires WIPI-1-regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy 2011, 7, 1448–1461. [Google Scholar] [CrossRef]
- Seo, G.; Kim, S.K.; Byun, Y.J.; Oh, E.; Jeong, S.W.; Chae, G.T.; Lee, S.B. Hydrogen peroxide induces Beclin 1-independent autophagic cell death by suppressing the mTOR pathway via promoting the ubiquitination and degradation of Rheb in GSH-depleted RAW 264.7 cells. Free Rad. Res. 2011, 45, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Horbinski, C.; Guo, F.; Watkins, S.; Uchiyama, Y.; Chu, C.T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol. 2007, 170, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Yang, M.; Kim, J.; Kang, S.; Kim, J.; Kim, J.C.; Jung, C.; Shin, T.; Kim, S.H.; Moon, C. Trimethyltin-induced hippocampal neurodegeneration: A mechanism-based review. Brain Res. Bull. 2016, 125, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Prabhakaran, K.; Borowitz, J.L.; Isom, G.E. Trimethyltin-induced apoptosis is associated with upregulation of inducible nitric oxide synthase and Bax in a hippocampal cell line. Toxicol. Appl. Pharmacol. 2006, 216, 34–43. [Google Scholar] [CrossRef]
- Morita, M.; Imai, H.; Liu, Y.; Xu, X.; Sadamatsu, M.; Nakagami, R.; Shirakawa, T.; Nakano, K.; Kita, Y.; Yoshida, K.; et al. FK506-protective effects against trimethyltin neurotoxicity in rats: Hippocampal expression analyses reveal the involvement of periarterial osteopontin. Neuroscience 2008, 153, 1135–1145. [Google Scholar] [CrossRef]
- Nilsberth, C.; Kostyszyn, B.; Luthman, J. Changes in APP, PS1 and other factors related to Alzheimer’s disease pathophysiology after trimethyltin-induced brain lesion in the rat. Neurotox. Res. 2002, 4, 625–636. [Google Scholar] [CrossRef]
- Joza, N.; Susin, S.A.; Daugas, E.; Stanford, W.L.; Cho, S.K.; Li, C.Y.J.; Sasaki, T.; Elia, A.; Al, E. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001, 410, 549–554. [Google Scholar] [CrossRef]
- Krantic, S.; Mechawar, N.; Reix, S.; Quirion, R. Apoptosis-inducing factor: A matter of neuron life and death. Prog. Neurobiol. 2007, 81, 179–196. [Google Scholar] [CrossRef]
- Vahsen, N.; Candé, C.; Brière, J.J.; Bénit, P.; Joza, N.; Larochette, N.; Mastroberardino, P.G.; Pequignot, M.O.; Casares, N.; Lazar, V.; et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004, 23, 4679–4689. [Google Scholar] [CrossRef] [Green Version]
- Susin, S.A.; Daugas, E.; Ravagnan, L.; Samejima, K.; Zamzami, N.; Loeffler, M.; Costantini, P.; Ferri, K.F.; Irinopoulou, T.; Prévost, M.-C.; et al. Two Distinct Pathways Leading to Nuclear Apoptosis. J. Exp. Med. 2000, 192, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Ohsakaya, S.; Nagaura, Z.I.; Ishihara, N.; Mihara, K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 2005, 24, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignotte, B.; Vayssiere, J. Review Mitochondria and apoptosis. Eur. J. Biochem. 1998, 252, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.G.; Janicke, R.U. Emerging roles of Caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Gasparova, Z.; Janega, P.; Stara, V.; Ujhazy, E. Early and late stage of neurodegeneration induced by trimethyltin in hippocampus and cortex of male Wistar rats. Neuro Endocrinol. Lett. 2012, 33, 689–696. [Google Scholar]
- Deniaud, A.; Sharaf El Dein, O.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Qu, M.; Zhou, Z.; Chen, C.; Li, M.; Pei, L.; Chu, F.; Yang, J.; Wang, Y.; Li, L.; Liu, C.; et al. Lycopene protects against trimethyltin-induced neurotoxicity in primary cultured rat hippocampal neurons by inhibiting the mitochondrial apoptotic pathway. Neurochem. Int. 2011, 59, 1095–1103. [Google Scholar] [CrossRef]
- Misiti, F.; Orsini, F.; Clementi, M.E.; Lattanzi, W.; Giardina, B.; Michetti, F. Mitochondrial oxygen consumption inhibition importance for TMT-dependent cell death in undifferentiated PC12 cells. Neurochem. Int. 2008, 52, 1092–1099. [Google Scholar] [CrossRef]
- Morita-Fujimura, Y.; Fujimura, M.; Kawase, M.; Chen, S.F.; Chan, P.H. Release of mitochondrial cytochrome c and DNA fragmentation after cold injury-induced brain trauma in mice: Possible role in neuronal apoptosis. Neurosci. Lett. 1999, 267, 201–205. [Google Scholar] [CrossRef]
- Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceccariglia, S.; Alvino, A.; Del Fà, A.; Parolini, O.; Michetti, F.; Gangitano, C. Autophagy is Activated In Vivo during Trimethyltin-Induced Apoptotic Neurodegeneration: A Study in the Rat Hippocampus. Int. J. Mol. Sci. 2020, 21, 175. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010175
Ceccariglia S, Alvino A, Del Fà A, Parolini O, Michetti F, Gangitano C. Autophagy is Activated In Vivo during Trimethyltin-Induced Apoptotic Neurodegeneration: A Study in the Rat Hippocampus. International Journal of Molecular Sciences. 2020; 21(1):175. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010175
Chicago/Turabian StyleCeccariglia, Sabrina, Alessandra Alvino, Aurora Del Fà, Ornella Parolini, Fabrizio Michetti, and Carlo Gangitano. 2020. "Autophagy is Activated In Vivo during Trimethyltin-Induced Apoptotic Neurodegeneration: A Study in the Rat Hippocampus" International Journal of Molecular Sciences 21, no. 1: 175. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010175