Similarity-Based Methods and Machine Learning Approaches for Target Prediction in Early Drug Discovery: Performance and Scope


Abstract
:1. Introduction
2. Results and Discussion
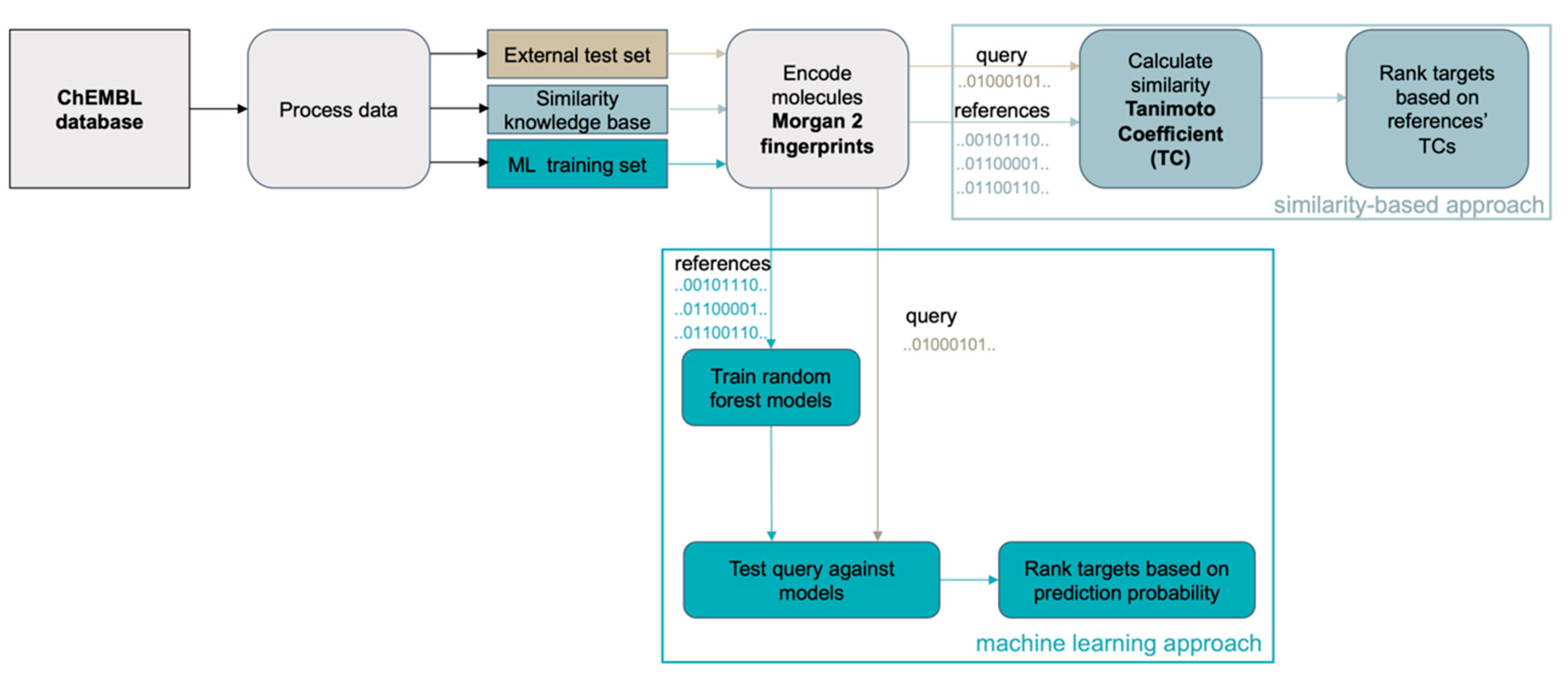
2.1. Similarity-Based Method and Machine Learning Approach for Target Prediction
2.2. Evaluation of the Scope and Performance of the Similarity-Based Method and Machine Learning Approach for Target Prediction
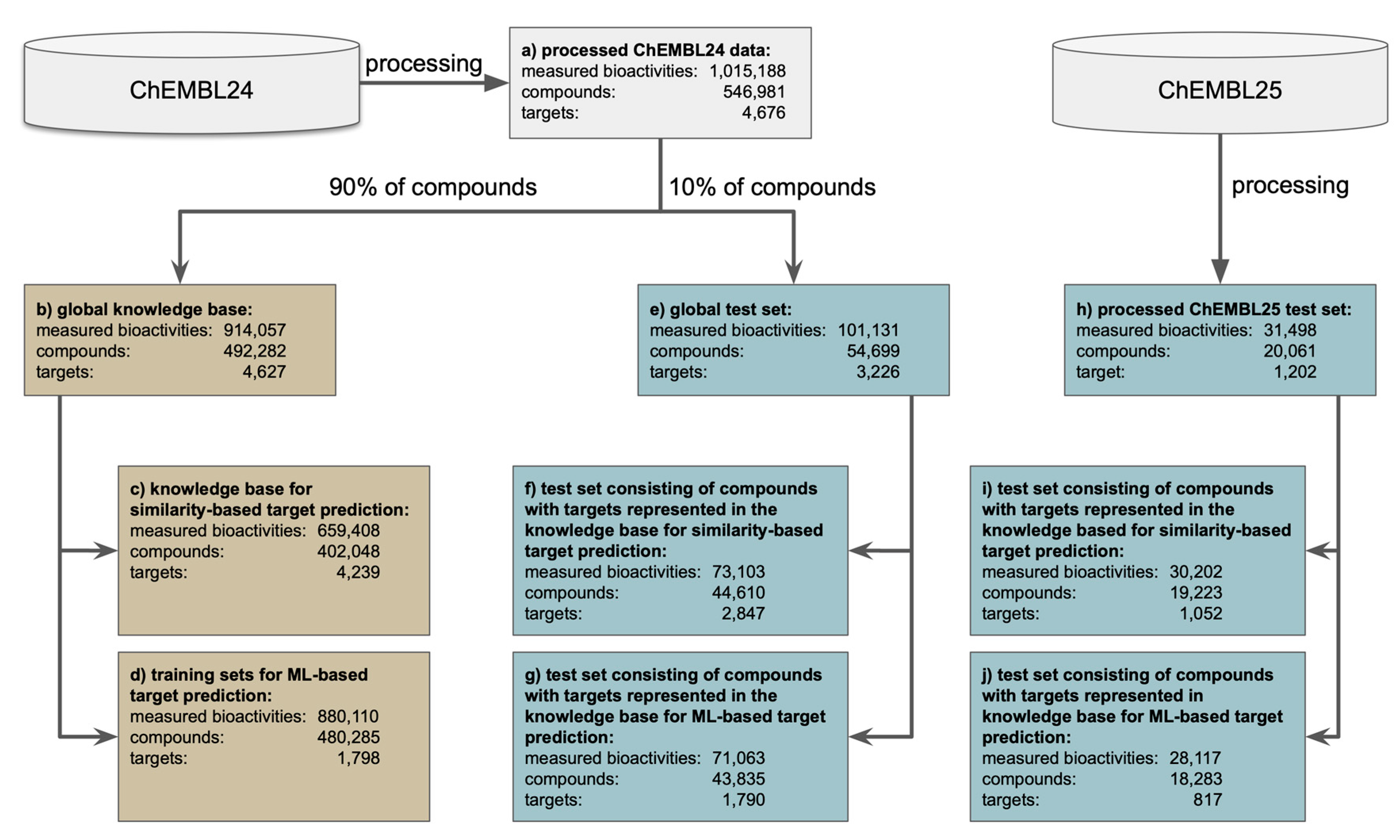
- Standard testing scenario with an external test set. Under this scenario, the approaches are tested for their ability to predict the targets of a set of approximately 44,000 query molecules (Figure 1f,g) obtained by a single random split of the processed ChEMBL24 database prior to model development.
- Standard time-split validation scenario within the target space covered by the approach. Under this scenario, the models are tested on the more than 18,000 molecules that have been newly introduced with version 25 of the ChEMBL database and have targets within the target space of the individual models (meaning that all compounds considered as queries have at least one known target that is covered by the approach’s knowledge base; Figure 1i,j). This test can give a sense of how model performance will change over time [39], with the increasing alienation of chemistry from that represented by the knowledge base (as observed in Figure 3).
- Close to real-world setting with an unbiased and comprehensive time-split dataset. Under this scenario the methods were tested on the full set of bioactive compounds newly introduced with version 25 of the ChEMBL database (20,061 compounds; Figure 1h), regardless of whether or not any of the annotated targets is covered by the approach’s knowledge base. This scenario comes closest to real-world applications of target prediction methods, as there is a good chance that the targets of the new compounds (in particular those based on new chemistry) are novel, not represented by the training data, and hence missed by the in silico models.
- “High similarity queries”: These queries share a high degree of molecular similarity with the closest ligand (of the same target) in the knowledge base (TC greater than 0.66). Chemists will identify queries of this class as structurally closely related to the nearest ligand (of the respective target) in the knowledge base.
- “Medium similarity queries”: These queries share a moderate degree of molecular similarity with the closest ligand (of the same target) in the knowledge base (TC between 0.33 and 0.66). Chemists will typically find it challenging to identify obvious similarities between a query molecule of this class and the nearest ligand (of the respective target) in the knowledge base.
- “Low similarity queries”: These queries share a low degree of molecular similarity with the closest ligand in the knowledge base (TC lower than 0.33). Chemists will unlikely identify a query molecule of this class as structurally related to any of the ligands (of the respective target) in the knowledge base.
2.2.1. Evaluation of the Scope and Performance of the Similarity-Based Method
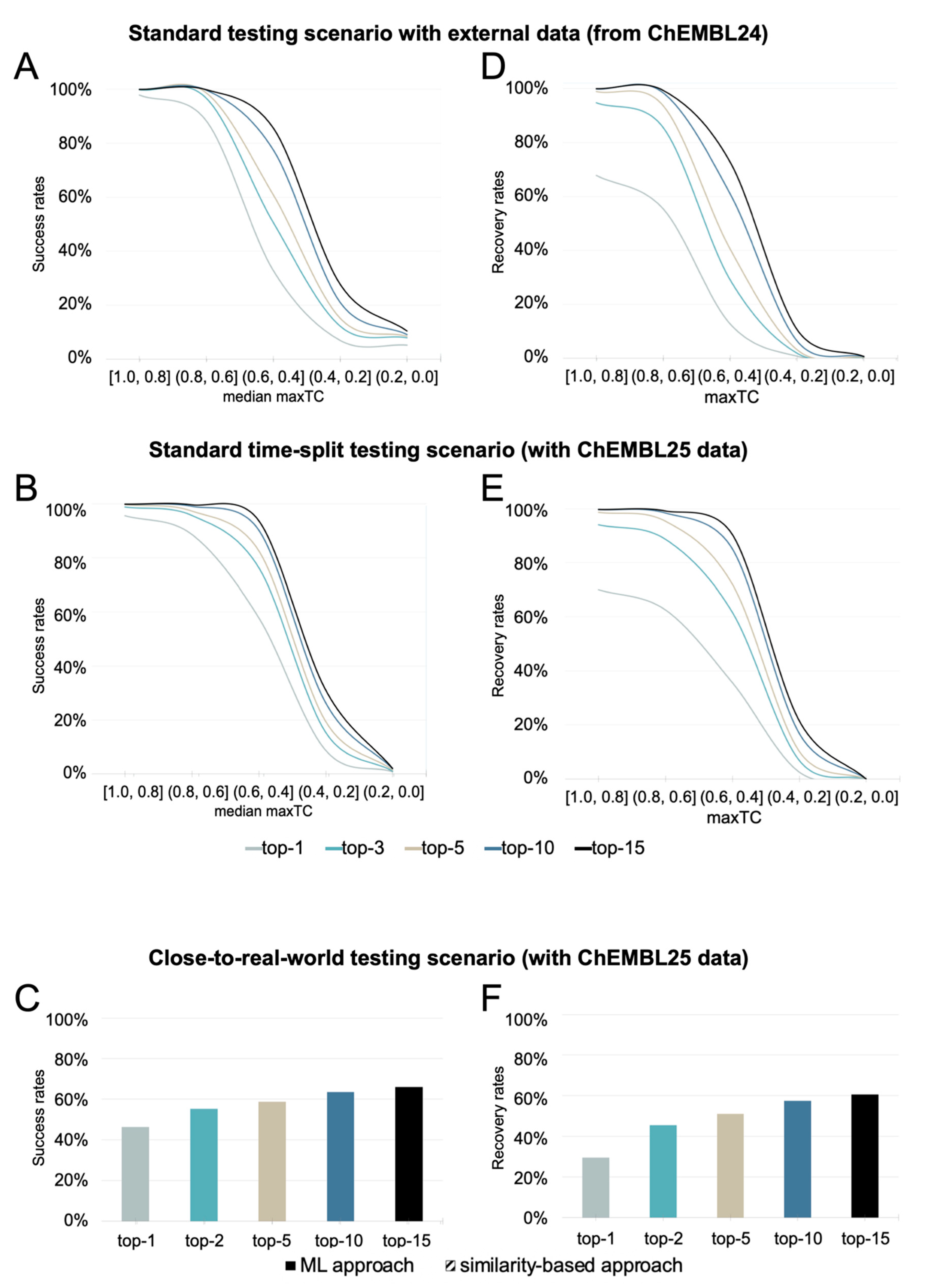
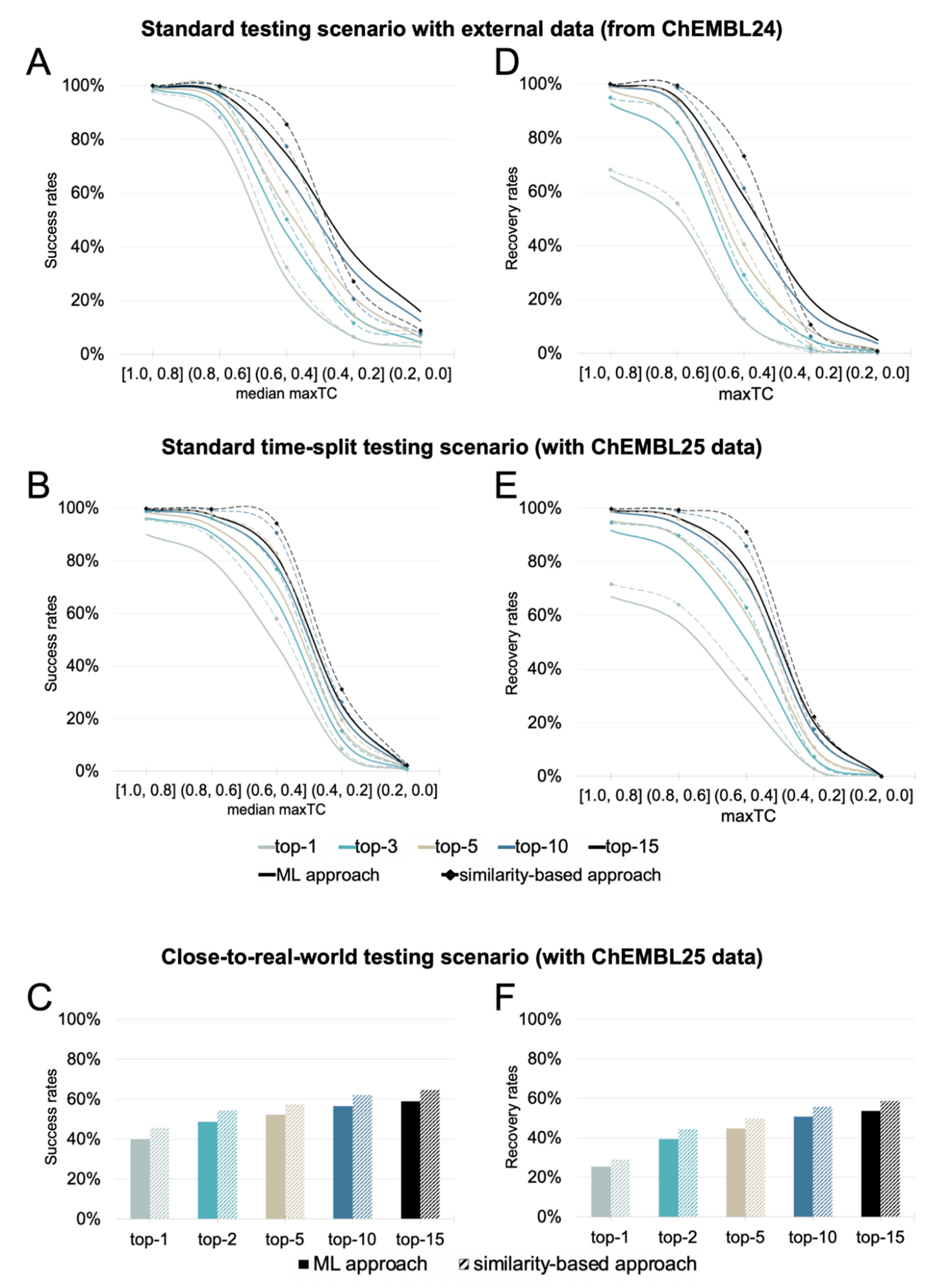
Performance in a Standard Testing Scenario with an External Test Set
Performance in a Standard Time-Split Testing Scenario
Performance in a Close-to-Real-World Testing Scenario
Scope of the Similarity-Based Approach
2.2.2. Evaluation of the Scope and Performance of the Machine-Learning Approach
Performance in a Standard Testing Scenario with an External Test Set
Performance in a Standard Time-Split Testing Scenario
Performance in a Close-to-Real-World Testing Scenario
Scope of the ML Approach
3. Methods
3.1. Data Preparation
- Assay covers a single protein or a protein complex (ChEMBL confidence_score is 7 or 9)
- data_validity_comment is null OR “manually validated”
- potential_duplicate is “0”
- standard_type is “Kd”, “Potency”, “AC50”, “IC50”, “Ki”, or “EC50”
- activity_comment is not “Inconclusive”, “inconclusive”, or “unspecified”
- NOT (standard_relation is null AND activity_comment is not “Active” or “active”)
- NOT (standard_relation “>”, “≥”, or “>>” AND standard_value less than 20,000)
3.2. Development of Target Prediction Models
3.2.1. Similarity-Based Approach
3.2.2. Machine Learning Approach for Target Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ML | Machine learning |
| RF | Random forest |
| SMILES | Simplified molecular-input line-entry system |
| TC | Tanimoto coefficient |
References
- Lauria, A.; Bonsignore, R.; Bartolotta, R.; Perricone, U.; Martorana, A.; Gentile, C. Drugs Polypharmacology by In Silico Methods: New Opportunities in Drug Discovery. Curr. Pharm. Des. 2016, 22, 3073–3081. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Cerchia, C. In Silico Methods to Address Polypharmacology: Current Status, Applications and Future Perspectives. Drug Discov. 2016, 21, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, R.; Tan, Z.; Huang, B.; Zhang, S. Computational Polypharmacology: A New Paradigm for Drug Discovery. Expert Opin. Drug Discov. 2017, 12, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug Discovery for the Future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A., Jr.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking Drug Design in the Artificial Intelligence Era. Nat. Rev. Drug Discov. 2019, 19, 353–364. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and Challenges in Phenotypic Drug Discovery: An Industry Perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Rodrigues, T.; Bernardes, G.J.L. Machine Learning for Target Discovery in Drug Development. Curr. Opin. Chem. Biol. 2019, 56, 16–22. [Google Scholar] [CrossRef]
- Ezzat, A.; Wu, M.; Li, X.-L.; Kwoh, C.-K. Computational Prediction of Drug–Target Interactions Using Chemogenomic Approaches: An Empirical Survey. Brief. Bioinform. 2019, 20, 1337–1357. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Ciriano, I.; Ain, Q.U.; Subramanian, V.; Lenselink, E.B.; Méndez-Lucio, O.; IJzerman, A.P.; Wohlfahrt, G.; Prusis, P.; Malliavin, T.E.; van Westen, G.J.P.; et al. Polypharmacology Modelling Using Proteochemometrics (PCM): Recent Methodological Developments, Applications to Target Families, and Future Prospects. MedChemComm 2015, 6, 24–50. [Google Scholar] [CrossRef] [Green Version]
- Reker, D.; Schneider, P.; Schneider, G.; Brown, J.B. Active Learning for Computational Chemogenomics. Future Med. Chem. 2017, 9, 381–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sydow, D.; Burggraaff, L.; Szengel, A.; van Vlijmen, H.W.T.; IJzerman, A.P.; van Westen, G.J.P.; Volkamer, A. Advances and Challenges in Computational Target Prediction. J. Chem. Inf. Model. 2019, 59, 1728–1742. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Cai, C.; Liu, X.; Ku, X.; Jiang, H.; Gao, D.; Li, H. ChemMapper: A Versatile Web Server for Exploring Pharmacology and Chemical Structure Association Based on Molecular 3D Similarity Method. Bioinformatics 2013, 29, 1827–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, J.; Gohlke, B.-O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Dunkel, M.; Preissner, R. SuperPred: Update on Drug Classification and Target Prediction. Nucleic Acids Res. 2014, 42, W26–W31. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, C.; Wipf, P.; Liu, H.; Su, W.; Xie, X.-Q. TargetHunter: An In Silico Target Identification Tool for Predicting Therapeutic Potential of Small Organic Molecules Based on Chemogenomic Database. AAPS J. 2013, 15, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peón, A.; Naulaerts, S.; Ballester, P.J. Predicting the Reliability of Drug-target Interaction Predictions with Maximum Coverage of Target Space. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peón, A.; Li, H.; Ghislat, G.; Leung, K.-S.; Wong, M.-H.; Lu, G.; Ballester, P.J. MolTarPred: A Web Tool for Comprehensive Target Prediction with Reliability Estimation. Chem. Biol. Drug Des. 2019, 94, 1390–1401. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Takigawa, I.; Mamitsuka, H.; Zhu, S. Similarity-Based Machine Learning Methods for Predicting Drug–Target Interactions: A Brief Review. Brief. Bioinform. 2014, 15, 734–747. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Kurgan, L. Review and Comparative Assessment of Similarity-Based Methods for Prediction of Drug-Protein Interactions in the Druggable Human Proteome. Brief. Bioinform. 2018, 20, 2066–2087. [Google Scholar] [CrossRef]
- Wang, C.; Kurgan, L. Survey of Similarity-based Prediction of Drug-Protein Interactions. Curr. Med. Chem. 2019, 26, 1. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallve, S. Tools for In Silico Target Fishing. Methods 2015, 71, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A Web Server for Target Prediction of Bioactive Small Molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Liu, S.; Chen, J.; Li, X.; Ma, Q.; Yu, B. Predicting Drug-Target Interactions Using Lasso with Random Forest Based on Evolutionary Information and Chemical Structure. Genomics 2019, 111, 1839–1852. [Google Scholar] [CrossRef] [PubMed]
- Bosc, N.; Atkinson, F.; Felix, E.; Gaulton, A.; Hersey, A.; Leach, A.R. Large Scale Comparison of QSAR and Conformal Prediction Methods and their Applications in Drug Discovery. J. Cheminform. 2019, 11, 4. [Google Scholar] [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Clevert, D.-A.; Hochreiter, S. Large-Scale Comparison of Machine Learning Methods for Drug Target Prediction on ChEMBL. Chem. Sci. 2018, 9, 5441–5451. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Tang, J.; Guo, F. Identification of Drug-Target Interactions via Multiple Information Integration. Inf. Sci. 2017, 418, 546–560. [Google Scholar] [CrossRef]
- Keum, J.; Nam, H. SELF-BLM: Prediction of Drug-Target Interactions via Self-Training SVM. PLoS ONE 2017, 12, e0171839. [Google Scholar] [CrossRef] [Green Version]
- Reker, D.; Rodrigues, T.; Schneider, P.; Schneider, G. Identifying the Macromolecular Targets of De Novo-Designed Chemical Entities Through Self-Organizing Map Consensus. Proc. Natl. Acad. Sci. USA 2014, 111, 4067–4072. [Google Scholar] [CrossRef] [Green Version]
- Gawehn, E.; Hiss, J.A.; Schneider, G. Deep Learning in Drug Discovery. Mol. Inform. 2016, 35, 3–14. [Google Scholar] [CrossRef]
- Zhang, H.; Liao, L.; Saravanan, K.M.; Yin, P.; Wei, Y. DeepBindRG: A Deep Learning Based Method for Estimating Effective Protein-Ligand Affinity. PeerJ 2019, 7, e7362. [Google Scholar] [CrossRef]
- Monteiro, N.R.C.; Ribeiro, B.; Arrais, J.P. Deep Neural Network Architecture for Drug-Target Interaction Prediction. In Artificial Neural Networks and Machine Learning—ICANN 2019: Workshop and Special Sessions. Lecture Notes in Computer Science, vol 11731.; Tetko, I.V., Kůrková, V., Karpov, P., Theis, F., Eds.; Springer: Cham, Germany, 2019; Volume 11731, pp. 804–809. ISBN 9783030304928. [Google Scholar] [CrossRef]
- Lee, K.; Kim, D. In-Silico Molecular Binding Prediction for Human Drug Targets Using Deep Neural Multi-Task Learning. Genes 2019, 10, 906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.-Y.; Zhang, Y.-F.; Wang, W.; Wang, X.-G.; Shan, X.-Q.; Xiong, Y.; Wei, D.-Q. DTI-CDF: A CDF Model Towards the Prediction of DTIs Based on Hybrid Features. bioRxiv 2019, 657973. [Google Scholar] [CrossRef]
- Lee, H.; Kim, W. Comparison of Target Features for Predicting Drug-Target Interactions by Deep Neural Network Based on Large-Scale Drug-Induced Transcriptome Data. Pharmaceutics 2019, 11, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boezio, B.; Audouze, K.; Ducrot, P.; Taboureau, O. Network-Based Approaches in Pharmacology. Mol. Inform. 2017, 36. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.-C.; Senese, S.; Damoiseaux, R.; Torres, J.Z. 3D Chemical Similarity Networks for Structure-Based Target Prediction and Scaffold Hopping. ACS Chem. Biol. 2016, 11, 2244–2253. [Google Scholar] [CrossRef]
- Carrella, D.; Napolitano, F.; Rispoli, R.; Miglietta, M.; Carissimo, A.; Cutillo, L.; Sirci, F.; Gregoretti, F.; Di Bernardo, D. Mantra 2.0: An Online Collaborative Resource for Drug Mode of Action and Repurposing by Network Analysis. Bioinformatics 2014, 30, 1787–1788. [Google Scholar] [CrossRef] [Green Version]
- Fu, G.; Ding, Y.; Seal, A.; Chen, B.; Sun, Y.; Bolton, E. Predicting Drug Target Interactions Using Meta-Path-Based Semantic Network Analysis. BMC Bioinform. 2016, 17, 160. [Google Scholar] [CrossRef] [Green Version]
- Mathai, N.; Chen, Y.; Kirchmair, J. Validation Strategies for Target Prediction Methods. Brief. Bioinform. 2019. [Google Scholar] [CrossRef]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating Protein Pharmacology by Ligand Chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting New Molecular Targets for Known Drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Côté, S.; et al. Large-scale Prediction and Testing of Drug Activity on Side-Effect Targets. Nature 2012, 486, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-L.; Li, Y.-K.; Liu, X.-Y.; Geng, X. Binary Relevance for Multi-Label Learning: An Overview. Front. Comput. Sci. 2018, 12, 191–202. [Google Scholar] [CrossRef]
- Cockroft, N.T.; Cheng, X.; Fuchs, J.R. STarFish: A Stacked Ensemble Target Fishing Approach and its Application to Natural Products. J. Chem. Inf. Model. 2019, 59, 4906–4920. [Google Scholar] [CrossRef]
- Hao, M.; Bryant, S.H.; Wang, Y. Open-Source Chemogenomic Data-Driven Algorithms for Predicting Drug-Target Interactions. Brief. Bioinform. 2019, 20, 1465–1474. [Google Scholar] [CrossRef]
- Stork, C.; Wagner, J.; Friedrich, N.-O.; de Bruyn Kops, C.; Šícho, M.; Kirchmair, J. Hit Dexter: A Machine-Learning Model for the Prediction of Frequent Hitters. ChemMedChem 2018, 13, 564–571. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Awale, M.; Reymond, J.-L. Polypharmacology Browser PPB2: Target Prediction Combining Nearest Neighbors with Machine Learning. J. Chem. Inf. Model. 2019, 59, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Riniker, S.; Landrum, G.A. Open-Source Platform to Benchmark Fingerprints for Ligand-Based Virtual Screening. J. Cheminform. 2013, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Success Rates | Recovery Rates | |||||||
|---|---|---|---|---|---|---|---|---|
| Minimum number of actives (number of targets represented) | 25 (1798) | 50 (1296) | 75 (1066) | 100 (899) | 25 (1798) | 50 (1296) | 75 (1066) | 100 (899) |
| top-1 | 74.23% (32,539/43,835) 1 | 74.27% (31,897/42,946) | 74.39% (31,369/42,170) | 74.40% (30,754/41,336) | 45.79% (32,539/71,063) | 46.18% (31,897/69,066) | 46.50% (31,369/67,457) | 46.68% (30,754/65,880) |
| top-3 | 82.37% (36,107/43,835) | 82.36% (35,370/42,946) | 82.36% (34,730/42,170) | 82.34% (34,035/41,336) | 68.61% (48,756/71,063) | 68.99% (47,649/69,066) | 69.20% (46,681/67,457) | 69.29% (45,651/65,880) |
| top-5 | 85.55% (37,503/43,835) | 85.51% (36,725/42,946) | 85.54% (36,072/42,170) | 85.53% (35,353/41,336) | 75.23% (53,460/71,063) | 75.58% (52,197/69,066) | 75.74% (51,095/67,457) | 75.85% (49,970/65,880) |
| top-10 | 89.16% (39,083/43,835) | 89.20% (38,308/42,946) | 89.24% (37,634/42,170) | 89.29% (36,909/41,336) | 80.98% (57,545/71,063) | 81.38% (56,205/69,066) | 81.58% (55,034/67,457) | 81.76% (53,863/65,880) |
| top-15 | 91.13% (39,946/43,835) | 91.22% (39,175/42,946) | 91.29% (38,496/42,170) | 91.35% (37,759/41,336) | 83.89% (59,617/71,063) | 84.37% (58,270/69,066) | 84.62% (57,083/67,457) | 84.80% (55,866/65,880) |
| Hyperparameter | Values Explored |
|---|---|
| n estimators: number of trees | 200, 500, 1000 |
| max depth: maximum depth of tree | 25, 45, 50, 75, 100 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathai, N.; Kirchmair, J. Similarity-Based Methods and Machine Learning Approaches for Target Prediction in Early Drug Discovery: Performance and Scope. Int. J. Mol. Sci. 2020, 21, 3585. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103585
Mathai N, Kirchmair J. Similarity-Based Methods and Machine Learning Approaches for Target Prediction in Early Drug Discovery: Performance and Scope. International Journal of Molecular Sciences. 2020; 21(10):3585. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103585
Chicago/Turabian StyleMathai, Neann, and Johannes Kirchmair. 2020. "Similarity-Based Methods and Machine Learning Approaches for Target Prediction in Early Drug Discovery: Performance and Scope" International Journal of Molecular Sciences 21, no. 10: 3585. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103585