Regulation of Vascular Function and Inflammation via Cross Talk of Reactive Oxygen and Nitrogen Species from Mitochondria or NADPH Oxidase—Implications for Diabetes Progression

 ,
,  ,
,  ,
, 
Abstract
:
1. Introduction
1.1. Reactive Oxygen and Nitrogen Species in the Organism
1.2. Sources of Reactive Oxygen and Nitrogen Species
1.3. Preclinical/Molecular Proof of a Role of Oxidative Stress for Cardiometabolic Diseases
1.4. Clinical Evidence for a Role of Oxidative Stress in Cardiovascular and Metabolic Diseases
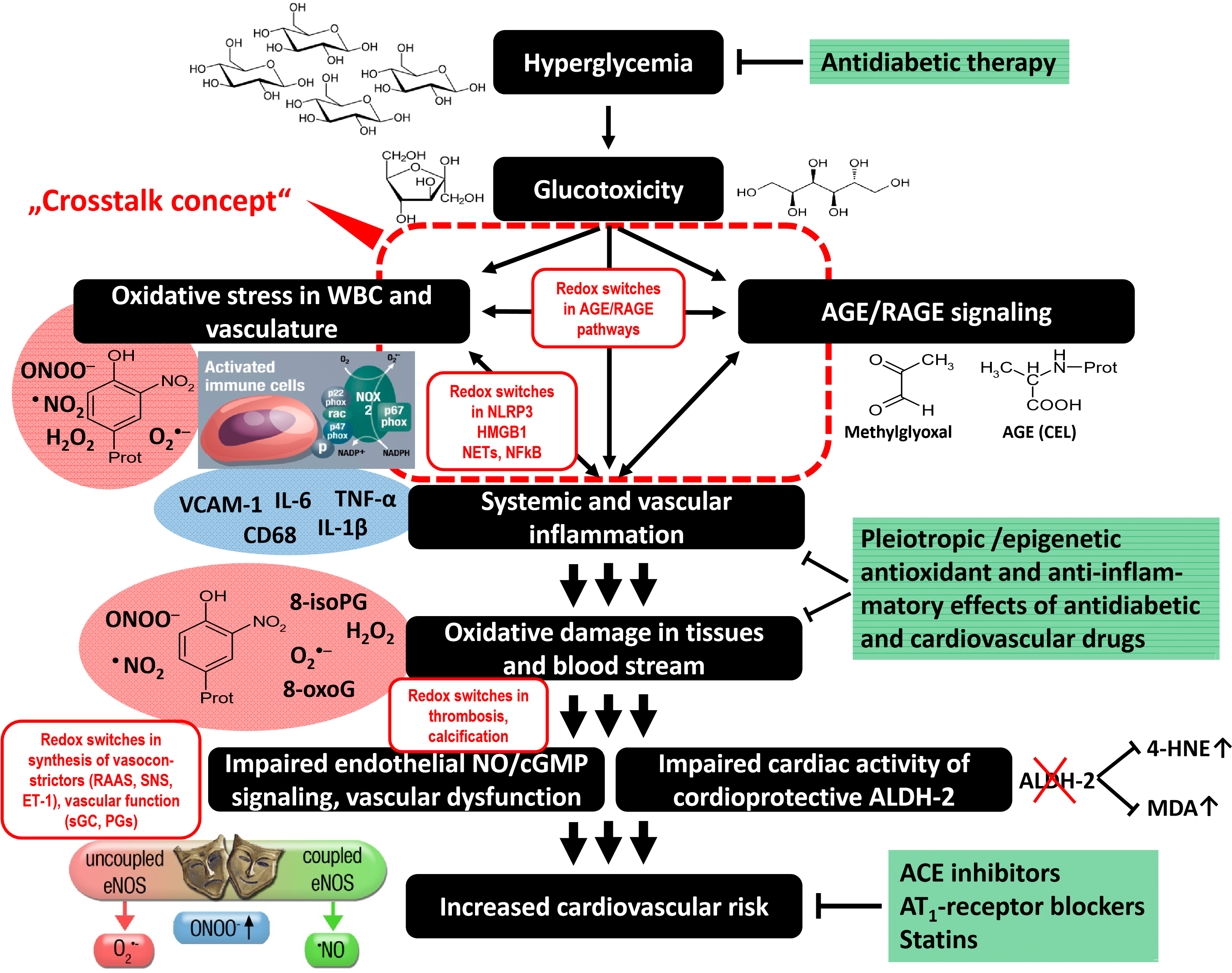
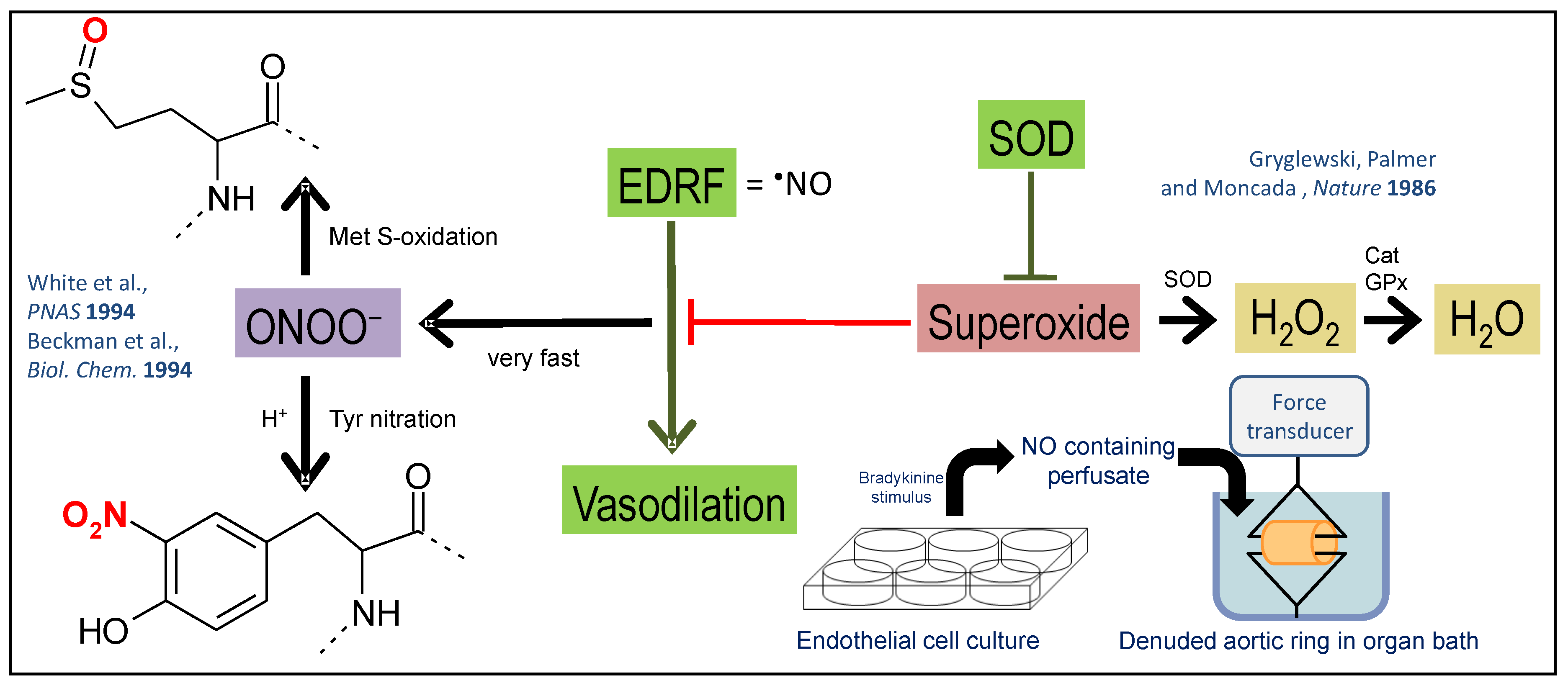
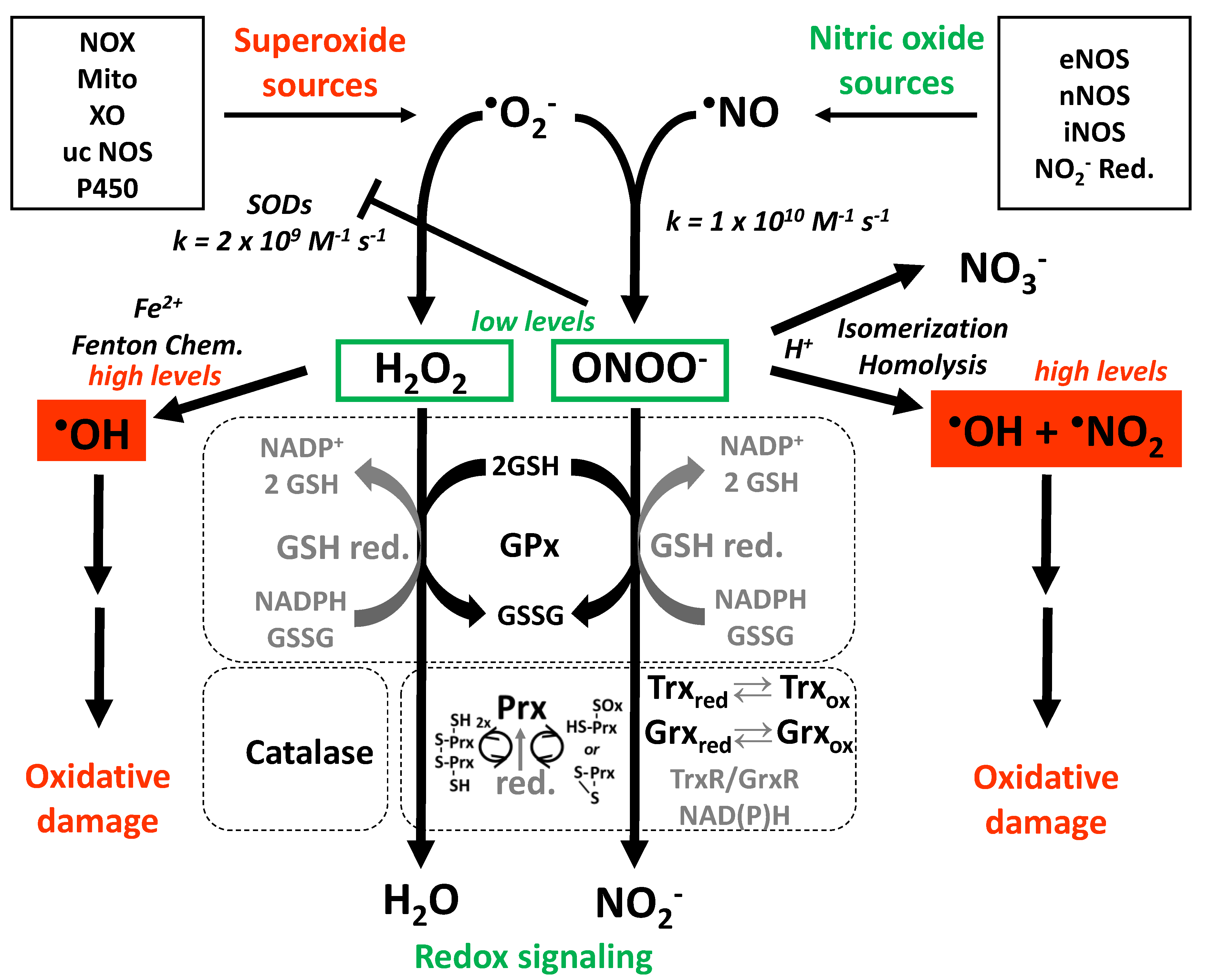
2. Cross talk between Different Sources of RONS
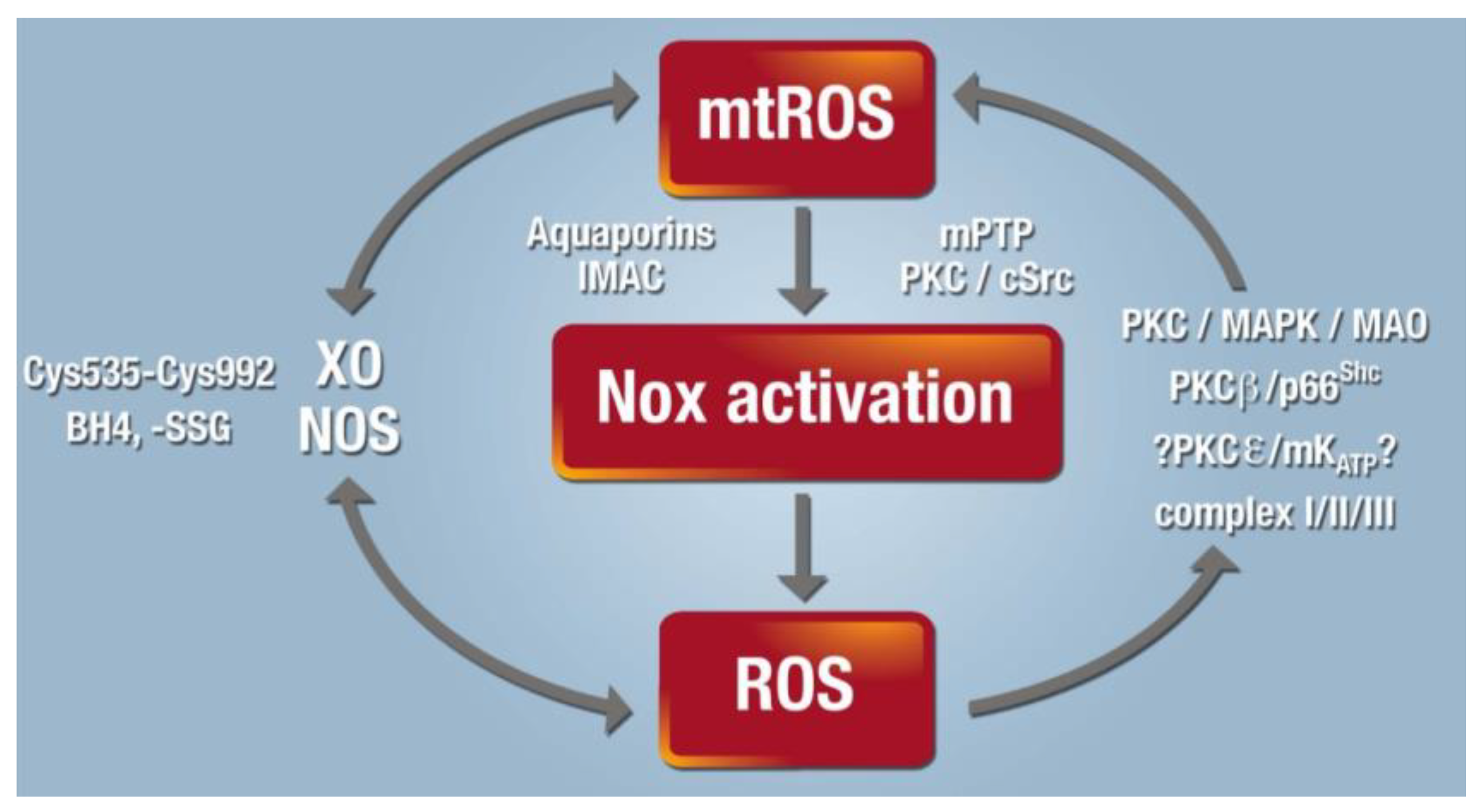
2.1. Interaction of Different RONS Sources—the Concept of “ROS-Triggered ROS Formation”

2.2. Cross Talk of Oxidative Stress and Inflammation
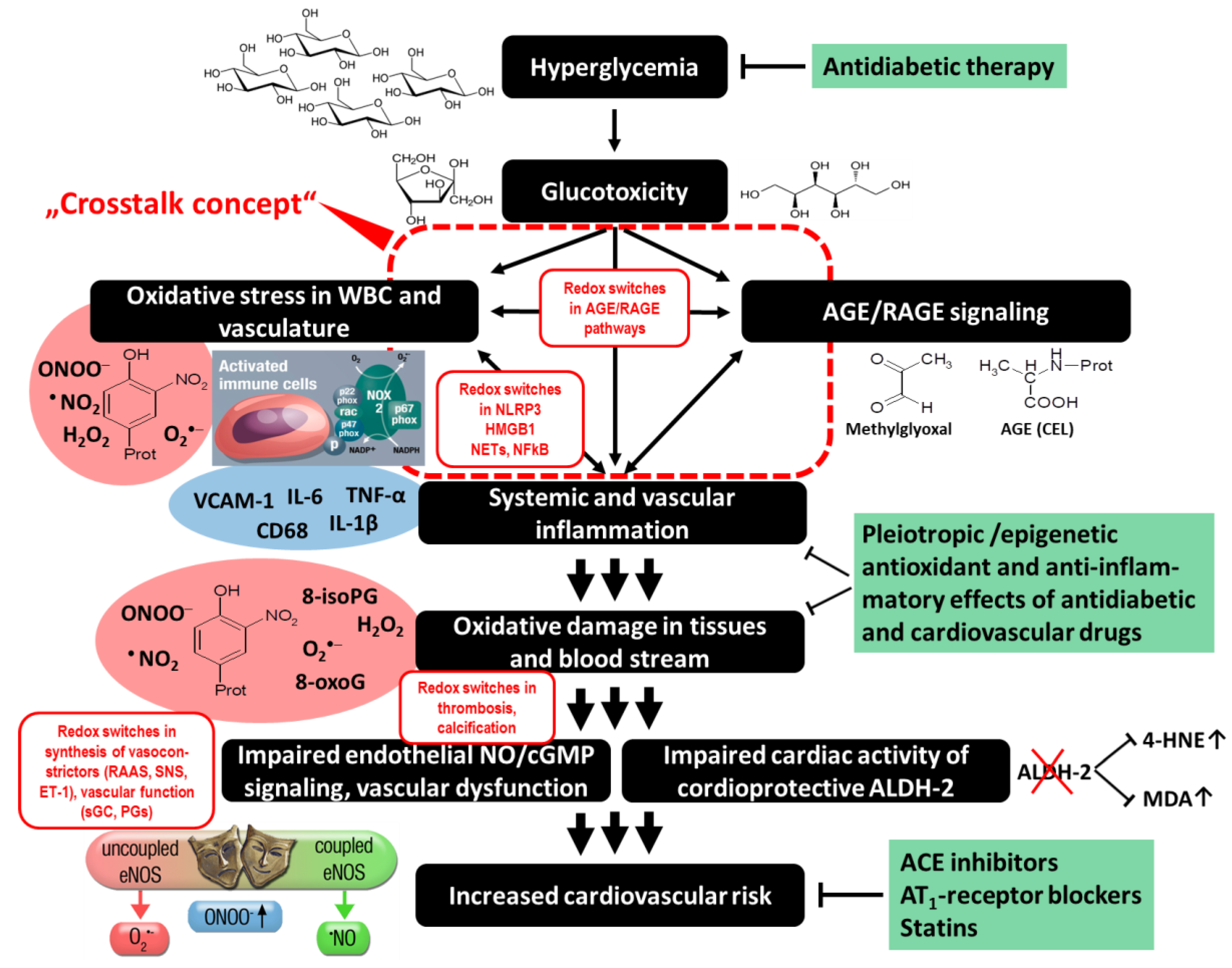
2.3. Glucotoxicity and AGE/RAGE Signaling
2.4. Other Redox Switches—Link between Oxidative stress, Inflammation, and Vascular Function
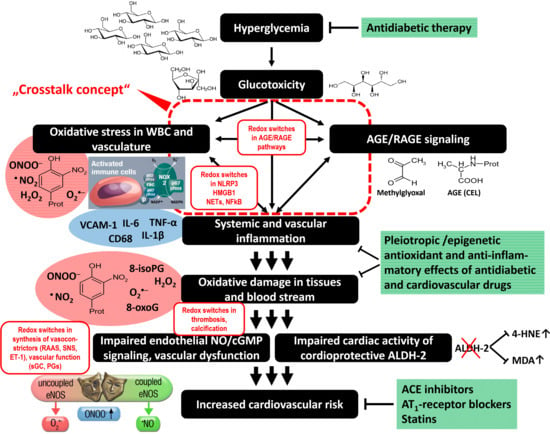
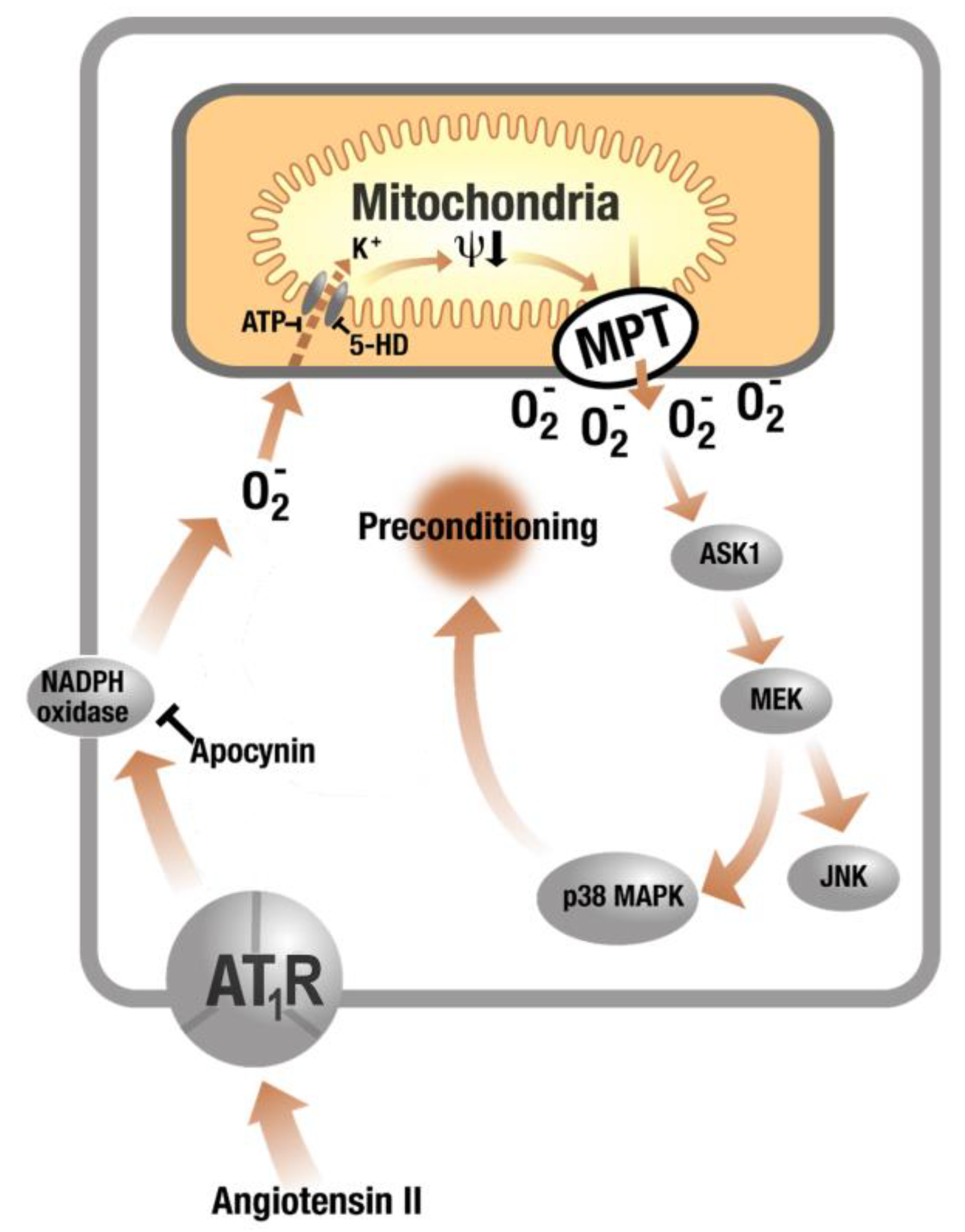
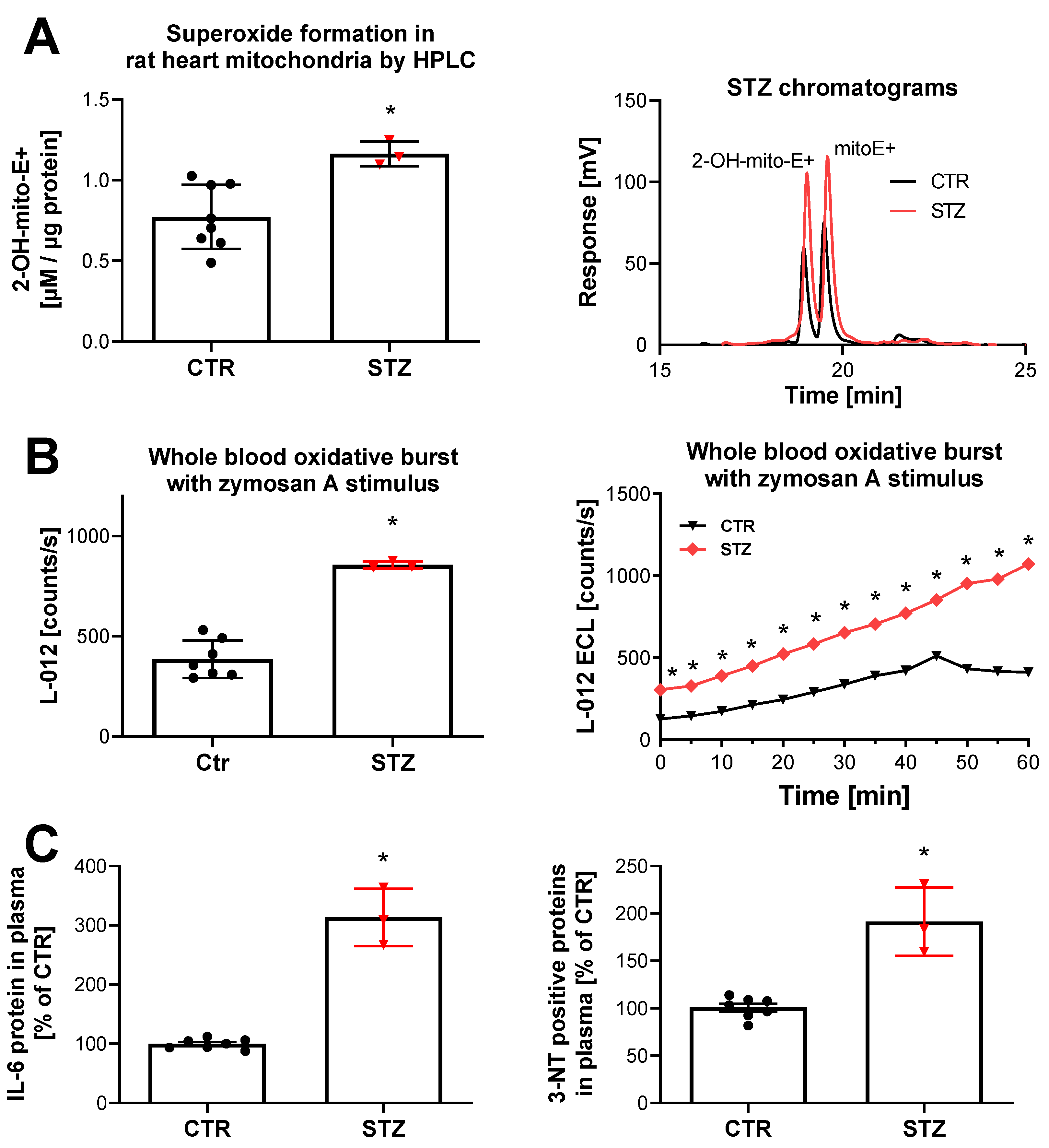
3. Evidence for a Cross Talk between Different Sources of ROS in the Setting of Diabetes
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end products |
| AT-II | Angiotensin II |
| BH4 | Tetrahydrobiopterin |
| CRP | C-reactive protein |
| CypD | Cyclophilin D |
| EDRF | Endothelium-derived relaxing factor |
| eNOS | Endothelial nitric oxide synthase |
| GPx | Glutathione peroxidases |
| H2O2 | Hydrogen peroxide |
| HO• | Hydroxyl radical |
| MDA | Malondialdehyde |
| MAO | Monoamine oxidase |
| MI | Myocardial infarction |
| Mn-SOD | Mitochondrial superoxide dismutase |
| mPTP | Mitochondrial permeability transition pore |
| mtKATP NOX | Mitochondrial ATP-sensitive potassium channels |
| •NO | NADPH oxidases Nitric oxide |
| NOS | Nitric oxide synthase |
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| O2•¯ | Superoxide anion radical, superoxide |
| PKC | protein kinase C |
| RONS | Reactive oxygen and nitrogen species |
| ROS | Reactive oxygen species |
| sGC | Soluble guanylyl cyclase |
| SOD | Superoxide dismutase |
| STZ | Streptozotocin |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| ucNOS | Uncoupled nitric oxide synthase |
| XO | Xanthine oxidase |
References
- McCord, J.M.; Keele, B.B., Jr.; Fridovich, I. An enzyme-based theory of obligate anaerobiosis: The physiological function of superoxide dismutase. Proc. Natl. Acad. Sci. USA 1971, 68, 1024–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Wandzioch, K.; Helmcke, I.; Brandes, R.P. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 239–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Kakihana, T.; Nagata, K.; Sitia, R. Peroxides and peroxidases in the endoplasmic reticulum: Integrating redox homeostasis and oxidative folding. Antioxid. Redox Signal. 2012, 16, 763–771. [Google Scholar] [CrossRef]
- Zito, E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 2015, 83, 299–304. [Google Scholar] [CrossRef]
- Schildknecht, S.; Bachschmid, M.; Ullrich, V. Peroxynitrite provides the peroxide tone for PGHS-2-dependent prostacyclin synthesis in vascular smooth muscle cells. FASEB J. 2005, 19, 1169–1171. [Google Scholar] [CrossRef]
- Ahsan, M.K.; Lekli, I.; Ray, D.; Yodoi, J.; Das, D.K. Redox regulation of cell survival by the thioredoxin superfamily: An implication of redox gene therapy in the heart. Antioxid. Redox Signal. 2009, 11, 2741–2758. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Gorlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in translational medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.P.; Standiford, T.J.; Rahman, A.; Newstead, M.; Holland, S.M.; Dinauer, M.C.; Liu, Q.H.; Malik, A.B. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: Studies in p47phox-/- and gp91phox-/- mice. J. Immunol. 2002, 168, 3974–3982. [Google Scholar] [CrossRef] [Green Version]
- Quie, P.G.; White, J.G.; Holmes, B.; Good, R.A. In vitro bactericidal capacity of human polymorphonuclear leukocytes: Diminished activity in chronic granulomatous disease of childhood. J. Clin. Investig. 1967, 46, 668–679. [Google Scholar] [CrossRef]
- Namgaladze, D.; Shcherbyna, I.; Kienhofer, J.; Hofer, H.W.; Ullrich, V. Superoxide targets calcineurin signaling in vascular endothelium. Biochem. Biophys. Res. Commun. 2005, 334, 1061–1067. [Google Scholar] [CrossRef]
- Ullrich, V.; Kissner, R. Redox signaling: Bioinorganic chemistry at its best. J. Inorg. Biochem. 2006, 100, 2079–2086. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [Green Version]
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric oxide activates guanylate cyclase and increases guanosine 3’:5’-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forstermann, U.; Schmidt, H.H.; Pollock, J.S.; Sheng, H.; Mitchell, J.A.; Warner, T.D.; Nakane, M.; Murad, F. Isoforms of nitric oxide synthase. Characterization and purification from different cell types. Biochem. Pharmacol. 1991, 42, 1849–1857. [Google Scholar] [CrossRef]
- Bian, K.; Murad, F. Nitric oxide (NO)—Biogeneration, regulation, and relevance to human diseases. Front. Biosci. J. Virtual Libr. 2003, 8, d264-78. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J. Nitric oxide: A unique endogenous signaling molecule in vascular biology. Biosci. Rep. 1999, 19, 51–71. [Google Scholar] [CrossRef]
- Ignarro, L.J. Nitric oxide as a unique signaling molecule in the vascular system: A historical overview. J. Physiol. Pharmacol. 2002, 53, 503–514. [Google Scholar]
- Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P.G.; Koppenol, W.H. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 1997, 10, 1285–1292. [Google Scholar] [CrossRef]
- Crow, J.P.; Beckman, J.S. Reaction between nitric oxide, superoxide, and peroxynitrite: Footprints of peroxynitrite in vivo. Adv. Pharmacol. 1995, 35, 17–43. [Google Scholar]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [Green Version]
- Ischiropoulos, H.; Zhu, L.; Chen, J.; Tsai, M.; Martin, J.C.; Smith, C.D.; Beckman, J.S. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem. Biophys. 1992, 298, 431–437. [Google Scholar] [CrossRef]
- Beckmann, J.S.; Ye, Y.Z.; Anderson, P.G.; Chen, J.; Accavitti, M.A.; Tarpey, M.M.; White, C.R. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol. Chem. Hoppe Seyler 1994, 375, 81–88. [Google Scholar] [CrossRef]
- White, C.R.; Brock, T.A.; Chang, L.Y.; Crapo, J.; Briscoe, P.; Ku, D.; Bradley, W.A.; Gianturco, S.H.; Gore, J.; Freeman, B.A.; et al. Superoxide and peroxynitrite in atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 1044–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.H.; Leist, M.; Ullrich, V. Selective nitration of prostacyclin synthase and defective vasorelaxation in atherosclerotic bovine coronary arteries. Am. J. Pathol. 1999, 154, 1359–1365. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denicola, A.; Freeman, B.A.; Trujillo, M.; Radi, R. Peroxynitrite reaction with carbon dioxide/bicarbonate: Kinetics and influence on peroxynitrite-mediated oxidations. Arch. Biochem. Biophys. 1996, 333, 49–58. [Google Scholar] [CrossRef]
- Daiber, A.; Herold, S.; Schoneich, C.; Namgaladze, D.; Peterson, J.A.; Ullrich, V. Nitration and inactivation of cytochrome P450BM-3 by peroxynitrite. Stopped-flow measurements prove ferryl intermediates. Eur. J. Biochem. FEBS 2000, 267, 6729–6739. [Google Scholar] [CrossRef]
- Daiber, A.; Schoneich, C.; Schmidt, P.; Jung, C.; Ullrich, V. Autocatalytic nitration of P450CAM by peroxynitrite. J. Inorg. Biochem. 2000, 81, 213–220. [Google Scholar] [CrossRef]
- Daiber, A.; Bachschmid, M.; Beckman, J.S.; Munzel, T.; Ullrich, V. The impact of metal catalysis on protein tyrosine nitration by peroxynitrite. Biochem. Biophys. Res. Commun. 2004, 317, 873–881. [Google Scholar] [CrossRef]
- MacMillan-Crow, L.A.; Crow, J.P.; Kerby, J.D.; Beckman, J.S.; Thompson, J.A. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc. Natl. Acad. Sci. USA 1996, 93, 11853–11858. [Google Scholar] [CrossRef] [Green Version]
- Quijano, C.; Hernandez-Saavedra, D.; Castro, L.; McCord, J.M.; Freeman, B.A.; Radi, R. Reaction of peroxynitrite with Mn-superoxide dismutase. Role of the metal center in decomposition kinetics and nitration. J. Biol. Chem. 2001, 276, 11631–11638. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.; Yesilkaya, A.; Ullrich, V. Peroxynitrite inactivates prostacyclin synthase by heme-thiolate-catalyzed tyrosine nitration. Drug Metab. Rev. 1999, 31, 343–349. [Google Scholar] [CrossRef]
- Daiber, A.; Oelze, M.; Steven, S.; Kroller-Schon, S.; Munzel, T. Taking up the cudgels for the traditional reactive oxygen and nitrogen species detection assays and their use in the cardiovascular system. Redox Biol. 2017, 12, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Cohen, R.; Ullrich, V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothel. J. Endothel. Cell Res. 2004, 11, 89–97. [Google Scholar] [CrossRef]
- Bachschmid, M.; Schildknecht, S.; Ullrich, V. Redox regulation of vascular prostanoid synthesis by the nitric oxide-superoxide system. Biochem. Biophys. Res. Commun. 2005, 338, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Gryglewski, R.J.; Palmer, R.M.; Moncada, S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986, 320, 454–456. [Google Scholar] [CrossRef]
- Munzel, T.; Daiber, A.; Ullrich, V.; Mulsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Lauer, N.; Mulsch, A.; Kojda, G. The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic. Biol. Med. 2001, 31, 1360–1367. [Google Scholar] [CrossRef]
- Zou, M.H.; Ullrich, V. Peroxynitrite formed by simultaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. FEBS Lett. 1996, 382, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Munzel, T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef]
- Munzel, T.; Sinning, C.; Post, F.; Warnholtz, A.; Schulz, E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann. Med. 2008, 40, 180–196. [Google Scholar] [CrossRef]
- Kimura, F.; Hasegawa, G.; Obayashi, H.; Adachi, T.; Hara, H.; Ohta, M.; Fukui, M.; Kitagawa, Y.; Park, H.; Nakamura, N.; et al. Serum extracellular superoxide dismutase in patients with type 2 diabetes: Relationship to the development of micro- and macrovascular complications. Diabetes Care 2003, 26, 1246–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.J. Metformin: Effects on micro and macrovascular complications in type 2 diabetes. Cardiovasc. Drugs Ther. 2008, 22, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Folli, F.; Corradi, D.; Fanti, P.; Davalli, A.; Paez, A.; Giaccari, A.; Perego, C.; Muscogiuri, G. The role of oxidative stress in the pathogenesis of type 2 diabetes mellitus micro- and macrovascular complications: Avenues for a mechanistic-based therapeutic approach. Curr. Diabetes Rev. 2011, 7, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Peroxynitrite, a stealthy biological oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Oelze, M.; Daub, S.; Steven, S.; Schuff, A.; Kroller-Schon, S.; Hausding, M.; Wenzel, P.; Schulz, E.; Gori, T.; et al. Vascular redox signaling, redox switches in endothelial nitric oxide synthase and endothelial dysfunction. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1177–1211. [Google Scholar]
- Casas, A.I.; Dao, V.T.; Daiber, A.; Maghzal, G.J.; Di Lisa, F.; Kaludercic, N.; Leach, S.; Cuadrado, A.; Jaquet, V.; Seredenina, T.; et al. Reactive oxygen-related diseases: Therapeutic targets and emerging clinical indications. Antioxid. Redox Signal. 2015, 23, 1171–1185. [Google Scholar] [CrossRef] [Green Version]
- Higgins, P.; Dawson, J.; Lees, K.R.; McArthur, K.; Quinn, T.J.; Walters, M.R. Xanthine oxidase inhibition for the treatment of cardiovascular disease: A systematic review and meta-analysis. Cardiovasc. Ther. 2012, 30, 217–226. [Google Scholar] [CrossRef]
- Bredemeier, M.; Lopes, L.M.; Eisenreich, M.A.; Hickmann, S.; Bongiorno, G.K.; d’Avila, R.; Morsch, A.L.B.; da Silva Stein, F.; Campos, G.G.D. Xanthine oxidase inhibitors for prevention of cardiovascular events: A systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2018, 18, 24. [Google Scholar] [CrossRef]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH oxidase inhibitors: Selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 2014, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Munzel, T.; Li, H. New therapeutic implications of endothelial nitric oxide synthase (eNOS) function/dysfunction in cardiovascular disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef] [Green Version]
- Robinson, B.H. The role of manganese superoxide dismutase in health and disease. J. Inherit. Metab. Dis. 1998, 21, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Drose, S.; Brandt, U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J. Biol. Chem. 2008, 283, 21649–21654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lisa, F.; Bernardi, P. Mitochondria and ischemia-reperfusion injury of the heart: Fixing a hole. Cardiovasc. Res. 2006, 70, 191–199. [Google Scholar] [CrossRef]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid. Redox Signal. 2007, 10, 601–620. [Google Scholar] [CrossRef]
- Smith, R.A.; Hartley, R.C.; Murphy, M.P. Mitochondria-targeted small molecule therapeutics and probes. Antioxid. Redox Signal. 2011, 15, 3021–3038. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta 2014, 1842, 1282–1294. [Google Scholar] [CrossRef] [Green Version]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell Cardiol. 2014, 73C, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Di Lisa, F.; Giorgio, M.; Ferdinandy, P.; Schulz, R. New aspects of p66Shc in ischaemia reperfusion injury and other cardiovascular diseases. Br. J. Pharmacol. 2017, 174, 1690–1703. [Google Scholar] [CrossRef] [Green Version]
- Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993, 91, 2546–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.G.; Ohara, Y. Physiologic consequences of increased vascular oxidant stresses in hypercholesterolemia and atherosclerosis: Implications for impaired vasomotion. Am. J. Cardiol. 1995, 75, 75B–81B. [Google Scholar] [CrossRef]
- Daiber, A.; Chlopicki, S. Revisiting pharmacology of oxidative stress and endothelial dysfunction in cardiovascular disease: Evidence for redox-based therapies. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Doerries, C.; Grote, K.; Hilfiker-Kleiner, D.; Luchtefeld, M.; Schaefer, A.; Holland, S.M.; Sorrentino, S.; Manes, C.; Schieffer, B.; Drexler, H.; et al. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ. Res. 2007, 100, 894–903. [Google Scholar] [CrossRef] [Green Version]
- Landmesser, U.; Cai, H.; Dikalov, S.; McCann, L.; Hwang, J.; Jo, H.; Holland, S.M.; Harrison, D.G. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002, 40, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 is involved in angiotensin II-mediated hypertension: A study in Nox1-deficient mice. Circulation 2005, 112, 2677–2685. [Google Scholar] [CrossRef]
- Dikalova, A.; Clempus, R.; Lassegue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, P.; Schuhmacher, S.; Kienhofer, J.; Muller, J.; Hortmann, M.; Oelze, M.; Schulz, E.; Treiber, N.; Kawamoto, T.; Scharffetter-Kochanek, K.; et al. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc. Res. 2008, 80, 280–289. [Google Scholar] [CrossRef]
- Daiber, A.; Oelze, M.; Sulyok, S.; Coldewey, M.; Schulz, E.; Treiber, N.; Hink, U.; Mulsch, A.; Scharffetter-Kochanek, K.; Munzel, T. Heterozygous deficiency of manganese superoxide dismutase in mice (Mn-SOD+/-): A novel approach to assess the role of oxidative stress for the development of nitrate tolerance. Mol. Pharmacol. 2005, 68, 579–588. [Google Scholar] [CrossRef]
- Torzewski, M.; Ochsenhirt, V.; Kleschyov, A.L.; Oelze, M.; Daiber, A.; Li, H.; Rossmann, H.; Tsimikas, S.; Reifenberg, K.; Cheng, F.; et al. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 850–857. [Google Scholar] [CrossRef] [Green Version]
- Oelze, M.; Kroller-Schon, S.; Steven, S.; Lubos, E.; Doppler, C.; Hausding, M.; Tobias, S.; Brochhausen, C.; Li, H.; Torzewski, M.; et al. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 2014, 63, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alp, N.J.; McAteer, M.A.; Khoo, J.; Choudhury, R.P.; Channon, K.M. Increased endothelial tetrahydrobiopterin synthesis by targeted transgenic GTP-cyclohydrolase I overexpression reduces endothelial dysfunction and atherosclerosis in ApoE-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 445–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.H.; Guan, Y.Y.; Alp, N.J.; Channon, K.M.; Chen, A.F. Endothelium-specific GTP cyclohydrolase I overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation 2008, 117, 1045–1054. [Google Scholar] [CrossRef]
- Alp, N.J.; Mussa, S.; Khoo, J.; Cai, S.; Guzik, T.; Jefferson, A.; Goh, N.; Rockett, K.A.; Channon, K.M. Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J. Clin. Investig. 2003, 112, 725–735. [Google Scholar] [CrossRef]
- Bendall, J.K.; Alp, N.J.; Warrick, N.; Cai, S.; Adlam, D.; Rockett, K.; Yokoyama, M.; Kawashima, S.; Channon, K.M. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: Insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 2005, 97, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.; Kroller-Schon, S.; Schonfelder, T.; Foretz, M.; Viollet, B.; Daiber, A.; Oelze, M.; Brandt, M.; Steven, S.; Kvandova, M.; et al. α1AMPK deletion in myelomonocytic cells induces a pro-inflammatory phenotype and enhances angiotensin II-induced vascular dysfunction. Cardiovasc. Res. 2018, 114, 1883–1893. [Google Scholar] [CrossRef] [Green Version]
- Kroller-Schon, S.; Jansen, T.; Tran, T.L.P.; Kvandova, M.; Kalinovic, S.; Oelze, M.; Keaney, J.F., Jr.; Foretz, M.; Viollet, B.; Daiber, A.; et al. Endothelial alpha1AMPK modulates angiotensin II-mediated vascular inflammation and dysfunction. Basic Res. Cardiol. 2019, 114, 8. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Schulz, E.; Wenzel, P.; Munzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2012, 20, 308–324. [Google Scholar] [CrossRef]
- Chen, A.F.; Chen, D.D.; Daiber, A.; Faraci, F.M.; Li, H.; Rembold, C.M.; Laher, I. Free radical biology of the cardiovascular system. Clin. Sci. 2012, 123, 73–91. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 2010, 1797, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7A–11A. [Google Scholar] [CrossRef]
- Blankenberg, S.; Rupprecht, H.J.; Bickel, C.; Torzewski, M.; Hafner, G.; Tiret, L.; Smieja, M.; Cambien, F.; Meyer, J.; Lackner, K.J. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N. Engl. J. Med. 2003, 349, 1605–1613. [Google Scholar] [CrossRef] [Green Version]
- Schottker, B.; Brenner, H.; Jansen, E.H.; Gardiner, J.; Peasey, A.; Kubinova, R.; Pajak, A.; Topor-Madry, R.; Tamosiunas, A.; Saum, K.U.; et al. Evidence for the free radical/oxidative stress theory of ageing from the CHANCES consortium: A meta-analysis of individual participant data. BMC Med. 2015, 13, 300. [Google Scholar] [CrossRef] [Green Version]
- Di Minno, A.; Turnu, L.; Porro, B.; Squellerio, I.; Cavalca, V.; Tremoli, E.; Di Minno, M.N. 8-hydroxy-2-deoxyguanosine levels and cardiovascular disease: A systematic review and meta-analysis of the literature. Antioxid. Redox Signal. 2016, 24, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Fratta Pasini, A.; Albiero, A.; Stranieri, C.; Cominacini, M.; Pasini, A.; Mozzini, C.; Vallerio, P.; Cominacini, L.; Garbin, U. Serum oxidative stress-induced repression of Nrf2 and GSH depletion: A mechanism potentially involved in endothelial dysfunction of young smokers. PLoS ONE 2012, 7, e30291. [Google Scholar] [CrossRef] [Green Version]
- Jurado-Gamez, B.; Fernandez-Marin, M.C.; Gomez-Chaparro, J.L.; Munoz-Cabrera, L.; Lopez-Barea, J.; Perez-Jimenez, F.; Lopez-Miranda, J. Relationship of oxidative stress and endothelial dysfunction in sleep apnoea. Eur. Respir. J. 2011, 37, 873–879. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstadter, J.; Kroller-Schon, S.; Munzel, T.; et al. Vascular inflammation and oxidative stress: Major triggers for cardiovascular disease. Oxidative Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [Green Version]
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953. [Google Scholar] [CrossRef] [Green Version]
- Jay, D.; Hitomi, H.; Griendling, K.K. Oxidative stress and diabetic cardiovascular complications. Free Radic. Biol. Med. 2006, 40, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Frenis, K.; Oelze, M.; Vujacic-Mirski, K.; Bayo Jimenez, M.T.; Kalinovic, S.; Kroller-Schon, S.; Munzel, T.; Daiber, A. SGLT2 inhibitors, diabetes and oxidative stress. In Diabetes—Oxidative Stress and Dietary Antioxidants, 2nd ed.; Preedy, V., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 1–12. [Google Scholar]
- Mansournia, M.A.; Ostadmohammadi, V.; Doosti-Irani, A.; Ghayour-Mobarhan, M.; Ferns, G.; Akbari, H.; Ghaderi, A.; Talari, H.R.; Asemi, Z. The effects of vitamin D supplementation on biomarkers of inflammation and oxidative stress in diabetic patients: A systematic review and meta-analysis of randomized controlled trials. Horm. Metab. Res. Horm. Und Stoffwechs. Horm. Et Metab. 2018, 50, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Haas, M.J.; Jafri, M.; Wehmeier, K.R.; Onstead-Haas, L.M.; Mooradian, A.D. Inhibition of endoplasmic reticulum stress and oxidative stress by vitamin D in endothelial cells. Free Radic. Biol. Med. 2016, 99, 1–10. [Google Scholar] [CrossRef]
- Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.A. Strategy to reduce free radical species in Alzheimer’s disease: An update of selected antioxidants. Expert Rev. Neurother. 2015, 15, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Chaiprasongsuk, A.; Janjetovic, Z.; Kim, T.K.; Jarrett, S.G.; D’Orazio, J.A.; Holick, M.F.; Tang, E.K.Y.; Tuckey, R.C.; Panich, U.; Li, W.; et al. Protective effects of novel derivatives of vitamin D3 and lumisterol against UVB-induced damage in human keratinocytes involve activation of Nrf2 and p53 defense mechanisms. Redox Biol. 2019, 24, 101206. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, J.; Xiang, J.; Li, Y.; Wu, D.; Xu, J. Calcitriol inhibits ROS-NLRP3-IL-1beta signaling axis via activation of Nrf2-antioxidant signaling in hyperosmotic stress stimulated human corneal epithelial cells. Redox Biol. 2019, 21, 101093. [Google Scholar] [CrossRef]
- Balbi, M.E.; Tonin, F.S.; Mendes, A.M.; Borba, H.H.; Wiens, A.; Fernandez-Llimos, F.; Pontarolo, R. Antioxidant effects of vitamins in type 2 diabetes: A meta-analysis of randomized controlled trials. Diabetol. Metab. Syndr. 2018, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.C.; Woodward, M.; Li, Q.; Pickering, R.; Tikellis, C.; Poulter, N.; Cooper, M.E.; Marre, M.; Zoungas, S.; Chalmers, J.; et al. Relationship between plasma 8-OH-deoxyguanosine and cardiovascular disease and survival in type 2 diabetes mellitus: Results from the ADVANCE trial. J. Am. Heart Assoc. 2018, 7, e008226. [Google Scholar] [CrossRef] [Green Version]
- Kjaer, L.K.; Cejvanovic, V.; Henriksen, T.; Petersen, K.M.; Hansen, T.; Pedersen, O.; Christensen, C.K.; Torp-Pedersen, C.; Gerds, T.A.; Brandslund, I.; et al. Cardiovascular and all-cause mortality risk associated with urinary excretion of 8-oxoGuo, a biomarker for RNA oxidation, in patients with type 2 diabetes: A prospective cohort study. Diabetes Care 2017, 40, 1771–1778. [Google Scholar] [CrossRef] [Green Version]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Ikehara, K.; Kanda, E.; Uchino, H.; Hirose, T. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc. Diabetol. 2017, 16, 84. [Google Scholar] [CrossRef] [Green Version]
- Heitzer, T.; Krohn, K.; Albers, S.; Meinertz, T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia 2000, 43, 1435–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzer, T.; Finckh, B.; Albers, S.; Krohn, K.; Kohlschutter, A.; Meinertz, T. Beneficial effects of alpha-lipoic acid and ascorbic acid on endothelium-dependent, nitric oxide-mediated vasodilation in diabetic patients: Relation to parameters of oxidative stress. Free Radic. Biol. Med. 2001, 31, 53–61. [Google Scholar] [CrossRef]
- De Jager, J.; Dekker, J.M.; Kooy, A.; Kostense, P.J.; Nijpels, G.; Heine, R.J.; Bouter, L.M.; Stehouwer, C.D. Endothelial dysfunction and low-grade inflammation explain much of the excess cardiovascular mortality in individuals with type 2 diabetes: The Hoorn Study. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1086–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef] [Green Version]
- Khaw, K.T.; Bingham, S.; Welch, A.; Luben, R.; Wareham, N.; Oakes, S.; Day, N. Relation between plasma ascorbic acid and mortality in men and women in EPIC-Norfolk prospective study: A prospective population study. European prospective investigation into cancer and nutrition. Lancet 2001, 357, 657–663. [Google Scholar] [CrossRef]
- Steven, S.; Munzel, T.; Daiber, A. Exploiting the pleiotropic antioxidant effects of established drugs in cardiovascular disease. Int. J. Mol. Sci. 2015, 16, 18185–18223. [Google Scholar] [CrossRef] [Green Version]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U.; et al. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007, 357, 562–571. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Zhang, G.X.; Nishiyama, A.; Shokoji, T.; Yao, L.; Fan, Y.Y.; Rahman, M.; Abe, Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: Comparison of angiotensin II and diazoxide. Hypertension 2005, 45, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P. Triggering mitochondrial radical release: A new function for NADPH oxidases. Hypertension 2005, 45, 847–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.X.; Chen, Y.F.; Campbell, W.B.; Zou, A.P.; Gross, G.J.; Li, P.L. Characteristics and superoxide-induced activation of reconstituted myocardial mitochondrial ATP-sensitive potassium channels. Circ. Res. 2001, 89, 1177–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhoutte, P.M. Cardiovascular pharmacology: Endothelial control. Adv. Pharmacol. 2010, 60, 13–14. [Google Scholar]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases—The role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Schulz, E.; Munzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, P.; Kossmann, S.; Munzel, T.; Daiber, A. Redox regulation of cardiovascular inflammation—Immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2017, 109, 48–60. [Google Scholar] [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Xu, J.; Xie, Z.; Reece, R.; Pimental, D.; Zou, M.H. Uncoupling of endothelial nitric oxidase synthase by hypochlorous acid: Role of NAD(P)H oxidase-derived superoxide and peroxynitrite. Arter. Thromb. Vasc. Biol. 2006, 26, 2688–2695. [Google Scholar] [CrossRef]
- Di Lisa, F.; Canton, M.; Menabo, R.; Kaludercic, N.; Bernardi, P. Mitochondria and cardioprotection. Heart Fail. Rev. 2007, 12, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Andrukhiv, A.; Costa, A.D.; West, I.C.; Garlid, K.D. Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2067–H2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzel, F.R.; Luo, Y.; Li, X.; Boengler, K.; Buechert, A.; Garcia-Dorado, D.; Di Lisa, F.; Schulz, R.; Heusch, G. Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ. Res. 2005, 97, 583–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pain, T.; Yang, X.M.; Critz, S.D.; Yue, Y.; Nakano, A.; Liu, G.S.; Heusch, G.; Cohen, M.V.; Downey, J.M. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ. Res. 2000, 87, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinkevich, N.S.; Gutterman, D.D. ROS-induced ROS release in vascular biology: Redox-redox signaling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H647–H653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wosniak, J., Jr.; Santos, C.X.; Kowaltowski, A.J.; Laurindo, F.R. Cross-talk between mitochondria and NADPH oxidase: Effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid. Redox Signal. 2009, 11, 1265–1278. [Google Scholar] [CrossRef]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Kroller-Schon, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, P.; Mollnau, H.; Oelze, M.; Schulz, E.; Wickramanayake, J.M.; Muller, J.; Schuhmacher, S.; Hortmann, M.; Baldus, S.; Gori, T.; et al. First evidence for a crosstalk between mitochondrial and NADPH oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid. Redox Signal. 2008, 10, 1435–1447. [Google Scholar] [CrossRef]
- Itani, H.A.; Dikalova, A.E.; McMaster, W.G.; Nazarewicz, R.R.; Bikineyeva, A.T.; Harrison, D.G.; Dikalov, S.I. Mitochondrial cyclophilin D in vascular oxidative stress and hypertension. Hypertension 2016, 67, 1218–1227. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Stevens, M.V.; Kohr, M.; Steenbergen, C.; Sack, M.N.; Murphy, E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 2011, 286, 40184–40192. [Google Scholar] [CrossRef] [Green Version]
- Nishino, T. The conversion from the dehydrogenase type to the oxidase type of rat liver xanthine dehydrogenase by modification of cysteine residues with fluorodinitrobenzene. J. Biol. Chem. 1997, 272, 29859–29864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, M.G.; Balendran, A.; Harrison, R.; Wolstenholme, A.; Bulkley, G.B. Xanthine oxidoreductase: Dehydrogenase to oxidase conversion. Biochem. Soc. Trans. 1997, 25, 530S. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.A.; Wang, T.Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.; Chen, Y.R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, e68–e75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loot, A.E.; Schreiber, J.G.; Fisslthaler, B.; Fleming, I. Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J. Exp. Med. 2009, 206, 2889–2896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H570–H578. [Google Scholar] [CrossRef] [Green Version]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef]
- Powell, C.S.; Jackson, R.M. Mitochondrial complex I, aconitase, and succinate dehydrogenase during hypoxia-reoxygenation: Modulation of enzyme activities by MnSOD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L189–L198. [Google Scholar] [CrossRef] [Green Version]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Bae, I.H.; Bae, Y.S.; Um, H.D. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J. Biol. Chem. 2006, 281, 36228–36235. [Google Scholar] [CrossRef]
- Daiber, A.; Kienhoefer, J.; Zee, R.; Ullrich, V.; Van der Loo, B.; Bachschmid, M. The Role of Mitochondrial Reactive Oxygen Species Formation for Age-Induced Vascular Dysfunction. In Aging and Age-Related Disorders, 3rd ed.; Bondy, S.C., Maiese, K., Eds.; Oxidative Stress in Applied Basic Research and Clinical Practice; Springer (Humana Press): Totowa, NJ, USA, 2010; pp. 237–257. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, K.A.; Sawa, T.; Ihara, H.; Kasamatsu, S.; Yoshitake, J.; Rahaman, M.M.; Okamoto, T.; Fujii, S.; Akaike, T. Regulation by mitochondrial superoxide and NADPH oxidase of cellular formation of nitrated cyclic GMP: Potential implications for ROS signalling. Biochem. J. 2012, 441, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.F.; Liang, S.S.; Thanasekaran, P.; Chang, H.W.; Wen, L.L.; Chen, C.H.; Liou, J.C.; Yeh, J.C.; Liu, S.H.; Dai, H.M.; et al. Translational medicine in pulmonary-renal crosstalk: Therapeutic targeting of p-cresyl sulfate triggered nonspecific ROS and chemoattractants in dyspneic patients with uremic lung injury. J. Clin. Med. 2018, 7, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.K.; Li, C.; Ahmad, A.; Abrams, O.; Gorbatyuk, M.S.; Harrod, K.S.; Wek, R.C.; Afaq, F.; Athar, M. ATF4 regulates arsenic trioxide-mediated NADPH oxidase, ER-mitochondrial crosstalk and apoptosis. Arch. Biochem. Biophys. 2016, 609, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veith, C.; Boots, A.W.; Idris, M.; van Schooten, F.J.; van der Vliet, A. Redox imbalance in idiopathic pulmonary fibrosis: A role for oxidant cross-talk between NADPH oxidase enzymes and mitochondria. Antioxid. Redox Signal. 2019, 31, 1092–1115. [Google Scholar] [CrossRef] [PubMed]
- Desouki, M.M.; Kulawiec, M.; Bansal, S.; Das, G.M.; Singh, K.K. Cross talk between mitochondria and superoxide generating NADPH oxidase in breast and ovarian tumors. Cancer Biol. Ther. 2005, 4, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.M.; Kim, S.J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 2017, 312, C749–C764. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, M.F.; Natali, A.J.; da Silva, E.; Gomes, G.J.; Teodoro, B.G.; Cunha, D.N.; Drummond, L.R.; Drummond, F.R.; Moura, A.G.; Belfort, F.G.; et al. Attenuation of Ca2+ homeostasis, oxidative stress, and mitochondrial dysfunctions in diabetic rat heart: Insulin therapy or aerobic exercise? J. Appl. Physiol. 2015, 119, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Hempel, N.; Trebak, M. Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium 2017, 63, 70–96. [Google Scholar] [CrossRef] [Green Version]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Li, W.; Doughan, A.K.; Blanco, R.R.; Zafari, A.M. Mitochondrial reactive oxygen species and calcium uptake regulate activation of phagocytic NADPH oxidase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1134–R1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, P.; Knorr, M.; Kossmann, S.; Stratmann, J.; Hausding, M.; Schuhmacher, S.; Karbach, S.H.; Schwenk, M.; Yogev, N.; Schulz, E.; et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011, 124, 1370–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kossmann, S.; Schwenk, M.; Hausding, M.; Karbach, S.H.; Schmidgen, M.I.; Brandt, M.; Knorr, M.; Hu, H.; Kroller-Schon, S.; Schonfelder, T.; et al. Angiotensin II-induced vascular dysfunction depends on interferon-gamma-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1313–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef]
- Itani, H.A.; McMaster, W.G., Jr.; Saleh, M.A.; Nazarewicz, R.R.; Mikolajczyk, T.P.; Kaszuba, A.M.; Konior, A.; Prejbisz, A.; Januszewicz, A.; Norlander, A.E.; et al. Activation of human T cells in hypertension: Studies of humanized mice and hypertensive humans. Hypertension 2016, 68, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Qiao, M.; Zhao, Q.; Lee, C.F.; Tannock, L.R.; Smart, E.J.; LeBaron, R.G.; Phelix, C.F.; Rangel, Y.; Asmis, R. Thiol oxidative stress induced by metabolic disorders amplifies macrophage chemotactic responses and accelerates atherogenesis and kidney injury in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1779–1786. [Google Scholar] [CrossRef] [Green Version]
- Tavakoli, S.; Asmis, R. Reactive oxygen species and thiol redox signaling in the macrophage biology of atherosclerosis. Antioxid Redox Signal. 2012, 17, 1785–1795. [Google Scholar] [CrossRef] [Green Version]
- Pittman, K.; Kubes, P. Damage-associated molecular patterns control neutrophil recruitment. J. Innate Immun. 2013, 5, 315–323. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Boeck, C.; Koenig, A.M.; Schury, K.; Geiger, M.L.; Karabatsiakis, A.; Wilker, S.; Waller, C.; Gundel, H.; Fegert, J.M.; Calzia, E.; et al. Inflammation in adult women with a history of child maltreatment: The involvement of mitochondrial alterations and oxidative stress. Mitochondrion 2016, 30, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.Y.; Park, H.H. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem. 2011, 286, 39528–39536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maugeri, N.; Rovere-Querini, P.; Baldini, M.; Baldissera, E.; Sabbadini, M.G.; Bianchi, M.E.; Manfredi, A.A. Oxidative stress elicits platelet/leukocyte inflammatory interactions via HMGB1: A candidate for microvessel injury in sytemic sclerosis. Antioxid. Redox Signal. 2014, 20, 1060–1074. [Google Scholar] [CrossRef]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; von Bruhl, M.L.; et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, K.; Deng, L.; Chen, Y.; Nice, E.C.; Huang, C. Redox regulation of inflammation: Old elements, a new story. Med. Res. Rev. 2015, 35, 306–340. [Google Scholar] [CrossRef]
- Janko, C.; Filipovic, M.; Munoz, L.E.; Schorn, C.; Schett, G.; Ivanovic-Burmazovic, I.; Herrmann, M. Redox modulation of HMGB1-related signaling. Antioxid. Redox Signal. 2014, 20, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, E.P.; Rochael, N.C.; Guimaraes-Costa, A.B.; de Souza-Vieira, T.S.; Ganilho, J.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced neutrophil extracellular trap (NET) formation. J. Biol. Chem. 2015, 290, 22174–22183. [Google Scholar] [CrossRef] [Green Version]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morshed, M.; Hlushchuk, R.; Simon, D.; Walls, A.F.; Obata-Ninomiya, K.; Karasuyama, H.; Djonov, V.; Eggel, A.; Kaufmann, T.; Simon, H.U.; et al. NADPH oxidase-independent formation of extracellular DNA traps by basophils. J. Immunol. 2014, 192, 5314–5323. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, T.; Moller, S.; Klinger, M.; Solbach, W.; Laskay, T.; Behnen, M. The impact of various reactive oxygen species on the formation of neutrophil extracellular traps. Mediat. Inflamm. 2012, 2012, 849136. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Apostolova, N.; Banuls, C.; Muntane, J.; Rocha, M.; Victor, V.M. Mitochondria, the NLRP3 inflammasome, and sirtuins in type 2 diabetes: New therapeutic targets. Antioxid. Redox Signal. 2018, 29, 749–791. [Google Scholar] [CrossRef] [Green Version]
- Uitto, J.; Perejda, A.J.; Grant, G.A.; Rowold, E.A.; Kilo, C.; Williamson, J.R. Glycosylation of human glomerular basement membrane collagen: Increased content of hexose in ketoamine linkage and unaltered hydroxylysine-O-glycosides in patients with diabetes. Connect. Tissue Res. 1982, 10, 287–296. [Google Scholar] [CrossRef]
- Bucala, R.; Tracey, K.J.; Cerami, A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J. Clin. Investig. 1991, 87, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Bucciarelli, L.G.; Wendt, T.; Qu, W.; Lu, Y.; Lalla, E.; Rong, L.L.; Goova, M.T.; Moser, B.; Kislinger, T.; Lee, D.C.; et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 2002, 106, 2827–2835. [Google Scholar] [CrossRef]
- Kumar, S.; Prasad, S.; Sitasawad, S.L. Multiple antioxidants improve cardiac complications and inhibit cardiac cell death in streptozotocin-induced diabetic rats. PLoS ONE 2013, 8, e67009. [Google Scholar] [CrossRef] [PubMed]
- Oelze, M.; Schuhmacher, S.; Daiber, A. Organic nitrates and nitrate resistance in diabetes: The role of vascular dysfunction and oxidative stress with emphasis on antioxidant properties of pentaerithrityl tetranitrate. Exp. Diabetes Res. 2010, 2010, 213176. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Godtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef]
- Von Leitner, E.C.; Klinke, A.; Atzler, D.; Slocum, J.L.; Lund, N.; Kielstein, J.T.; Maas, R.; Schmidt-Haupt, R.; Pekarova, M.; Hellwinkel, O.; et al. Pathogenic cycle between the endogenous nitric oxide synthase inhibitor asymmetrical dimethylarginine and the leukocyte-derived hemoprotein myeloperoxidase. Circulation 2011, 124, 2735–2745. [Google Scholar] [CrossRef] [Green Version]
- Burnouf, C.; Pruniaux, M.P. Recent advances in PDE4 inhibitors as immunoregulators and anti-inflammatory drugs. Curr. Pharm. Des. 2002, 8, 1255–1296. [Google Scholar] [CrossRef]
- Stasch, J.P.; Schlossmann, J.; Hocher, B. Renal effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Curr. Opin. Pharmacol. 2015, 21, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Mikhed, Y.; Gorlach, A.; Knaus, U.G.; Daiber, A. Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 2015, 5, 275–289. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.; Zhong, H.; Yanamadala, S.; Campese, V.M. Oxidative stress mediates the stimulation of sympathetic nerve activity in the phenol renal injury model of hypertension. Hypertension 2006, 48, 309–315. [Google Scholar] [CrossRef]
- Campos, R.R.; Oliveira-Sales, E.B.; Nishi, E.E.; Paton, J.F.; Bergamaschi, C.T. Mechanisms of renal sympathetic activation in renovascular hypertension. Exp. Physiol. 2015, 100, 496–501. [Google Scholar] [CrossRef] [Green Version]
- Oliveira-Sales, E.B.; Nishi, E.E.; Carillo, B.A.; Boim, M.A.; Dolnikoff, M.S.; Bergamaschi, C.T.; Campos, R.R. Oxidative stress in the sympathetic premotor neurons contributes to sympathetic activation in renovascular hypertension. Am. J. Hypertens. 2009, 22, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Rajamohan, S.B.; Raghuraman, G.; Prabhakar, N.R.; Kumar, G.K. NADPH oxidase-derived H2O2 contributes to angiotensin II-induced aldosterone synthesis in human and rat adrenal cortical cells. Antioxid. Redox Signal. 2012, 17, 445–459. [Google Scholar] [CrossRef] [Green Version]
- Keidar, S.; Kaplan, M.; Pavlotzky, E.; Coleman, R.; Hayek, T.; Hamoud, S.; Aviram, M. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: A possible role for angiotensin-converting enzyme and the receptors for angiotensin II and aldosterone. Circulation 2004, 109, 2213–2220. [Google Scholar] [CrossRef] [Green Version]
- Emmerson, A.; Trevelin, S.C.; Mongue-Din, H.; Becker, P.D.; Ortiz, C.; Smyth, L.A.; Peng, Q.; Elgueta, R.; Sawyer, G.; Ivetic, A.; et al. Nox2 in regulatory T cells promotes angiotensin II-induced cardiovascular remodeling. J. Clin. Investig. 2018, 128, 3088–3101. [Google Scholar] [CrossRef] [Green Version]
- Kahler, J.; Mendel, S.; Weckmuller, J.; Orzechowski, H.D.; Mittmann, C.; Koster, R.; Paul, M.; Meinertz, T.; Munzel, T. Oxidative stress increases synthesis of big endothelin-1 by activation of the endothelin-1 promoter. J. Mol. Cell Cardiol. 2000, 32, 1429–1437. [Google Scholar] [CrossRef]
- Hink, U.; Oelze, M.; Kolb, P.; Bachschmid, M.; Zou, M.H.; Daiber, A.; Mollnau, H.; August, M.; Baldus, S.; Tsilimingas, N.; et al. Role for peroxynitrite in the inhibition of prostacyclin synthase in nitrate tolerance. J. Am. Coll. Cardiol. 2003, 42, 1826–1834. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, P.; Youhnovski, N.; Daiber, A.; Balan, A.; Arsic, M.; Bachschmid, M.; Przybylski, M.; Ullrich, V. Specific nitration at tyrosine 430 revealed by high resolution mass spectrometry as basis for redox regulation of bovine prostacyclin synthase. J. Biol. Chem. 2003, 278, 12813–12819. [Google Scholar] [CrossRef] [Green Version]
- Bachschmid, M.; Thurau, S.; Zou, M.H.; Ullrich, V. Endothelial cell activation by endotoxin involves superoxide/NO-mediated nitration of prostacyclin synthase and thromboxane receptor stimulation. FASEB J. 2003, 17, 914–916. [Google Scholar] [CrossRef]
- Schildknecht, S.; Karreman, C.; Daiber, A.; Zhao, C.; Hamacher, J.; Perlman, D.; Jung, B.; van der Loo, B.; O’Connor, P.; Leist, M.; et al. Autocatalytic nitration of prostaglandin endoperoxide synthase-2 by nitrite inhibits prostanoid formation in rat alveolar macrophages. Antioxid. Redox Signal. 2012, 17, 1393–1406. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.; Martin, C.; Ullrich, V. Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biol. Chem. 1997, 378, 707–713. [Google Scholar] [CrossRef]
- Zou, M.H.; Bachschmid, M. Hypoxia-reoxygenation triggers coronary vasospasm in isolated bovine coronary arteries via tyrosine nitration of prostacyclin synthase. J. Exp. Med. 1999, 190, 135–139. [Google Scholar] [CrossRef]
- Zou, M.H.; Shi, C.; Cohen, R.A. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H2 receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes 2002, 51, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Brune, B.; Schmidt, K.U.; Ullrich, V. Activation of soluble guanylate cyclase by carbon monoxide and inhibition by superoxide anion. Eur. J. Biochem. Febs 1990, 192, 683–688. [Google Scholar] [CrossRef]
- Riego, J.A.; Broniowska, K.A.; Kettenhofen, N.J.; Hogg, N. Activation and inhibition of soluble guanylyl cyclase by S-nitrosocysteine: Involvement of amino acid transport system L. Free Radic. Biol. Med. 2009, 47, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Crassous, P.A.; Couloubaly, S.; Huang, C.; Zhou, Z.; Baskaran, P.; Kim, D.D.; Papapetropoulos, A.; Fioramonti, X.; Duran, W.N.; Beuve, A. Soluble guanylyl cyclase is a target of angiotensin II-induced nitrosative stress in a hypertensive rat model. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H597–H604. [Google Scholar] [CrossRef] [Green Version]
- Beuve, A. Thiol-based redox modulation of soluble guanylyl cyclase, the nitric oxide receptor. Antioxid. Redox Signal. 2017, 26, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, T.S.; Savithri, H.S.; Rao, N.A.; Venkitasubramanian, T.A. Temperature and thiol-induced desensitization of a Ca2+-sensitive cyclic-3’,5’-nucleotide phosphodiesterase from sheep lung. Biochem. Int. 1988, 17, 927–933. [Google Scholar]
- Rossig, L.; Fichtlscherer, B.; Breitschopf, K.; Haendeler, J.; Zeiher, A.M.; Mulsch, A.; Dimmeler, S. Nitric oxide inhibits caspase-3 by S-nitrosation in vivo. J. Biol. Chem. 1999, 274, 6823–6826. [Google Scholar] [CrossRef] [Green Version]
- Benhar, M.; Stamler, J.S. A central role for S-nitrosylation in apoptosis. Nat. Cell Biol. 2005, 7, 645–646. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cocheme, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Espey, M.G.; Miranda, K.M.; Thomas, D.D.; Xavier, S.; Citrin, D.; Vitek, M.P.; Wink, D.A. A chemical perspective on the interplay between NO, reactive oxygen species, and reactive nitrogen oxide species. Ann. N. Y. Acad. Sci. 2002, 962, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Schildknecht, S.; Muller, J.; Kamuf, J.; Bachschmid, M.M.; Ullrich, V. Chemical model systems for cellular nitros(yl)ation reactions. Free Radic. Biol. Med. 2009, 47, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolhuter, K.; Whitwell, H.J.; Switzer, C.H.; Burgoyne, J.R.; Timms, J.F.; Eaton, P. Evidence against stable protein S-nitrosylation as a widespread mechanism of post-translational regulation. Mol. Cell 2018, 69, 438–450. [Google Scholar] [CrossRef]
- Sieve, I.; Munster-Kuhnel, A.K.; Hilfiker-Kleiner, D. Regulation and function of endothelial glycocalyx layer in vascular diseases. Vasc. Pharmacol. 2018, 100, 26–33. [Google Scholar] [CrossRef]
- Barnett, A.H.; Bain, S.C.; Bouter, P.; Karlberg, B.; Madsbad, S.; Jervell, J.; Mustonen, J. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N. Engl. J. Med. 2004, 351, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Candido, R.; Allen, T.J.; Lassila, M.; Cao, Z.; Thallas, V.; Cooper, M.E.; Jandeleit-Dahm, K.A. Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation 2004, 109, 1536–1542. [Google Scholar] [CrossRef]
- Takahashi, K.; Ghatei, M.A.; Lam, H.C.; O’Halloran, D.J.; Bloom, S.R. Elevated plasma endothelin in patients with diabetes mellitus. Diabetologia 1990, 33, 306–310. [Google Scholar] [CrossRef] [Green Version]
- Wendt, M.C.; Daiber, A.; Kleschyov, A.L.; Mulsch, A.; Sydow, K.; Schulz, E.; Chen, K.; Keaney, J.F., Jr.; Lassegue, B.; Walter, U.; et al. Differential effects of diabetes on the expression of the gp91(phox) homologues nox1 and nox4. Free Radic. Biol. Med. 2005, 39, 381–391. [Google Scholar] [CrossRef]
- Schuhmacher, S.; Oelze, M.; Bollmann, F.; Kleinert, H.; Otto, C.; Heeren, T.; Steven, S.; Hausding, M.; Knorr, M.; Pautz, A.; et al. Vascular dysfunction in experimental diabetes is improved by pentaerithrityl tetranitrate but not isosorbide-5-mononitrate therapy. Diabetes 2011, 60, 2608–2616. [Google Scholar] [CrossRef] [Green Version]
- Manea, S.A.; Antonescu, M.L.; Fenyo, I.M.; Raicu, M.; Simionescu, M.; Manea, A. Epigenetic regulation of vascular NADPH oxidase expression and reactive oxygen species production by histone deacetylase-dependent mechanisms in experimental diabetes. Redox Biol. 2018, 16, 332–343. [Google Scholar] [CrossRef]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [Green Version]
- Oelze, M.; Kroller-Schon, S.; Welschof, P.; Jansen, T.; Hausding, M.; Mikhed, Y.; Stamm, P.; Mader, M.; Zinssius, E.; Agdauletova, S.; et al. The sodium-glucose co-transporter 2 inhibitor empagliflozin improves diabetes-induced vascular dysfunction in the streptozotocin diabetes rat model by interfering with oxidative stress and glucotoxicity. PLoS ONE 2014, 9, e112394. [Google Scholar] [CrossRef]
- Kalinovic, S.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Vujacic-Mirski, K.; Kvandova, M.; Schmal, I.; Al Zuabi, A.; Munzel, T.; Daiber, A. Comparison of mitochondrial superoxide detection ex vivo/in vivo by mitoSOX HPLC method with classical assays in three different animal models of oxidative stress. Antioxidants 2019, 8, 514. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, P.; Schulz, E.; Oelze, M.; Muller, J.; Schuhmacher, S.; Alhamdani, M.S.; Debrezion, J.; Hortmann, M.; Reifenberg, K.; Fleming, I.; et al. AT1-receptor blockade by telmisartan upregulates GTP-cyclohydrolase I and protects eNOS in diabetic rats. Free Radic. Biol. Med. 2008, 45, 619–626. [Google Scholar] [CrossRef]
- Fakhruddin, S.; Alanazi, W.; Jackson, K.E. Diabetes-induced reactive oxygen species: Mechanism of their generation and role in renal injury. J. Diabetes Res. 2017, 2017, 8379327. [Google Scholar] [CrossRef]
- Lindblom, R.S.J.; Higgins, G.C.; Nguyen, T.V.; Arnstein, M.; Henstridge, D.C.; Granata, C.; Snelson, M.; Thallas-Bonke, V.; Cooper, M.E.; Forbes, J.M.; et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clin. Sci. (Lond.) 2020, 134, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, C.M.; Lutsey, P.L.; Ballantyne, C.M.; Folsom, A.R.; Pankow, J.S.; Selvin, E. Six-year change in high-sensitivity C-reactive protein and risk of diabetes, cardiovascular disease, and mortality. Am. Heart J. 2015, 170, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Ofstad, A.P.; Gullestad, L.; Orvik, E.; Aakhus, S.; Endresen, K.; Ueland, T.; Aukrust, P.; Fagerland, M.W.; Birkeland, K.I.; Johansen, O.E. Interleukin-6 and activin A are independently associated with cardiovascular events and mortality in type 2 diabetes: The prospective Asker and Baerum Cardiovascular Diabetes (ABCD) cohort study. Cardiovasc. Diabetol. 2013, 12, 126. [Google Scholar] [CrossRef] [Green Version]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.; Warnholtz, A.; et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001, 88, E14–E22. [Google Scholar] [CrossRef]
- Rezende, F.; Moll, F.; Walter, M.; Helfinger, V.; Hahner, F.; Janetzko, P.; Ringel, C.; Weigert, A.; Fleming, I.; Weissmann, N.; et al. The NADPH organizers NoxO1 and p47phox are both mediators of diabetes-induced vascular dysfunction in mice. Redox Biol. 2018, 15, 12–21. [Google Scholar] [CrossRef]
- Gray, S.P.; Jha, J.C.; Kennedy, K.; van Bommel, E.; Chew, P.; Szyndralewiez, C.; Touyz, R.M.; Schmidt, H.; Cooper, M.E.; Jandeleit-Dahm, K.A.M. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 2017, 60, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Kakehi, T.; Matsumoto, M.; Iwata, K.; Ibi, M.; Ohshima, Y.; Zhang, J.; Liu, J.; Wen, X.; Taye, A.; et al. NADPH oxidase NOX1 is involved in activation of protein kinase C and premature senescence in early stage diabetic kidney. Free Radic. Biol. Med. 2015, 83, 21–30. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [Green Version]
- Sukumar, P.; Viswambharan, H.; Imrie, H.; Cubbon, R.M.; Yuldasheva, N.; Gage, M.; Galloway, S.; Skromna, A.; Kandavelu, P.; Santos, C.X.; et al. Nox2 NADPH oxidase has a critical role in insulin resistance-related endothelial cell dysfunction. Diabetes 2013, 62, 2130–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, F.L.; Lu, X.; Strutt, B.; Hill, D.J.; Feng, Q. NOX2 deficiency protects against streptozotocin-induced beta-cell destruction and development of diabetes in mice. Diabetes 2010, 59, 2603–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Menini, S.; Amadio, L.; Oddi, G.; Ricci, C.; Pesce, C.; Pugliese, F.; Giorgio, M.; Migliaccio, E.; Pelicci, P.; Iacobini, C.; et al. Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress. Diabetes 2006, 55, 1642–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, S.C.; Fusco, S.; Panieri, E.; Labate, V.; Mele, M.; Tesori, V.; Ferrara, A.M.; Maulucci, G.; De Spirito, M.; Martorana, G.E.; et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 13420–13425. [Google Scholar] [CrossRef] [Green Version]
- Deshwal, S.; Forkink, M.; Hu, C.H.; Buonincontri, G.; Antonucci, S.; Di Sante, M.; Murphy, M.P.; Paolocci, N.; Mochly-Rosen, D.; Krieg, T.; et al. Monoamine oxidase-dependent endoplasmic reticulum-mitochondria dysfunction and mast cell degranulation lead to adverse cardiac remodeling in diabetes. Cell Death Differ. 2018, 25, 1671–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshwal, S.; Di Sante, M.; Di Lisa, F.; Kaludercic, N. Emerging role of monoamine oxidase as a therapeutic target for cardiovascular disease. Curr. Opin. Pharmacol. 2017, 33, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Desco, M.C.; Asensi, M.; Marquez, R.; Martinez-Valls, J.; Vento, M.; Pallardo, F.V.; Sastre, J.; Vina, J. Xanthine oxidase is involved in free radical production in type 1 diabetes: Protection by allopurinol. Diabetes 2002, 51, 1118–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhao, J.; Qiu, J.; Li, J.; Liang, X.; Zhang, Z.; Zhang, X.; Fu, H.; Korantzopoulos, P.; Letsas, K.P.; et al. Xanthine oxidase inhibitor allopurinol prevents oxidative stress-mediated atrial remodeling in alloxan-induced diabetes mellitus rabbits. J. Am. Heart Assoc. 2018, 7, e008807. [Google Scholar] [CrossRef]
- Mizuno, Y.; Yamamotoya, T.; Nakatsu, Y.; Ueda, K.; Matsunaga, Y.; Inoue, M.K.; Sakoda, H.; Fujishiro, M.; Ono, H.; Kikuchi, T.; et al. Xanthine oxidase inhibitor febuxostat exerts an anti-inflammatory action and protects against diabetic nephropathy development in KK-Ay obese diabetic mice. Int. J. Mol. Sci. 2019, 20, 4680. [Google Scholar] [CrossRef] [Green Version]
- Elrod, J.W.; Duranski, M.R.; Langston, W.; Greer, J.J.; Tao, L.; Dugas, T.R.; Kevil, C.G.; Champion, H.C.; Lefer, D.J. eNOS gene therapy exacerbates hepatic ischemia-reperfusion injury in diabetes: A role for eNOS uncoupling. Circ. Res. 2006, 99, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Morota, S.; Mansson, R.; Hansson, M.J.; Kasuya, K.; Shimazu, M.; Hasegawa, E.; Yanagi, S.; Omi, A.; Uchino, H.; Elmer, E. Evaluation of putative inhibitors of mitochondrial permeability transition for brain disorders--specificity vs. toxicity. Exp. Neurol. 2009, 218, 353–362. [Google Scholar] [CrossRef]
- Widlansky, M.E.; Hill, R.B. Mitochondrial regulation of diabetic vascular disease: An emerging opportunity. Transl. Res. 2018, 202, 83–98. [Google Scholar] [CrossRef]
- Zinkevich, N.S.; Fancher, I.S.; Gutterman, D.D.; Phillips, S.A. Roles of NADPH oxidase and mitochondria in flow-induced vasodilation of human adipose arterioles: ROS-induced ROS release in coronary artery disease. Microcirculation 2017, 24, e12380. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studies and Major Outcomes | Ref. |
|---|---|
| T1DM rats (STZ model) show sevenfold increase in gp91phox (Nox2) mRNA and uncoupled eNOS—diabetic complications were partially normalized by inhibition of PKC by chelerythrine | [242] |
| Genetic deletion of NoxO1 or p47phox reduced blood pressure and prevented diabetes-induced vascular dysfunction | [243] |
| Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease | [244] |
| Nox1 deficiency normalized diabetic glomerular DNA damage | [245] |
| NOX1 plays a key role in diabetes mellitus-accelerated atherosclerosis, which can be prevented by siRNA against Nox1 and GKT137831 therapy | [246] |
| Critical role for NOX2 in insulin resistance-related endothelial cell dysfunction as demonstrated by genetic deletion of Nox2 | [247] |
| Nox2 deficiency protects against STZ-induced beta-cell destruction and development of diabetes in mice | [248] |
| Normalization of mitochondrial ROS formation prevents several diabetic complications (glucose-induced activation of protein kinase C, formation of AGEs, sorbitol accumulation, and NFkB activation) | [89] |
| Blocking mitochondrial ROS formation with mitoTEMPO prevented diabetic cardiomyopathy | [249] |
| The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via NRF2/PINK1 | [250] |
| Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress | [251] |
| Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66Shc gene | [252] |
| Mammalian life-span determinant p66ShcA mediates obesity-induced insulin resistance | [253] |
| MAO-dependent endoplasmic reticulum-mitochondria dysfunction and mast cell degranulation lead to adverse cardiac remodeling in diabetes, which was supported by protective effects of MAO inhibito rpargyline | [254] |
| Emerging role of MAO as a therapeutic target for cardiovascular disease including treatment of diabetes | [255] |
| XO is activated in human and experimental T1DM, and the XO inhibitor allopurinol normalizes major diabetic complications | [256] |
| Atrial remodeling was prevented by allopurinol in diabetic rabbits (alloxan model) | [257] |
| XO inhibitor febuxostat exerts an anti-inflammatory action and protects against diabetic nephropathy development in KK-Ay obese diabetic mice | [258] |
| eNOS gene therapy exacerbates hepatic ischemia-reperfusion injury in diabetes, which was normalized by BH4 or the BH4 precursor sepiapterin providing evidence for eNOS uncoupling | [259] |
| Oxidation of the zinc–thiolate complex and uncoupling of eNOS by ONOO¯ as an explanation for endothelial dysfunction in the setting of diabetes | [260] |
| Overexpression of the BH4-generating enzyme GTP-cyclohydrolase-1 rescues eNOS function in diabetic mice indicating oxidative BH4 depletion in this model | [85] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daiber, A.; Steven, S.; Vujacic-Mirski, K.; Kalinovic, S.; Oelze, M.; Di Lisa, F.; Münzel, T. Regulation of Vascular Function and Inflammation via Cross Talk of Reactive Oxygen and Nitrogen Species from Mitochondria or NADPH Oxidase—Implications for Diabetes Progression. Int. J. Mol. Sci. 2020, 21, 3405. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103405
Daiber A, Steven S, Vujacic-Mirski K, Kalinovic S, Oelze M, Di Lisa F, Münzel T. Regulation of Vascular Function and Inflammation via Cross Talk of Reactive Oxygen and Nitrogen Species from Mitochondria or NADPH Oxidase—Implications for Diabetes Progression. International Journal of Molecular Sciences. 2020; 21(10):3405. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103405
Chicago/Turabian StyleDaiber, Andreas, Sebastian Steven, Ksenija Vujacic-Mirski, Sanela Kalinovic, Matthias Oelze, Fabio Di Lisa, and Thomas Münzel. 2020. "Regulation of Vascular Function and Inflammation via Cross Talk of Reactive Oxygen and Nitrogen Species from Mitochondria or NADPH Oxidase—Implications for Diabetes Progression" International Journal of Molecular Sciences 21, no. 10: 3405. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103405