Targeting Mitochondria during Cold Storage to Maintain Proteasome Function and Improve Renal Outcome after Transplantation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Ischemia/Reperfusion Injury
1.2. Mitochondrial Injury
1.2.1. Fission and Fusion
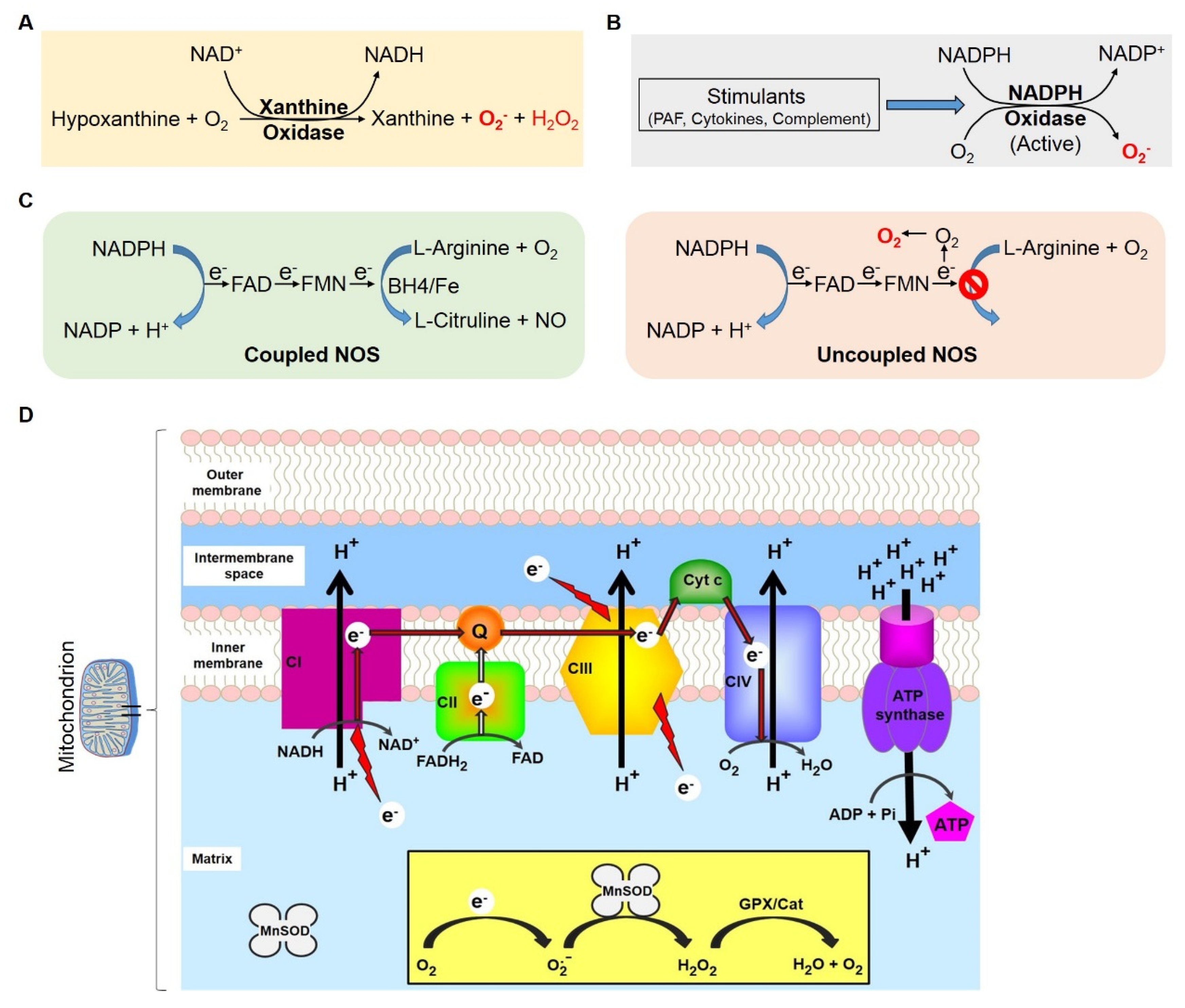
1.2.2. Mitochondrial Respiratory Complex and ROS
1.2.3. Mitophagy
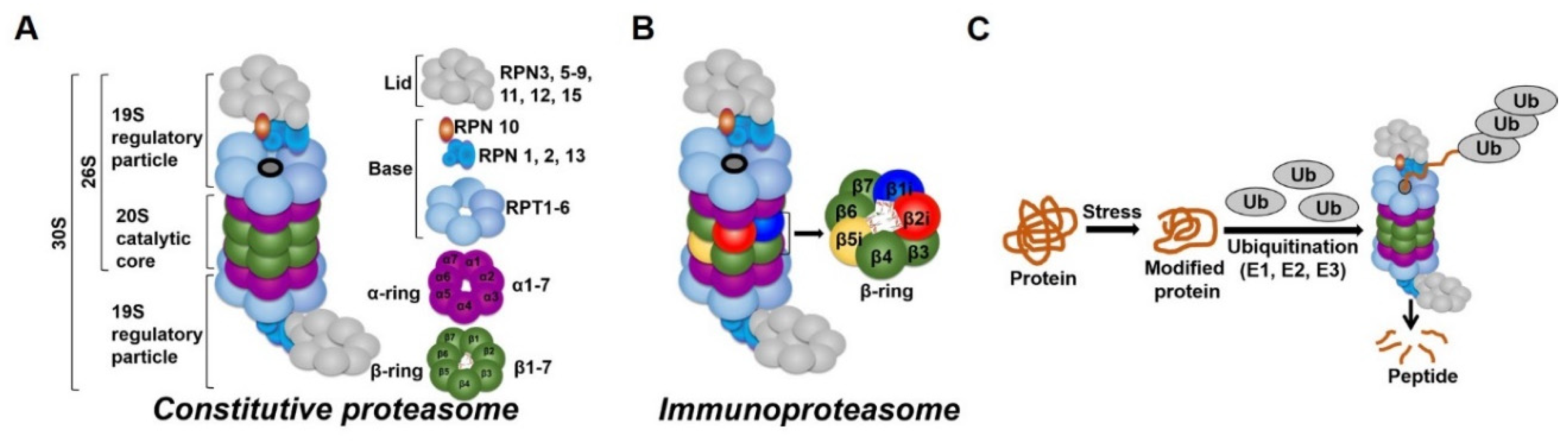
1.3. The Proteasome
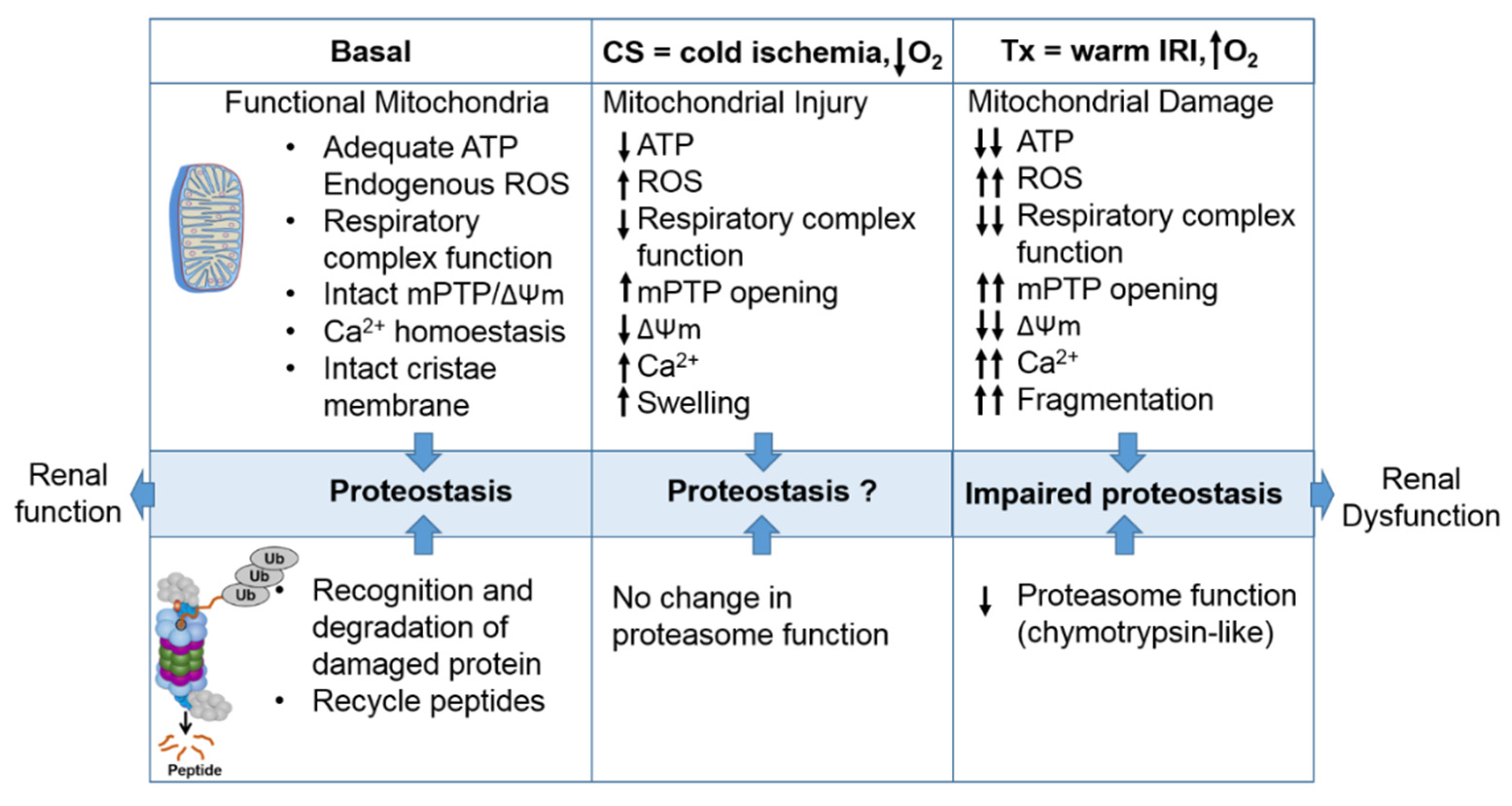
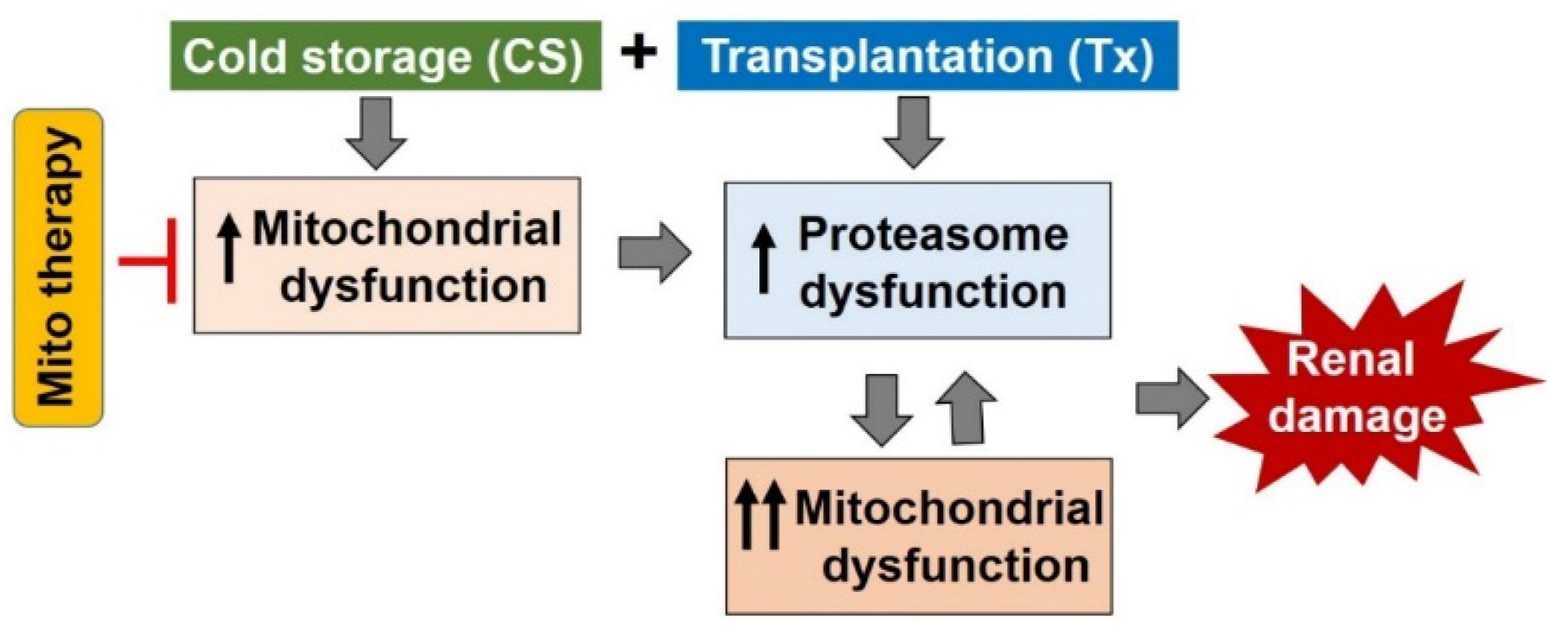
1.4. Interactions between the Mitochondria and Proteasome and the Potential Role of this Interaction during CS/Tx
2. Therapeutics Targeting Mitochondria to Improve Outcome after Transplantation
3. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sawhney, S.; Mitchell, M.; Marks, A.; Fluck, N.; Black, C. Long-term prognosis after acute kidney injury (AKI): What is the role of baseline kidney function and recovery? A systematic review. BMJ Open 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debout, A.; Foucher, Y.; Trébern-Launay, K.; Legendre, C.; Kreis, H.; Mourad, G.; Garrigue, V.; Morelon, E.; Buron, F.; Rostaing, L.; et al. Each additional hour of cold ischemia time significantly increases the risk of graft failure and mortality following renal transplantation. Kidney Int. 2015, 87, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosgood, S.A.; Bagul, A.; Nicholson, M.L. Minimising cold ischaemic injury in an experimental model of kidney transplantation. Eur. J. Clin. Investig. 2010, 41, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.K.; May, W. Reduction in Cold Ischemia Time of Renal Allografts in the United States Over the Last Decade. Transplant. Proc. 2008, 40, 1285–1289. [Google Scholar] [CrossRef] [Green Version]
- Kay, M.D.; Hosgood, S.A.; Bagul, A.; Nicholson, M. Comparison of preservation solutions in an experimental model of organ cooling in kidney transplantation. BJS 2009, 96, 1215–1221. [Google Scholar] [CrossRef]
- O’Callaghan, J.; Morgan, R.D.; Knight, S.; Morris, P.J. Systematic review and meta-analysis of hypothermic machine perfusion versus static cold storage of kidney allografts on transplant outcomes. BJS 2013, 100, 991–1001. [Google Scholar] [CrossRef]
- Mohan, S.; Foley, K.F.; Chiles, M.C.; Dube, G.K.; Patzer, R.E.; Pastan, S.O.; Crew, R.J.; Cohen, D.J.; Ratner, L.E. The weekend effect alters the procurement and discard rates of deceased donor kidneys in the United States. Kidney Int. 2016, 90, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D.; Garcia, V.C.; Rosendale, J.D.; Klassen, D.; Carrico, B.J. Diagnosing the Decades-Long Rise in the Deceased Donor Kidney Discard Rate in the United States. Transplantation 2017, 101, 575–587. [Google Scholar] [CrossRef]
- Oxburgh, L. Kidney Nephron Determination. Ann. Rew. Cell Dev. Biol. 2018, 34, 427–450. [Google Scholar] [CrossRef]
- M’Donnell, R. Observations on the Anatomy and Physiology of the Kidney. Glas. Med. J. 2009, 101, 285–295. [Google Scholar]
- Kriz, W.; Sanchez-Lozada, L.G.; Bulger, R.E.; Burg, M.B.; Goncharevskaya, O.A.; Imai, M.; Kaissling, B.; Maunsbach, A.B.; Moffat, D.B.; Morgan, T.O.; et al. A standard nomenclature for structures of the kidney. Pflüger, Archiv für die Gesammte Physiologie des Menschen und der Thiere 1988, 411, 113–120. [Google Scholar] [CrossRef]
- Kumaran, G.K.; Hanukoglu, I. Identification and classification of epithelial cells in nephron segments by actin cytoskeleton patterns. FEBS J. 2019, 287, 1176–1194. [Google Scholar] [CrossRef]
- Wallace, M.A. Anatomy, and physiology of the kidney. AORN J. 1998, 68, 799–820. [Google Scholar] [CrossRef]
- Hansell, P.; Welch, W.J.; Blantz, R.C.; Palm, F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 2013, 40, 123–137. [Google Scholar] [CrossRef] [Green Version]
- Persson, P.B.; Ehmke, H.; Kirchheim, H.R.; Janssen, B.; E Baumann, J.; Just, A.; Nafz, B. Autoregulation and non-homeostatic behaviour of renal blood flow in conscious dogs. J. Physiol. 1993, 462, 261–273. [Google Scholar] [CrossRef]
- Levy, M.N. Effect of variations of blood flow on renal oxygen extraction. Am. J. Physiol. Content 1960, 199, 13–18. [Google Scholar] [CrossRef]
- Bonventre, J.V. Mediators of ischemic renal injury. Ann. Rev. Med. 1988, 39, 531–544. [Google Scholar] [CrossRef]
- Land, W.G. Impact of the reperfusion injury on acute and chronic rejection events following clinical cadaveric renal transplantation. J. Mol. Med. 1994, 72, 719. [Google Scholar] [CrossRef]
- Zhao, H.; Yoshida, A.; Xiao, W.; Ologunde, R.; O’Dea, K.P.; Takata, M.; Tralau-Stewart, C.; George, A.J.; Ma, D. Xenon treatment attenuates early renal allograft injury associated with prolonged hypothermic storage in rats. FASEB J. 2013, 27, 4076–4088. [Google Scholar] [CrossRef]
- Tullius, S.G.; Reutzel-Selke, A.; Egermann, F.; Nieminen-Kelhä, M.; Jonas, S.; Bechstein, W.O.; Volk, H.D.; Neuhaus, P. Contribution of prolonged ischemia and donor age to chronic renal allograft dysfunction. J. Am. Soc. Nephrol. 2000, 11, 1317–1324. [Google Scholar] [PubMed]
- Wagner, M.; Cadetg, P.; Ruf, R.; Mazzucchelli, L.; Ferrari, P.; Redaelli, C.A. Heme oxygenase-1 attenuates ischemia/reperfusion-induced apoptosis and improves survival in rat renal allografts. Kidney Int. 2003, 63, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- Engerson, T.D.; McKelvey, T.G.; Rhyne, D.B.; Boggio, E.B.; Snyder, S.J.; Jones, H.P. Conversion of xanthine dehydrogenase to oxidase in ischemic rat tissues. J. Clin. Investig. 1987, 79, 1564–1570. [Google Scholar] [CrossRef]
- Koyama, I.; Bulkley, G.B.; Williams, G.M.; IM, M.J. The Role of Oxygen Free Radivals in Mediating the Reperfusion Injury of Cold-Preserved Ischemic Kidneys1,2. Transplantation 1985, 40, 590–595. [Google Scholar] [CrossRef]
- Paller, M.S.; Hoidal, J.R.; Ferris, T.F. Oxygen free radicals in ischemic acute renal failure in the rat. J. Clin. Investig. 1984, 74, 1156–1164. [Google Scholar] [CrossRef] [Green Version]
- Fridovich, I. Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J. Boil. Chem. 1970, 245, 4053–4057. [Google Scholar]
- Sun, K.; Kiss, E.; Bedke, J.; Stojanovic, T.; Li, Y.; Gwinner, W.; Gröne, H.-J. Role of Xanthine Oxidoreductase in Experimental Acute Renal-Allograft Rejection. Transplantation 2004, 77, 1683–1692. [Google Scholar] [CrossRef]
- Mangnall, D.; Bartley, W. Changes in the Enzymic Capacity of the Rat Kidney and Heart Due to Unilateral Nephrectomy and Deoxycorticosterone Induced Hypertension. Br. J. Exp. Pathol. 1970, 51, 464–476. [Google Scholar]
- Simone, S.; Rascio, F.; Castellano, G.; Divella, C.; Chieti, A.; Ditonno, P.; Battaglia, M.; Crovace, A.; Staffieri, F.; Oortwijn, B.; et al. Complement-dependent NADPH oxidase enzyme activation in renal ischemia/reperfusion injury. Free Radic. Boil. Med. 2014, 74, 263–273. [Google Scholar] [CrossRef]
- Schramm, L.; La, M.; Heidbreder, E.; Hecker, M.; Beckman, J.S.; Lopau, K.; Zimmermann, J.; Rendl, J.; Reiners, C.; Winderl, S.; et al. L-Arginine deficiency and supplementation in experimental acute renal failure and in human kidney transplantation. Kidney Int. 2002, 61, 1423–1432. [Google Scholar] [CrossRef] [Green Version]
- Mandegary, A.; Rahmanian-Koshkaki, S.; Mohammadifar, M.-A.; Pourgholi, L.; Mehdipour, M.; Etminan, A.; Ebadzadeh, M.-R.; Fazeli, F.; Azmandian, J. Investigation of association between donors’ and recipients’ NADPH oxidase p22phox C242T polymorphism and acute rejection, delayed graft function and blood pressure in renal allograft recipients. Transpl. Immunol. 2015, 32, 46–50. [Google Scholar] [CrossRef]
- Kleikers, P.W.M.; Wingler, K.; Hermans, J.J.R.; Diebold, I.; Altenhöfer, S.; Radermacher, K.A.; Janssen, B.; Görlach, A.; Schmidt, H.H.H.W. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J. Mol. Med. 2012, 90, 1391–1406. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Hear. J. 2011, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Münzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Masano, T.; Kawashima, S.; Toh, R.; Satomi-Kobayashi, S.; Shinohara, M.; Takaya, T.; Sasaki, N.; Takeda, M.; Tawa, H.; Yamashita, T.; et al. Beneficial effects of exogenous tetrahydrobiopterin on left ventricular remodeling after myocardial infarction in rats: The possible role of oxidative stress caused by uncoupled endothelial nitric oxide synthase. Circ. J. 2008, 72, 1512–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkaitis, M.S.; Crabtree, M.J. Recoupling the Cardiac Nitric Oxide Synthases: Tetrahydrobiopterin Synthesis and Recycling. Curr. Hear. Fail. Rep. 2012, 9, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Salahudeen, A.; Nawaz, M.; Poovala, V.; Kanji, V.; Wang, C.; Morrow, J.; Roberts, J. Cold storage induces time-dependent F2-isoprostane formation in renal tubular cells and rat kidneys. Kidney Int. 1999, 55, 1759–1762. [Google Scholar] [CrossRef] [Green Version]
- Salahudeen, A.K. Free radicals in kidney disease and transplantation. Saudi J. Kidney Dis. Transplant. 1999, 10, 137–143. [Google Scholar]
- Salin, M.L.; Mccord, J.M. Free-Radicals, and Inflammation-Protection of Phagocytosing Leukocytes by Superoxide-Dismutase. J. Clin. Investig. 1975, 56, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.J.; Weinberg, J.M. Postischemic renal injury due to oxygen radicals. Curr. Opin. Nephrol. Hypertens. 1993, 2, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Hypoxia-inducible factors in the kidney. Am. J. Physiol. Physiol. 2006, 291, F271–F281. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of Oxygen Homeostasis by Hypoxia-Inducible Factor 1. Physiology 2009, 24, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberger, C.; Heyman, S.N.; Rosen, S.; Shina, A.; Goldfarb, M.; Griethe, W.; Frei, U.; Reinke, P.; Bachmann, S.; Eckardt, K.-U. Up-regulation of HIF in experimental acute renal failure: Evidence for a protective transcriptional response to hypoxia. Kidney Int. 2005, 67, 531–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell, U.R.; Kvietikova, I.; Scheid, A.; Bauer, C.; Wenger, R.H.; Gassmann, M. Induction of HIF–1α in response to hypoxia is instantaneous. FASEB J. 2001, 15, 1312–1314. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, A.; Gaskell, S.J.; Von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, I.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef] [Green Version]
- Gunaratnam, L.; Bonventre, J.V. HIF in Kidney Disease and Development. J. Am. Soc. Nephrol. 2008, 20, 1877–1887. [Google Scholar] [CrossRef] [Green Version]
- Sethi, K.; Rao, K.; Bolton, D.; Patel, O.; Ischia, J. Targeting HIF-1α to Prevent Renal Ischemia-Reperfusion Injury: Does It Work? Int. J. Cell Boil. 2018, 2018, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Legrand, M.; Mik, E.G.; Johannes, T.; Payen, D.; Ince, C. Renal Hypoxia and Dysoxia After Reperfusion of the Ischemic Kidney. Mol. Med. 2008, 14, 502–516. [Google Scholar] [CrossRef]
- Thiel, G.; De Rougemont, D.; Kriz, W.; Mason, J.; Torhorst, J.; Wolgast, M. The role of reduced medullary perfusion in the genesis of acute ischemic renal failure. Summary of a round-table discussion. Nephron 1982, 31, 321–323. [Google Scholar] [CrossRef]
- Mason, J.; Torhorst, J.; Welsch, J. Role of the medullary perfusion defect in the pathogenesis of ischemic renal failure. Kidney Int. 1984, 26, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Hellberg, P.O.A.; Källskog, Ö.; Wolgast, M. Red cell trapping and postischemic renal blood flow. Differences between the cortex, outer and inner medulla. Kidney Int. 1991, 40, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Schrier, R.W.; Wang, W.; Poole, B.; Mitra, A. Acute renal failure: Definitions, diagnosis, pathogenesis, and therapy. J. Clin. Investig. 2004, 114, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Rosen, S.; Epstein, F.H.; Brezis, M. Determinants of Intrarenal Oxygenation: Factors in Acute Renal Failure. Ren. Fail. 1992, 14, 321–325. [Google Scholar] [CrossRef]
- Cohen, J.J. Is the function of the renal papilla coupled exclusively to an anaerobic pattern of metabolism? Am. J. Physiol. Physiol. 1979, 236, F423–F433. [Google Scholar] [CrossRef]
- Nangaku, M. Chronic Hypoxia and Tubulointerstitial Injury: A Final Common Pathway to End-Stage Renal Failure. J. Am. Soc. Nephrol. 2005, 17, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, A.S.; Rothstein, D.M.; Safa, K.; Riella, L.V.; Kassem, S. Outstanding questions in transplantation: B cells, alloantibodies, and humoral rejection. Arab. Archaeol. Epigr. 2019, 19, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- Barnett, N.; Dorling, A.; Mamode, N. B cells in renal transplantation: Pathological aspects and therapeutic interventions. Nephrol. Dial. Transplant. 2010, 26, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Cucchiari, D.; Podestà, M.A.; Ponticelli, C. The Critical Role of Innate Immunity in Kidney Transplantation. Nephron 2016, 132, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Biglarnia, A.-R.; Huber-Lang, M.; Mohlin, C.; Ekdahl, K.N.; Nilsson, B. The multifaceted role of complement in kidney transplantation. Nat. Rev. Nephrol. 2018, 14, 767–781. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; Mastellos, D.; Tudoran, R.; DeAngelis, R.A.; Strey, C.W.; Franchini, S.; Wetsel, R.A.; Erdei, A.; Lambris, J.D. C3a and C3b activation products of the third component of complement (C3) are critical for normal liver recovery after toxic injury. J. Immunol. 2004, 173, 747–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Li, H.; Feng, J.; Zhang, Y.; Feng, J.; Wang, Q.; Zhao, S.; Meng, P.; Li, J. Mst1 deletion attenuates renal ischaemia-reperfusion injury: The role of microtubule cytoskeleton dynamics, mitochondrial fission and the GSK3β-p53 signalling pathway. Redox Boil. 2018, 20, 261–274. [Google Scholar] [CrossRef]
- Xiao, X.; Hu, Y.; Quiros, P.M.; Wei, Q.; Lopez-Otin, C.; Dong, Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Physiol. 2014, 306, 1318–1326. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.M. Maintaining mitochondrial morphology in AKI: Looks matter. J. Am. Soc. Nephrol. 2013, 24, 1185–1187. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.M.; Rhodes, G.J.; Sandoval, R.M.; Corridon, P.R.; Molitoris, B.A. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 2012, 83, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.-Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhu, P.; Li, R.; Ren, J.; Zhou, H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Boil. 2020, 30, 101415. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, J.; Li, X.; Liu, Z.; Han, Y.; Xu, X.; Li, X.; Tang, Y.; Liu, Y.; Yu, T.; et al. Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway. J. Cell. Physiol. 2019, 234, 23495–23506. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; I Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Boil. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman1, R.L. Reciprocal Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.J.; A Lampe, P.; Stojanovski, D.; Korwitz, A.; Anand1 ‡, R.; Tatsuta, T.; Langer, T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014, 33, 578–593. [Google Scholar] [CrossRef] [Green Version]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman1, R.L. YME 1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Rep. 2014, 16, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.; Currin, R.T.; Peng, X.-X.; Mekeel, H.E.; Schoonhoven, R.; Lemasters, J.J. Different Patterns of Renal Cell Killing After Warm and Cold schemia. Ren. Fail. 2002, 24, 147–163. [Google Scholar] [CrossRef]
- Salahudeen, A.K.; Joshi, M.; Jenkins, J.K. Apoptosis Versus Necrosis During Cold Storage and Rewarming of Human Renal Proximal Tubular Cellis1. Transplantation 2001, 72, 798–804. [Google Scholar] [CrossRef]
- Parajuli, N.; Shrum, S.; Tobacyk, J.; Harb, A.; Arthur, J.; MacMillan-Crow, L.A. Renal cold storage followed by transplantation impairs expression of key mitochondrial fission and fusion proteins. PLoS ONE 2017, 12, e0185542. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhang, G.; Song, Z.; Xiang, X.; Shu, S.; Liu, Z.; Yang, D.; Wei, Q.; Dong, Z. Protein Kinase C-delta Mediates Kidney Tubular Injury in Cold Storage-Associated Kidney Transplantation. J. Am. Soc. Nephrol. JASN 2020, 31, 1050–1065. [Google Scholar] [CrossRef] [PubMed]
- Ralto, K.M.; Parikh, S.M. Mitochondria in Acute Kidney Injury. Semin. Nephrol. 2016, 36, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatefi, Y. The mitochondrial electron transport and oxidative phosphorylation system. Ann. Rew. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.; Rotaru, D.; Saba, H.; Smith, R.A.J.; Murphy, M.P.; MacMillan-Crow, L.A. The mitochondria-targeted antioxidant mitoquinone protects against cold storage injury of renal tubular cells and rat kidneys. J. Pharmacol. Exp. Ther. 2010, 336, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Salahudeen, A.K.; Huang, H.; Joshi, M.; Moore, N.A.; Jenkins, J.K. Involvement of the mitochondrial pathway in cold storage and rewarming-associated apoptosis of human renal proximal tubular cells. Arab. Archaeol. Epigr. 2003, 3, 273–280. [Google Scholar] [CrossRef]
- Salahudeen, A.K.; Huang, H.; Patel, P.; Jenkins, J.K. Mechanism and Prevention of Cold Storage-Induced Human Renal Tubular Cell Injury12. Transplantation 2000, 70, 1424–1431. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Lindell, S.L.; Henderson, S.; Mangino, M. Protective Effects of Ezrin on Cold Storage Preservation Injury in the Pig Kidney Proximal Tubular Epithelial Cell Line (LLC-PK1). Transplantation 2009, 87, 1488–1496. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.; Saba, H.; Laakman, J.; Parajuli, N.; MacMillan-Crow, L.A. Role of mitochondrial-derived oxidants in renal tubular cell cold-storage injury. Free Radic. Boil. Med. 2010, 49, 1273–1282. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, N.; Campbell, L.H.; Marine, A.; Brockbank, K.G.M.; MacMillan-Crow, L.A. MitoQ Blunts Mitochondrial and Renal Damage during Cold Preservation of Porcine Kidneys. PLoS ONE 2012, 7, e48590. [Google Scholar] [CrossRef] [Green Version]
- Shrum, S.; MacMillan-Crow, L.A.; Parajuli, N. Cold Storage Exacerbates Renal and Mitochondrial Dysfunction Following Transplantation. J. Kidney 2016, 2. [Google Scholar] [CrossRef]
- Pajuri, N.; MacMillan-Crow, L.A. Role of reduced manganese superoxide dismutase in ischemia-reperfusion injury: A possible trigger for autophagy and mitochondrial biogenesis? Am. J. Physiol. Physiol. 2012, 304, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, N.; Marine, A.; Simmons, S.; Saba, H.; Mitchell, T.; Shimizu, T.; Shirasawa, T.; MacMillan-Crow, L.A. Generation and characterization of a novel kidney-specific manganese superoxide dismutase knockout mouse. Free Radic. Boil. Med. 2011, 51, 406–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosieradzki, M.; Rowiński, W. Ischemia/Reperfusion Injury in Kidney Transplantation: Mechanisms and Prevention. Transplant. Proc. 2008, 40, 3279–3288. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P. Cellular and molecular derangements in acute tubular necrosis. Curr. Opin. Pediatr. 2005, 17, 193–199. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Boil. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Wang, X.; Ding, R.; Tao, T.-Q.; Mao, H.-M.; Liu, M.; Xie, Y.-S.; Liu, X. Myofibrillogenesis Regulator 1 Rescues Renal Ischemia/Reperfusion Injury by Recruitment of PI3K-Dependent P-AKT to Mitochondria. Shock 2016, 46, 531–540. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Nakagawa, T.; Shimizu, S. Mitochondrial membrane permeability transition and cell death. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 1297–1300. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, S.J.; Hosford, M.A.; Molitoris, B.A. Mechanism of Action Polymerization in Cellular ATP Depletion. J. Boil. Chem. 2003, 279, 5194–5199. [Google Scholar] [CrossRef] [Green Version]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Morrison, A.R.; Pascoe, N.; Tauk, N.; Kennerly, D. Biochemical alterations of membrane lipids associated with renal injury. Fed. Proc. 1984, 43, 2811–2814. [Google Scholar]
- Arnarez, C.; Mazat, J.-P.; Elezgaray, J.; Marrink, S.J.; Periole, X. Evidence for Cardiolipin Binding Sites on the Membrane-Exposed Surface of the Cytochrome bc1. J. Am. Chem. Soc. 2013, 135, 3112–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, W.B.; Neff, J.K.; Gohil, V.M. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett. 2017, 592, 1273–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Boil. Chem. 2005, 280, 29403–29408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehdipour, A.R.; Hummer, G. Cardiolipin puts the seal on ATP synthase. Proc. Nat. Acad. Sci. USA 2016, 113, 8568–8570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, A.; Robinson, A.J.; Walker, J.E. Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Nat. Acad. Sci. USA 2016, 113, 8687–8692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasdev, S.C.; Biro, G.P.; Narbaitz, R.; Kako, K.J. Membrane changes induced by early myocardial ischemia in the dog. Can. J. Biochem. 1980, 58, 1112–1119. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Minkler, P.; Hoppel, C.L. Enhanced modification of cardiolipin during ischemia in the aged heart. J. Mol. Cell. Cardiol. 2009, 46, 1008–1015. [Google Scholar] [CrossRef]
- Wiswedel, I.; Gardemann, A.; Storch, A.; Peter, D.; Schild, L. Degradation of phospholipids by oxidative stress--exceptional significance of cardiolipin. Free Radic. Res. 2010, 44, 135–145. [Google Scholar] [CrossRef]
- Petrosillo, G.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Paradies, G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: Induction of permeability transition and cytochromecrelease. FEBS Lett. 2006, 580, 6311–6316. [Google Scholar] [CrossRef] [Green Version]
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Ca2+-induced Reactive Oxygen Species Production Promotes CytochromecRelease from Rat Liver Mitochondria via Mitochondrial Permeability Transition (MPT)-dependent and MPT-independent Mechanisms. J. Boil. Chem. 2004, 279, 53103–53108. [Google Scholar] [CrossRef] [Green Version]
- Silvestro, L.; Ruikun, C.; Sommer, F.; Duc, T.; Biancone, L.; Montrucchio, G.; Camussi, G. Platelet-Activating Factor-Induced Endothelial Cell Expression of Adhesion Molecules and Modulation of Surface Glycocalyx, Evaluated by Electron Spectroscopy for Chemical Analysis. Semin. Thromb. Hemost. 1994, 20, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimoto, A.; Matsukuma, Y.; Ueki, K.; Nishiki, T.; Doi, A.; Okabe, Y.; Nakamura, M.; Tsuruya, K.; Nakano, T.; Kitazono, T.; et al. Thrombotic microangiopathy associated with anticardiolipin antibody in a kidney transplant recipient with polycythemia. CEN Case Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, J.; Abeliovich, H.; Acevedo-Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Boil. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Li, H.; Zhang, Y.; Wang, Q.; Zhao, S.; Meng, P.; Li, J. Mammalian STE20-Like Kinase 1 Deletion Alleviates Renal Ischaemia-Reperfusion Injury via Modulating Mitophagy and the AMPK-YAP Signalling Pathway. Cell. Physiol. Biochem. 2018, 51, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Han, F.; Xiang, H.; Wang, Y.; Dou, M.; Xia, X.; Li, Y.; Zheng, J.; Ding, X.; Xue, W.; et al. Role of prostaglandin E2 receptor 4 in the modulation of apoptosis and mitophagy during ischemia/reperfusion injury in the kidney. Mol. Med. Rep. 2019, 20, 3337–3346. [Google Scholar] [CrossRef]
- Ang, C.T.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia–reperfusion injury. Cell Death Dis. 2019, 10, 677. [Google Scholar] [CrossRef]
- Li, N.; Wang, H.; Jiang, C.; Zhang, M. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp. Cell Res. 2018, 369, 27–33. [Google Scholar] [CrossRef]
- Jackson, E.K.; Menshikova, E.V.; Mi, Z.; Verrier, J.D.; Bansal, R.; Janesko-Feldman, K.; Jackson, T.C.; Kochanek, P.M. Renal 2’,3’-Cyclic Nucleotide 3’-Phosphodiesterase Is an Important Determinant of AKI Severity after Ischemia-Reperfusion. J. Am. Soc. Nephrol. JASN 2016, 27, 2069–2081. [Google Scholar] [CrossRef]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.-M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Deken, J.; Rex, S.; Lerut, E.; Martinet, W.; Monbaliu, D.; Pirenne, J.; Jochmans, I. Postconditioning effects of argon or xenon on early graft function in a porcine model of kidney autotransplantation. BJS 2018, 105, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Keys, D.; Nydam, T.; Plenter, R.J.; Edelstein, C.L.; Jani, A. Inhibition of autophagy increases apoptosis during re-warming after cold storage in renal tubular epithelial cells. Transpl. Int. 2014, 28, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Pallet, N. Emerging Roles of Autophagy in the Stressed Kidney Allograft. Semin. Nephrol. 2014, 34, 34–41. [Google Scholar] [CrossRef]
- Pallet, N.; Livingston, M.; Dong, Z. Emerging Functions of Autophagy in Kidney Transplantation. Arab. Archaeol. Epigr. 2013, 14, 13–20. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Damage-associated molecular patterns derived from mitochondria may contribute to the hemodialysis-associated inflammation. Int. Urol. Nephrol. 2013, 46, 107–112. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Ren, J.; Wu, J.; Li, G.; Wu, X.; Liu, S.; Wang, G.; Gu, G.; Ren, H.; Hong, Z.; et al. Urinary Mitochondrial DNA Levels Identify Acute Kidney Injury in Surgical Critical Illness Patients. Shock 2017, 48, 11–17. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, J.; Ren, H.; Wu, J.; Wu, X.; Liu, S.; Wang, G.; Gu, G.; Guo, K.; Li, J. Urinary Mitochondrial DNA Identifies Renal Dysfunction and Mitochondrial Damage in Sepsis-Induced Acute Kidney Injury. Oxidative Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef]
- Kim, K.; Moon, H.; Lee, Y.H.; Seo, J.-W.; Kim, Y.G.; Moon, J.-Y.; Kim, J.S.; Jeong, K.-H.; Lee, T.W.; Ihm, C.-G.; et al. Clinical relevance of cell-free mitochondrial DNA during the early postoperative period in kidney transplant recipients. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Soares, F.; Tattoli, I.; Girardin, S.E. Mitochondria in innate immunity. EMBO Rep. 2011, 12, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2010, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2015, 68, 20–48. [Google Scholar] [CrossRef]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin-dependent proteolytic pathway: Specificity of recognition of the proteolytic substrates. Revis. Sobre Boil. Cel. RBC 1989, 20, 217–234. [Google Scholar]
- Glickman, M.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Zwickl, P.; Voges, D.; Baumeister, W. The proteasome: A macromolecular assembly designed for controlled proteolysis. Philos. Trans. R. Soc. B: Boil. Sci. 1999, 354, 1501–1511. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The Ubiquitin Pathway for the Degradation of Intracellular Proteins. Prog. Nucl. Acid Res. Mol. Biol. 1986, 33, 19–56. [Google Scholar] [CrossRef]
- Jung, T.; Grune, T. Structure of the Proteasome. Prog. Mol. Biol. Transl. Sci. 2012, 109, 1–39. [Google Scholar] [CrossRef]
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basler, M.; Mundt, S.; Bitzer, A.; Schmidt, C.; Groettrup, M. The immunoproteasome: A novel drug target for autoimmune diseases. Clin. Exp. Rheumatol. 2015, 33. [Google Scholar]
- Kaur, G.; Batra, S. Emerging role of immunoproteasomes in pathophysiology. Immunol. Cell Boil. 2016, 94, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Sjöstrand, J.; Karlsson, J.-O. Differential Inhibition of Three Peptidase Activities of the Proteasome in Human Lens Epithelium by Heat and Oxidation. Exp. Eye Res. 1999, 69, 129–138. [Google Scholar] [CrossRef]
- Reinheckel, T.; Sitte, N.; Ullrich, O.; Kuckelkorn, U.; Davies, K.E.; Grune, T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem. J. 1998, 335, 637–642. [Google Scholar] [CrossRef]
- Reinheckel, T.; Ullrich, O.; Sitte, N.; Grune, T. Differential Impairment of 20S and 26S Proteasome Activities in Human Hematopoietic K562 Cells during Oxidative Stress. Arch. Biochem. Biophys. 2000, 377, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.; MacMillan-Crow, L.A.; Parajuli, N. Renal cold storage followed by transplantation impairs proteasome function and mitochondrial protein homeostasis. Am. J. Physiol. 2019, 316, 42–53. [Google Scholar] [CrossRef]
- Huber, J.M.; Tagwerker, A.; Heininger, D.; Mayer, G.; Rosenkranz, A.R.; Eller, K. The proteasome inhibitor Bortezomib aggravates renal ischemia-reperfusion injury. Am. J. Physiol. 2009, 297, 451–460. [Google Scholar] [CrossRef]
- Parajuli, N. A Cycle of Altered Proteasome and Reactive Oxygen Species Production in Renal Proximal Tubular Cells. Toxicol. Forensic Med. Open J. 2019, 4, 13–17. [Google Scholar] [CrossRef]
- Scruggs, S.B.; Zong, N.C.; Wang, D.; Stefani, E.; Ping, P. Post-translational modification of cardiac proteasomes: Functional delineation enabled by proteomics. Am. J. Physiol. Circ. Physiol. 2012, 303, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Kors, S.; Geijtenbeek, K.; Reits, E.; Schipper-Krom, S. Regulation of Proteasome Activity by (Post-)transcriptional Mechanisms. Front. Mol. Biosci. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherukuri, A.; Mehta, R.; Sood, P.; Hariharan, S. Early allograft inflammation and scarring associate with graft dysfunction and poor outcomes in renal transplant recipients with delayed graft function: A prospective single center cohort study. Transpl. Int. 2018, 31, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Aiken, C.T.; Kaake, R.M.; Wang, X.; Huang, L. Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteom. MCP 2011, 10. [Google Scholar]
- Seifert, U.; Bialy, L.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes Preserve Protein Homeostasis upon Interferon-Induced Oxidative Stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Basler, M.; Alvarez, G.; Brunner, T.; Kirk, C.J.; Groettrup, M. Immunoproteasome inhibition prevents chronic antibody-mediated allograft rejection in renal transplantation. Kidney Int. 2018, 93, 670–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, X.; Wang, Y.; Lei, H.; Su, H.; Zeng, J.; Pei, Z.; Huang, R. Inhibition of immunoproteasome reduces infarction volume and attenuates inflammatory reaction in a rat model of ischemic stroke. Cell Death Dis. 2015, 6, 1626. [Google Scholar] [CrossRef] [Green Version]
- Karreci, E.S.; Fan, H.; Uehara, M.; Mihali, A.B.; Singh, P.K.; Kurdi, A.T.; Solhjou, Z.; Riella, L.V.; Ghobrial, I.; Laragione, T.; et al. Brief treatment with a highly selective immunoproteasome inhibitor promotes long-term cardiac allograft acceptance in mice. Proc. Nat. Acad. Sci. USA 2016, 113, 8425–8432. [Google Scholar] [CrossRef] [Green Version]
- Bellavista, E.; Andreoli, F.; Parenti, M.D.; Martucci, M.; Santoro, A.; Salvioli, S.; Capri, M.; Baruzzi, A.; Del Rio, A.; Franceschi, C.; et al. Immunoproteasome in Cancer and Neuropathologies: A New Therapeutic Target? Curr. Pharm. Des. 2013, 19, 702–718. [Google Scholar] [CrossRef]
- Miller, Z.; Ao, L.; Kim, K.B.; Lee, W. Inhibitors of the immunoproteasome: Current status and future directions. Curr. Pharm. Des. 2013, 19, 4140–4151. [Google Scholar] [CrossRef] [Green Version]
- Paul, S. Dysfunction of the ubiquitin-proteasome system in multiple disease conditions: Therapeutic approaches. BioEssays 2008, 30, 1172–1184. [Google Scholar] [CrossRef]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. et Biophys. Acta Bioenerg. 2013, 1843, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livnat-Levanon, N.; Glickman, M. Ubiquitin–Proteasome System and mitochondria—Reciprocity. Biochim. Biophys. Acta Bioenerg. 2011, 1809, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Bragoszewski, P.; Gornicka, A.; Sztolsztener, M.E.; Chacinska, A. The Ubiquitin-Proteasome System Regulates Mitochondrial Intermembrane Space Proteins. Mol. Cell. Boil. 2013, 33, 2136–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, S.; Chander, H.; Schäfer, P.; Meiss, G.; Krüger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Boil. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.B.; Choi, E.S.; Yoon, J.H.; Hwang, J.H.; Chang, J.W.; Lee, E.K.; Choi, H.W.; Park, Z.-Y.; Yoo, Y.J. A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem. Biophys. Res. Commun. 2007, 357, 731–736. [Google Scholar] [CrossRef]
- Altmann, K.; Westermann, B. Role of Essential Genes in Mitochondrial Morphogenesis In Saccharomyces cerevisiae. Mol. Boil. Cell 2005, 16, 5410–5417. [Google Scholar] [CrossRef] [Green Version]
- Azzu, V.; Brand, M.D. Degradation of an intramitochondrial protein by the cytosolic proteasome. J. Cell Sci. 2010, 123, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Neutzner, A.; Youle, R.J.; Karbowski, M. Outer mitochondrial membrane protein degradation by the proteasome. Ciba Found. Sym. Bilharz. 2007, 287, 4–20. [Google Scholar] [CrossRef]
- Karbowski, M.; Youle, R.J. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Boil. 2011, 23, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Chatenay-Lapointe, M.; Shadel, G.S. Stressed-Out Mitochondria Get MAD. Cell Metab. 2010, 12, 559–560. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Peng, G.; Wang, Y.; Fang, S.; Karbowski, M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Boil. Cell 2011, 22, 291–300. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Brand, M.D. Characteristics of the turnover of uncoupling protein 3 by the ubiquitin proteasome system in isolated mitochondria. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 1474–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzu, V.; Mookerjee, S.A.; Brand, M.D. Rapid turnover of mitochondrial uncoupling protein 3. Biochem. J. 2010, 426, 13–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Q.; Romero, J.; Saini, V.; Baker, T.A.; Picken, M.M.; Gamelli, R.L.; Majetschak, M. A subset of 26S proteasomes is activated at critically low ATP concentrations and contributes to myocardial injury during cold ischemia. Biochem. Biophys. Res. Commun. 2009, 390, 1136–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Zhang, X.; Li, S.; Liu, N.; Lian, W.; McDowell, E.; Zhou, P.; Zhao, C.; Guo, H.; Zhang, C.; et al. Physiological levels of ATP negatively regulate proteasome function. Cell Res. 2010, 20, 1372–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, S.R.; Davies, K.E.; Divald, A. Optimal determination of heart tissue 26S-proteasome activity requires maximal stimulating ATP concentrations. J. Mol. Cell. Cardiol. 2006, 42, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Groll, M.; Bajorek, M.; Köhler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.; Finley, D. A gated channel into the proteasome core particle. Nat. Genet. 2000, 7, 1062–1067. [Google Scholar] [CrossRef]
- Bech-Otschir, D.; Helfrich, A.; Enenkel, C.; Consiglieri, G.; Seeger, M.; Holzhütter, H.-G.; Dahlmann, B.; Kloetzel, P.-M. Polyubiquitin substrates allosterically activate their own degradation by the 26S proteasome. Nat. Struct. Mol. Boil. 2009, 16, 219–225. [Google Scholar] [CrossRef]
- Smith, D.; Benaroudj, N.; Goldberg, A.L. Proteasomes and their associated ATPases: A destructive combination. J. Struct. Boil. 2006, 156, 72–83. [Google Scholar] [CrossRef]
- Smith, D.; Fraga, H.; Reis, C.; Kafri, G.; Goldberg, A.L. ATP Binds to Proteasomal ATPases in Pairs with Distinct Functional Effects, Implying an Ordered Reaction Cycle. Cell 2011, 144, 526–538. [Google Scholar] [CrossRef] [Green Version]
- Kron, P.; Schlegel, A.; de Rougemont, O.; Oberkofler, C.E.; Clavien, P.A.; Dutkowski, P. Short Cool, and Well Oxygenated-HOPE for Kidney Transplantation in a Rodent Model. Ann. Surg. 2016, 264, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kron, P.; Schlegel, A.; Muller, X.; Gaspert, A.; Clavien, P.-A.; Dutkowski, P. Hypothermic Oxygenated Perfusion. Transplantation 2019, 103, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yen, J.; Kaiser, P.; Huang, L. Regulation of the 26S Proteasome Complex During Oxidative Stress. Sci. Signal. 2010, 3, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.; Grune, T. The proteasome and the degradation of oxidized proteins: Part I-structure of proteasomes. Redox Boil. 2013, 1, 178–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.; Höhn, A.; Grune, T. The proteasome and the degradation of oxidized proteins: Part II -protein oxidation and proteasomal degradation. Redox Boil. 2013, 2, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Sitte, N.; Merker, K.; Grune, T. Proteasome-dependent degradation of oxidized proteins in MRC-5 fibroblasts. FEBS Lett. 1998, 440, 399–402. [Google Scholar] [CrossRef] [Green Version]
- Dare, A.J.; Logan, A.; Prime, T.A.; Rogatti, S.; Goddard, M.; Bolton, E.; Bradley, J.A.; Pettigrew, G.; Murphy, M.P.; Saeb-Parsy, K. The mitochondria-targeted antioxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J. Hear. Lung Transplant. 2015, 34, 1471–1480. [Google Scholar] [CrossRef] [Green Version]
- Dare, A.J.; Bolton, E.; Pettigrew, G.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Boil. 2015, 5, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Schlame, M.; Ren, M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta Bioenerg. 2009, 1788, 2080–2083. [Google Scholar] [CrossRef] [Green Version]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.K.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [Green Version]
- Talbert, E.E.; Smuder, A.J.; Hudson, M.; Nelson, W.; Min, K.; Szeto, H.H.; Kavazis, A.N.; Powers, S.K. A Mitochondrial-targeted Antioxidant Protects against Mechanical Ventilation-induced Diaphragm Weakness. Med. Sci. Sports Exerc. 2010, 42, 17–18. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Liu, S.; Soong, Y.; Szeto, H.H. Disruption of cytochrome c heme coordination is responsible for mitochondrial injury during ischemia. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1847, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochromec/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [Green Version]
- Saad, A.; Herrmann, S.M.; Eirin, A.; Ferguson, C.M.; Glockner, J.F.; Bjarnason, H.; McKusick, M.A.; Misra, S.; Lerman, L.O.; Textor, S.C. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) During Stent Revascularization in Patients With Atherosclerotic Renal Artery Stenosis. Circ. Cardiovasc. Interv. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology 2003, 124, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates someznecrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Kim, S.Y.; Shim, M.S.; Kim, K.-Y.; Weinreb, R.N.; A Wheeler, L.; Ju, W.-K. Inhibition of cyclophilin D by cyclosporin A promotes retinal ganglion cell survival by preventing mitochondrial alteration in ischemic injury. Cell Death Dis. 2014, 5, 1105. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Chander, V.; Chopra, K. Cyclosporine protects against ischemia/reperfusion injury in rat kidneys. Toxicology 2005, 207, 339–347. [Google Scholar] [CrossRef]
- De Arriba, G.; Calvino, M.; Benito, S.; Parra, T. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicol. Lett. 2013, 218, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Puigmulé, M.; Lopez-Hellin, J.; Suñè, G.; Tornavaca, O.; Camano, S.; Tejedor, A.; Meseguer, A. Differential proteomic analysis of cyclosporine A-induced toxicity in renal proximal tubule cells. Nephrol. Dial. Transplant. 2009, 24, 2672–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, N.; Kukla, A.; Ibrahim, H.N. Calcineurin Inhibitor Nephrotoxicity: A Review and Perspective of the Evidence. Am. J. Nephrol. 2013, 37, 602–612. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo, S.B.; Blaszak, R.T.; Parajuli, N. Targeting Mitochondria during Cold Storage to Maintain Proteasome Function and Improve Renal Outcome after Transplantation. Int. J. Mol. Sci. 2020, 21, 3506. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103506
Lo SB, Blaszak RT, Parajuli N. Targeting Mitochondria during Cold Storage to Maintain Proteasome Function and Improve Renal Outcome after Transplantation. International Journal of Molecular Sciences. 2020; 21(10):3506. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103506
Chicago/Turabian StyleLo, Sorena B., Richard T. Blaszak, and Nirmala Parajuli. 2020. "Targeting Mitochondria during Cold Storage to Maintain Proteasome Function and Improve Renal Outcome after Transplantation" International Journal of Molecular Sciences 21, no. 10: 3506. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103506