Clinical and Genetic Analysis of a European Cohort with Pericentral Retinitis Pigmentosa

 and
and
Abstract
:1. Introduction
2. Results
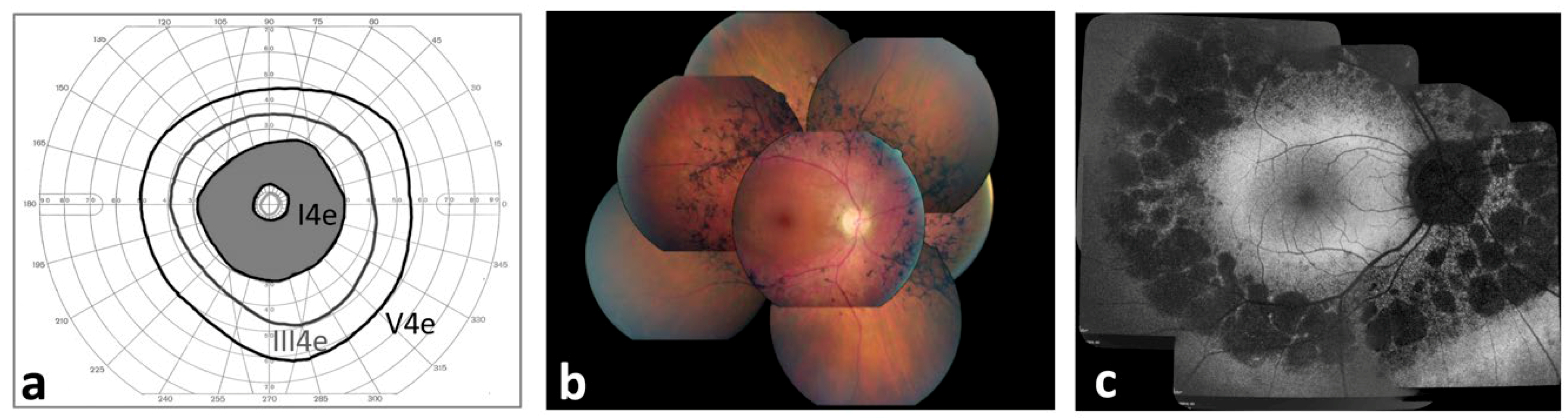
2.1. Ophthalmological Characterization of Pericentral Retinitis Pigmentosa (RP) Patients
2.2. Genetic Analysis
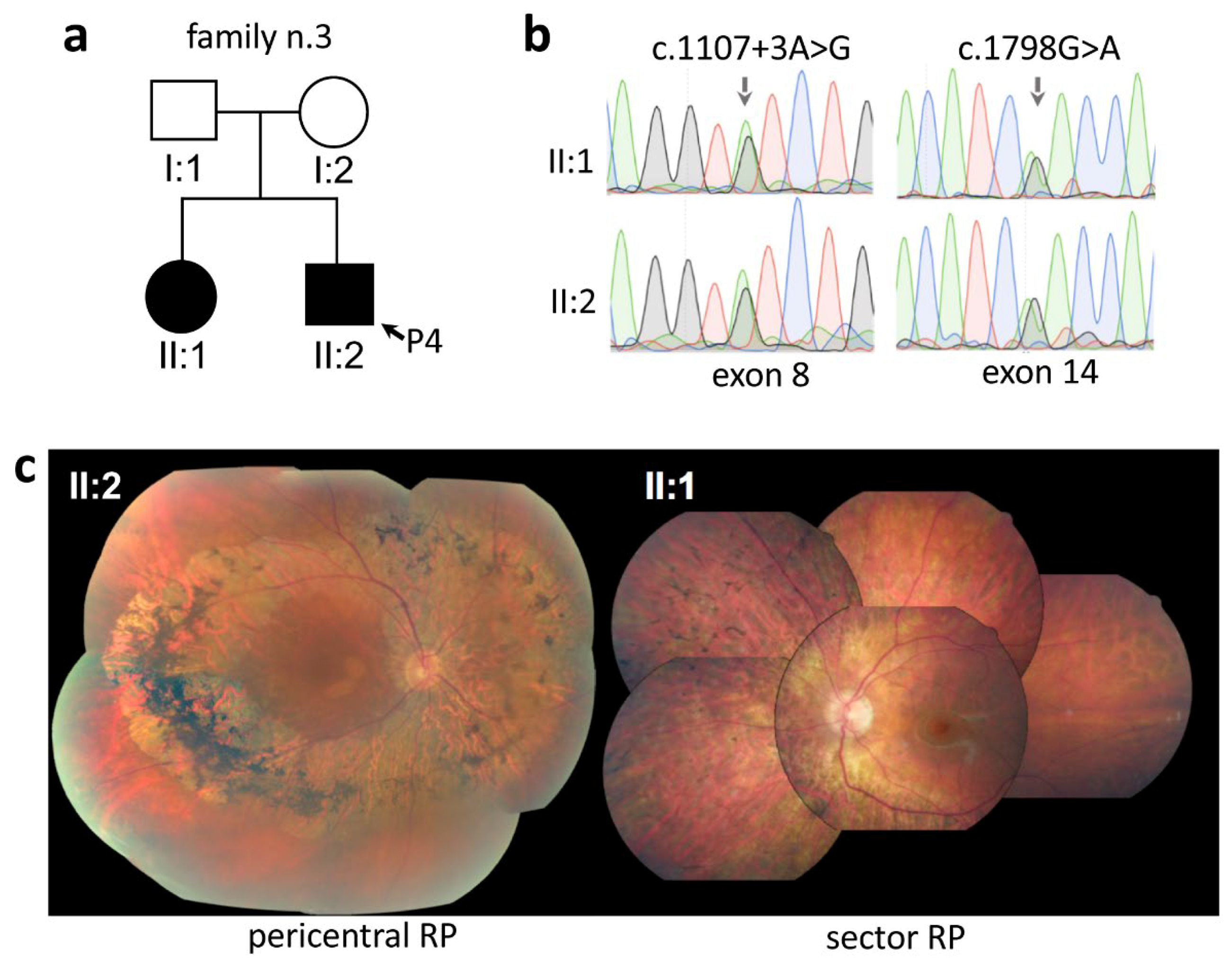
2.3. Intrafamilial Variability of Pericentral RP
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Patients Inclusion Criteria
4.3. Ophthalmological Examination
4.4. Next-Generation Sequencing
4.5. Variant Analysis and Interpretation
4.6. Variant Validation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BCVA | Best corrected visual acuity |
| ERG | Electroretinogram |
| HGMD | Human Gene Mutation Database |
| LOVD | Leiden Open Variation Database |
| NGS | Next generation sequencing |
| OCT | Optical coherence tomography |
| RP | Retinitis pigmentosa |
References
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Wright, A.F.; Chakarova, C.F.; Abd El-Aziz, M.M.; Bhattacharya, S.S. Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010, 11, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.A.; Gaudio, A.R.; Berson, E.L. Disease course of patients with pericentral retinitis pigmentosa. Am. J. Ophthalmol. 2005, 140, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Comander, J.; Weigel-DiFranco, C.; Maher, M.; Place, E.; Wan, A.; Harper, S.; Sandberg, M.A.; Navarro-Gomez, D.; Pierce, E.A. The Genetic Basis of Pericentral Retinitis Pigmentosa-A Form of Mild Retinitis Pigmentosa. Genes (Basel) 2017, 8, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, R.; Cideciyan, A.V.; Schwartz, S.B.; Sumaroka, A.; Roman, A.J.; Swider, M.; Huang, W.C.; Sheplock, R.; Jacobson, S.G. Molecular heterogeneity within the clinical diagnosis of pericentral retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6007–6018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat 2012, 33, 963–972. [Google Scholar] [CrossRef]
- Tsang, S.H.; Tsui, I.; Chou, C.L.; Zernant, J.; Haamer, E.; Iranmanesh, R.; Tosi, J.; Allikmets, R. A novel mutation and phenotypes in phosphodiesterase 6 deficiency. Am. J. Ophthalmol. 2008, 146, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Shevach, E.; Ali, M.; Mizrahi-Meissonnier, L.; McKibbin, M.; El-Asrag, M.; Watson, C.M.; Inglehearn, C.F.; Ben-Yosef, T.; Blumenfeld, A.; Jalas, C.; et al. Association between missense mutations in the BBS2 gene and nonsyndromic retinitis pigmentosa. JAMA Ophthalmol. 2015, 133, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Baux, D.; Blanchet, C.; Hamel, C.; Meunier, I.; Larrieu, L.; Faugere, V.; Vache, C.; Castorina, P.; Puech, B.; Bonneau, D.; et al. Enrichment of LOVD-USHbases with 152 USH2A genotypes defines an extensive mutational spectrum and highlights missense hotspots. Hum. Mutat. 2014, 35, 1179–1186. [Google Scholar] [CrossRef]
- Herrera, W.; Aleman, T.S.; Cideciyan, A.V.; Roman, A.J.; Banin, E.; Ben-Yosef, T.; Gardner, L.M.; Sumaroka, A.; Windsor, E.A.; Schwartz, S.B.; et al. Retinal disease in Usher syndrome III caused by mutations in the clarin-1 gene. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2651–2660. [Google Scholar] [CrossRef]
- Pennings, R.J.; Te Brinke, H.; Weston, M.D.; Claassen, A.; Orten, D.J.; Weekamp, H.; Van Aarem, A.; Huygen, P.L.; Deutman, A.F.; Hoefsloot, L.H.; et al. USH2A mutation analysis in 70 Dutch families with Usher syndrome type II. Hum. Mutat. 2004, 24, 185. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Casteels, I.; Meire, F.; De Jaegere, S.; Hooghe, S.; van Regemorter, N.; Van Esch, H.; Matuleviciene, A.; Nunes, L.; Meersschaut, V.; et al. Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum. Mutat. 2010, 31, E1709–E1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, C.; Riahi, Z.; Chantot-Bastaraud, S.; Smagghe, L.; Letexier, M.; Marcaillou, C.; Lefevre, G.M.; Hardelin, J.P.; El-Amraoui, A.; Singh-Estivalet, A.; et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 2016, 24, 1730–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aller, E.; Jaijo, T.; Beneyto, M.; Najera, C.; Oltra, S.; Ayuso, C.; Baiget, M.; Carballo, M.; Antinolo, G.; Valverde, D.; et al. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J. Med. Genet. 2006, 43, e55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosewich, H.; Ohlenbusch, A.; Gartner, J. Genetic and clinical aspects of Zellweger spectrum patients with PEX1 mutations. J. Med. Genet. 2005, 42, e58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glockle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Krawitz, P.M.; Schiska, D.; Kruger, U.; Appelt, S.; Heinrich, V.; Parkhomchuk, D.; Timmermann, B.; Millan, J.M.; Robinson, P.N.; Mundlos, S.; et al. Screening for single nucleotide variants, small indels and exon deletions with a next-generation sequencing based gene panel approach for Usher syndrome. Mol. Genet. Genomic Med. 2014, 2, 393–401. [Google Scholar] [CrossRef]
- Sodi, A.; Mariottini, A.; Passerini, I.; Murro, V.; Tachyla, I.; Bianchi, B.; Menchini, U.; Torricelli, F. MYO7A and USH2A gene sequence variants in Italian patients with Usher syndrome. Mol. Vis. 2014, 20, 1717–1731. [Google Scholar]
- Haider, N.B.; Jacobson, S.G.; Cideciyan, A.V.; Swiderski, R.; Streb, L.M.; Searby, C.; Beck, G.; Hockey, R.; Hanna, D.B.; Gorman, S.; et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000, 24, 127–131. [Google Scholar] [CrossRef]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef] [Green Version]
- Testa, F.; Rossi, S.; Colucci, R.; Gallo, B.; Di Iorio, V.; della Corte, M.; Azzolini, C.; Melillo, P.; Simonelli, F. Macular abnormalities in Italian patients with retinitis pigmentosa. Br. J. Ophthalmol. 2014, 98, 946–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grondahl, J.; Riise, R.; Heiberg, A.; Leren, T.; Christoffersen, T.; Bragadottir, R. Autosomal dominant retinitis pigmentosa in Norway: A 20-year clinical follow-up study with molecular genetic analysis. Two novel rhodopsin mutations: 1003delG and I179F. Acta Ophthalmol. Scand. 2007, 85, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Selmer, K.K.; Grondahl, J.; Riise, R.; Brandal, K.; Braaten, O.; Bragadottir, R.; Undlien, D.E. Autosomal dominant pericentral retinal dystrophy caused by a novel missense mutation in the TOPORS gene. Acta Ophthalmol. 2010, 88, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L.; Rosner, B.; Weigel-DiFranco, C.; Dryja, T.P.; Sandberg, M.A. Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3027–3036. [Google Scholar]
- Haer-Wigman, L.; Newman, H.; Leibu, R.; Bax, N.M.; Baris, H.N.; Rizel, L.; Banin, E.; Massarweh, A.; Roosing, S.; Lefeber, D.J.; et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum. Mol. Genet. 2015, 24, 3742–3751. [Google Scholar] [CrossRef]
- Sandberg, M.A.; Rosner, B.; Weigel-DiFranco, C.; McGee, T.L.; Dryja, T.P.; Berson, E.L. Disease Course in Patients with Autosomal Recessive Retinitis Pigmentosa due to the USH2A Gene. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5532–5539. [Google Scholar] [CrossRef] [Green Version]
- Grover, S.; Fishman, G.A.; Alexander, K.R.; Anderson, R.J.; Derlacki, D.J. Visual acuity impairment in patients with retinitis pigmentosa. Ophthalmology 1996, 103, 1593–1600. [Google Scholar] [CrossRef]
- Grover, S.; Fishman, G.A.; Anderson, R.J.; Tozatti, M.S.; Heckenlively, J.R.; Weleber, R.G.; Edwards, A.O.; Brown, J., Jr. Visual acuity impairment in patients with retinitis pigmentosa at age 45 years or older. Ophthalmology 1999, 106, 1780–1785. [Google Scholar] [CrossRef]
- Berson, E.L.; Sandberg, M.A.; Rosner, B.; Birch, D.G.; Hanson, A.H. Natural course of retinitis pigmentosa over a three-year interval. Am. J. Ophthalmol. 1985, 99, 240–251. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Hayes, K.C.; Nicholson, B.W.; Weigel-DiFranco, C.; Willett, W. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch. Ophthalmol. 1993, 111, 761–772. [Google Scholar] [CrossRef]
- Sandberg, M.A.; Rosner, B.; Weigel-DiFranco, C.; Dryja, T.P.; Berson, E.L. Disease Course of Patients with X-linked Retinitis Pigmentosa due to RPGR Gene Mutations. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1298–1304. [Google Scholar] [CrossRef] [Green Version]
- Popovic, P.; Jarc-Vidmar, M.; Hawlina, M. Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefes Arch. Clin. Exp. Ophthalmol. 2005, 243, 1018–1027. [Google Scholar] [CrossRef]
- Sujirakul, T.; Lin, M.K.; Duong, J.; Wei, Y.; Lopez-Pintado, S.; Tsang, S.H. Multimodal Imaging of Central Retinal Disease Progression in a 2-Year Mean Follow-up of Retinitis Pigmentosa. Am. J. Ophthalmol. 2015, 160, 786–798.e784. [Google Scholar] [CrossRef] [Green Version]
- Burnight, E.R.; Wiley, L.A.; Drack, A.V.; Braun, T.A.; Anfinson, K.R.; Kaalberg, E.E.; Halder, J.A.; Affatigato, L.M.; Mullins, R.F.; Stone, E.M.; et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. 2014, 21, 662–672. [Google Scholar] [CrossRef] [Green Version]
- Chacon-Camacho, O.F.; Zenteno, J.C. Review and update on the molecular basis of Leber congenital amaurosis. World J. Clin. Cases 2015, 3, 112–124. [Google Scholar] [CrossRef]
- Nair, A.A.; Marmor, M.F. ERG and other discriminators between advanced hydroxychloroquine retinopathy and retinitis pigmentosa. Doc. Ophthalmol. 2017, 134, 175–183. [Google Scholar] [CrossRef]
- Iannaccone, A.; Kritchevsky, S.B.; Ciccarelli, M.L.; Tedesco, S.A.; Macaluso, C.; Kimberling, W.J.; Somes, G.W. Kinetics of visual field loss in Usher syndrome Type II. Investig. Ophthalmol. Vis. Sci. 2004, 45, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Aboshiha, J.; Dubis, A.M.; Cowing, J.; Fahy, R.T.; Sundaram, V.; Bainbridge, J.W.; Ali, R.R.; Dubra, A.; Nardini, M.; Webster, A.R.; et al. A prospective longitudinal study of retinal structure and function in achromatopsia. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5733–5743. [Google Scholar] [CrossRef] [Green Version]
- Fakin, A.; Jarc-Vidmar, M.; Glavac, D.; Bonnet, C.; Petit, C.; Hawlina, M. Fundus autofluorescence and optical coherence tomography in relation to visual function in Usher syndrome type 1 and 2. Vis. Res. 2012, 75, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Musacchia, F.; Ciolfi, A.; Mutarelli, M.; Bruselles, A.; Castello, R.; Pinelli, M.; Basu, S.; Banfi, S.; Casari, G.; Tartaglia, M.; et al. VarGenius executes cohort-level DNA-seq variant calling and annotation and allows to manage the resulting data through a PostgreSQL database. BMC Bioinform. 2018, 19, 477. [Google Scholar] [CrossRef]
- Di Iorio, V.; Karali, M.; Brunetti-Pierri, R.; Filippelli, M.; Di Fruscio, G.; Pizzo, M.; Mutarelli, M.; Nigro, V.; Testa, F.; Banfi, S.; et al. Clinical and Genetic Evaluation of a Cohort of Pediatric Patients with Severe Inherited Retinal Dystrophies. Genes (Basel) 2017, 8, 280. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Family | Gene Mutation | Patient | Age of Onset (yrs) | Age (yrs) | BCVA RE | BCVA LE | V4e Field Description RE | V4e Field Description LE |
|---|---|---|---|---|---|---|---|---|
| F1 | PRPF31 | P1 | 8 | 15 | 1 | 1 | pericentral field loss | pericentral field loss |
| P2 | 12 | 49 | 1 | 1 | constricted with peripheral scotoma | constricted with peripheral scotoma | ||
| F2 | P3 | 30 | 55 | 0.8 | 1 | constricted with ring scotoma | constricted with ring scotoma | |
| F3 | PDE6B | P4 | 49 | 52 | 0.9 | 0.9 | constricted with ring scotoma | constricted with ring scotoma |
| F4 | BBS2 | P5 | 15 | 20 | 1 | 1 | pericentral field loss | pericentral field loss |
| F5 | P6 | 50 | 84 | 0.2 | 0.4 | n.a. | n.a. | |
| F6 | P7 | 31 | 51 | 1 | 1 | pericentral field loss | pericentral field loss | |
| P8 | 25 | 55 | 0.7 | 0.9 | constricted with ring scotoma | constricted with ring scotoma | ||
| F7 | RP1 | P9 | 47 | 52 | 1 | 1 | constricted with ring scotoma | constricted with ring scotoma |
| F8 | USH2A | P10 | 11 | 47 | 1 | 1 | constricted with ring scotoma | constricted with ring scotoma |
| P11 | 38 | 58 | 0.7 | 0.6 | constricted with ring scotoma | constricted with ring scotoma | ||
| F9 | USH2A | P12 | 14 | 40 | 0.8 | 0.9 | concentric constricted | concentric constricted |
| F10 | CEP290 | P13 | 5 | 34 | 1 | 1 | constricted with ring scotoma | constricted with ring scotoma |
| F11 | USH2A | P14 | 20 | 44 | 0.9 | 0.9 | constricted with peripheral scotoma | constricted with peripheral scotoma |
| F12 | PRPF31 | P15 | 4 | 22 | 1 | 1 | pericentral field loss | pericentral field loss |
| P16 | 5 | 27 | 1 | 1 | constricted with peripheral scotoma | concentric constricted | ||
| F13 | USH2A | P17 | 11 | 44 | 0.7 | 0.5 | concentric constricted | concentric constricted |
| F14 | PRPH2 | P18 | 27 | 45 | 1 | 0.8 | constricted with ring scotoma | constricted with ring scotoma |
| F15 | P19 | 57 | 76 | 0.0016 | 0.0016 | n.a. | n.a. | |
| F16 | P20 | 47 | 59 | 1 | 1 | constricted with ring scotoma | constricted with ring scotoma | |
| F17 | P21 | 9 | 27 | 0.1 | 0.1 | pericentral field loss | pericentral field loss | |
| F18 | P22 | 36 | 47 | 0.8 | 1 | constricted with ring scotoma | constricted with ring scotoma | |
| F19 | P23 | 30 | 53 | 1 | 0.9 | constricted with peripheral scotoma | constricted with peripheral scotoma | |
| F20 | CEP290 | P24 | 12 | 19 | 1 | 1 | pericentral field loss | constricted with peripheral scotoma |
| F21 | P25 | 41 | 43 | 1 | 1 | pericentral field loss | pericentral field loss | |
| F22 | RHO | P26 | 9 | 14 | 1 | 1 | pericentral field loss | pericentral field loss |
| F23 | PEX1 | P27 | 10 | 11 | 0.3 | 0.3 | pericentral field loss | pericentral field loss |
| F24 | P28 | 34 | 50 | 0.2 | 0.1 | n.a. | n.a. | |
| F25 | USH2A | P29 | 43 | 49 | 0.1 | 0.7 | n.a. | n.a. |
| F26 | USH2A | P30 | 15 | 56 | 0.8 | 0.4 | pericentral field loss | pericentral field loss |
| F27 | P31 | 53 | 59 | 0.6 | 1 | constricted with ring scotoma | constricted with ring scotoma | |
| F28 | USH2A | P32 | 42 | 48 | 0.7 | 0.8 | pericentral field loss | pericentral field loss |
| F29 | P33 | 13 | 20 | 0.9 | 0.6 | pericentral field loss | pericentral field loss | |
| F30 | P34 | 5 | 58 | 0.8 | 0.8 | concentric constricted | concentric constricted | |
| F31 | PDE6B | P35 | 19 | 41 | 0.8 | 1 | constricted with ring scotoma | constricted with ring scotoma |
| P36 | 15 | 43 | 0.05 | 0.05 | constricted with ring scotoma | constricted with ring scotoma | ||
| F32 | NR2E3 | P37 | 0.67 | 22 | 0.6 | 0.9 | pericentral field loss | pericentral field loss |
| P38 | 3 | 27 | 0.6 | 0.6 | pericentral field loss | pericentral field loss | ||
| F33 | USH2A | P39 | 40 | 69 | 0.6 | 0.8 | central scotoma | central scotoma |
| F34 | P40 | 49 | 58 | 0.7 | 0.9 | pericentral field loss | pericentral field loss | |
| F35 | P41 | 45 | 64 | 0.6 | 0.5 | concentric constricted | concentric constricted | |
| F36 | P42 | 5 | 40 | 0.7 | 0.7 | pericentral field loss | pericentral field loss | |
| F37 | P43 | 50 | 78 | 0.1 | 0.05 | concentric constricted | concentric constricted | |
| F38 | P44 | 55 | 60 | 0.9 | 1 | pericentral field loss | pericentral field loss | |
| F39 | P45 | 27 | 71 | 1 | 0.4 | n.a. | n.a. | |
| F40 | RP1 | P46 | 38 | 42 | 0.9 | 0.6 | pericentral field loss | pericentral field loss |
| F41 | USH2A | P47 | 24 | 45 | 0.6 | 0.6 | concentric constricted | concentric constricted |
| F42 | P48 | 38 | 44 | 1 | 0.9 | constricted with ring scotoma | constricted with ring scotoma | |
| F43 | P49 | 14 | 68 | 1 | 1 | pericentral field loss | pericentral field loss | |
| F44 | P50 | 64 | 80 | 0.8 | 0.8 | concentric constricted | concentric constricted | |
| F45 | P51 | 6 | 92 | 0.3 | 0.4 | constricted with peripheral scotoma | constricted with ring scotoma | |
| F46 | P52 | 50 | 77 | 0.6 | 0.5 | pericentral field loss | pericentral field loss | |
| F47 | P53 | 18 | 39 | 0.8 | 0.7 | pericentral field loss | pericentral field loss | |
| F48 | P54 | 15 | 33 | 0.9 | 0.9 | pericentral field loss | pericentral field loss |
| Patient | Family | Gene | RefSeq | Allele 1 | Allele 2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Protein | Reference | Nucleotide | Protein | Reference | |||||
| P1 | F1 | PRPF31 | NM_015629 | c.3G > A | p.(Met1Ile) | this study | - | - | - | |
| P4 | F3 | PDE6B | NM_000283 | c.1107 + 3A > G | p.(?) | [6] | c.1798G > A | p.(Asp600Asn) | [7] | |
| P5 | F4 | BBS2 | NM_031885.3 | c.401C > G | p.(Pro134Arg) | [8] | c.401C > G | p.(Pro134Arg) | [8] | |
| P9 | F7 | RP1 | NM_006269 | c.2219C > G | p.(Ser740 * ) | this study | - | - | - | |
| P46 | F40 | RP1 | NM_006269 | c.2978delC | p.(Ser993Phefs * 20) | this study | - | - | - | |
| P10 | F8 | USH2A | NM_206933 | c.497A > G | p.(Glu166Gly) | this study | c.14977_14978del | p.(Phe4993Profs * 7) | [9] | |
| P12 | F9 | USH2A | NM_206933 | c.9815C > T | p.(Pro3272Leu) | [10] | c.949C > A | p.(Arg317Arg) | [11] | |
| P13 | F10 | CEP290 | NM_025114 | c.5709 + 2T > G | p.(?) | this study | c.384_385del | p.(Asp128Glufs * 17) | [12] | |
| P14 | F11 | USH2A | NM_206933 | c.4732C > T | p.(Arg1578Cys) | [9] | c.14885dup | p.(Glu4963Glyfs * 38) | [13] | |
| P15 | F12 | PRPF31 | NM_015629 | c.690delG | p.(Ile231Serfs*8) | this study | - | - | - | |
| P17 | F13 | USH2A | NM_206933 | c.4717C > T | p.(Gln1573*) | [13] | c.10712C > T | p.(Thr3571Met) | [14] | |
| P18 | F14 | PRPH2 | NM_000322 | c.458A > G | p.(Lys153Arg) | LOVD ‡ | - | - | - | |
| P24 | F20 | CEP290 | NM_025114 | c.1664A > T | p.(Lys555Ile) | this study | c.1092T > G | p.(Ile364Met) | this study | |
| P26 | F22 | RHO | NM_000539 | c.560G > T | p.(Cys187Phe) | this study | - | - | - | |
| P27 | F23 | PEX1 | NM_000466 | c.274G > C | p.(Val92Leu) | [15] | c.2145_2146insTCTCAG | p.(Gln716delinsSerGlnGln) | this study | |
| P29 | F25 | USH2A | NM_206933 | c.3045C > G | p.(His1015Gln) | this study | c.6992G > A | p.(Gly2331Glu) | this study | |
| P30 | F26 | USH2A | NM_206933 | c.2296T > C | p.(Cys766Arg) | [16] | c.2296T > C | p.(Cys766Arg) | [16] | |
| P32 | F28 | USH2A | NM_206933 | c.11713C > T | p.(Arg3905Cys) | [17] | c.9959-1G > C | p.(?) | [18] | |
| P35 | F31 | PDE6B | NM_000283 | c.1798G > A | p.(Asp600Asn) | [7] | c.1798G > A | p.(Asp600Asn) | [7] | |
| P37 | F32 | NR2E3 | NM_014249 | c.119-2A > C | p.(?) | [19] | c.119-2A > C | p.(?) | [19] | |
| P39 | F33 | USH2A | NM_206933 | c.2276G > T | p.(Cys759Phe) | [20] | c.3684T > A | p.(Cys1228 *) | [13] | |
| P47 | F41 | USH2A | NM_206933 | c.953A > G | p.(Tyr318Cys) | LOVD ‡ | c.5776 + 1G > C | p.(?) | this study | |
| Gene | RefSeq | Nucleotide | Protein | In Silico Pathogenicity Analysis | |||
|---|---|---|---|---|---|---|---|
| MutationTaster † | PolyPhen-2 ‡ | SIFT * | Cadd13 # | ||||
| CEP290 | NM_025114 | c.1664A > T | p.(Lys555Ile) | LP | LP | P | 27.8 |
| CEP290 | NM_025114 | c.1092T > G | p.(Ile364Met) | LP | P | P | 25.3 |
| PRPF31 | NM_015629 | c.3G > A | p.(Met1Ile) | LP | B | P | 23.8 |
| RHO | NM_000539 | c.560G > T | p.(Cys187Phe) | LP | P | P | 24.8 |
| USH2A | NM_206933 | c.497A > G | p.(Glu166Gly) | LP | P | P | 26.2 |
| USH2A | NM_206933 | c.3045C > G | p.(His1015Gln) | LP | LP | P | 22.8 |
| USH2A | NM_206933 | c.6992G > A | p.(Gly2331Glu) | P | LP | P | 28.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karali, M.; Testa, F.; Brunetti-Pierri, R.; Di Iorio, V.; Pizzo, M.; Melillo, P.; Barillari, M.R.; Torella, A.; Musacchia, F.; D’Angelo, L.; et al. Clinical and Genetic Analysis of a European Cohort with Pericentral Retinitis Pigmentosa. Int. J. Mol. Sci. 2020, 21, 86. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010086
Karali M, Testa F, Brunetti-Pierri R, Di Iorio V, Pizzo M, Melillo P, Barillari MR, Torella A, Musacchia F, D’Angelo L, et al. Clinical and Genetic Analysis of a European Cohort with Pericentral Retinitis Pigmentosa. International Journal of Molecular Sciences. 2020; 21(1):86. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010086
Chicago/Turabian StyleKarali, Marianthi, Francesco Testa, Raffaella Brunetti-Pierri, Valentina Di Iorio, Mariateresa Pizzo, Paolo Melillo, Maria Rosaria Barillari, Annalaura Torella, Francesco Musacchia, Luigi D’Angelo, and et al. 2020. "Clinical and Genetic Analysis of a European Cohort with Pericentral Retinitis Pigmentosa" International Journal of Molecular Sciences 21, no. 1: 86. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010086