Lon Protease Is Important for Growth under Stressful Conditions and Pathogenicity of the Phytopathogen, Bacterium Dickeya solani

 , ,
, ,
Abstract
:1. Introduction
2. Results
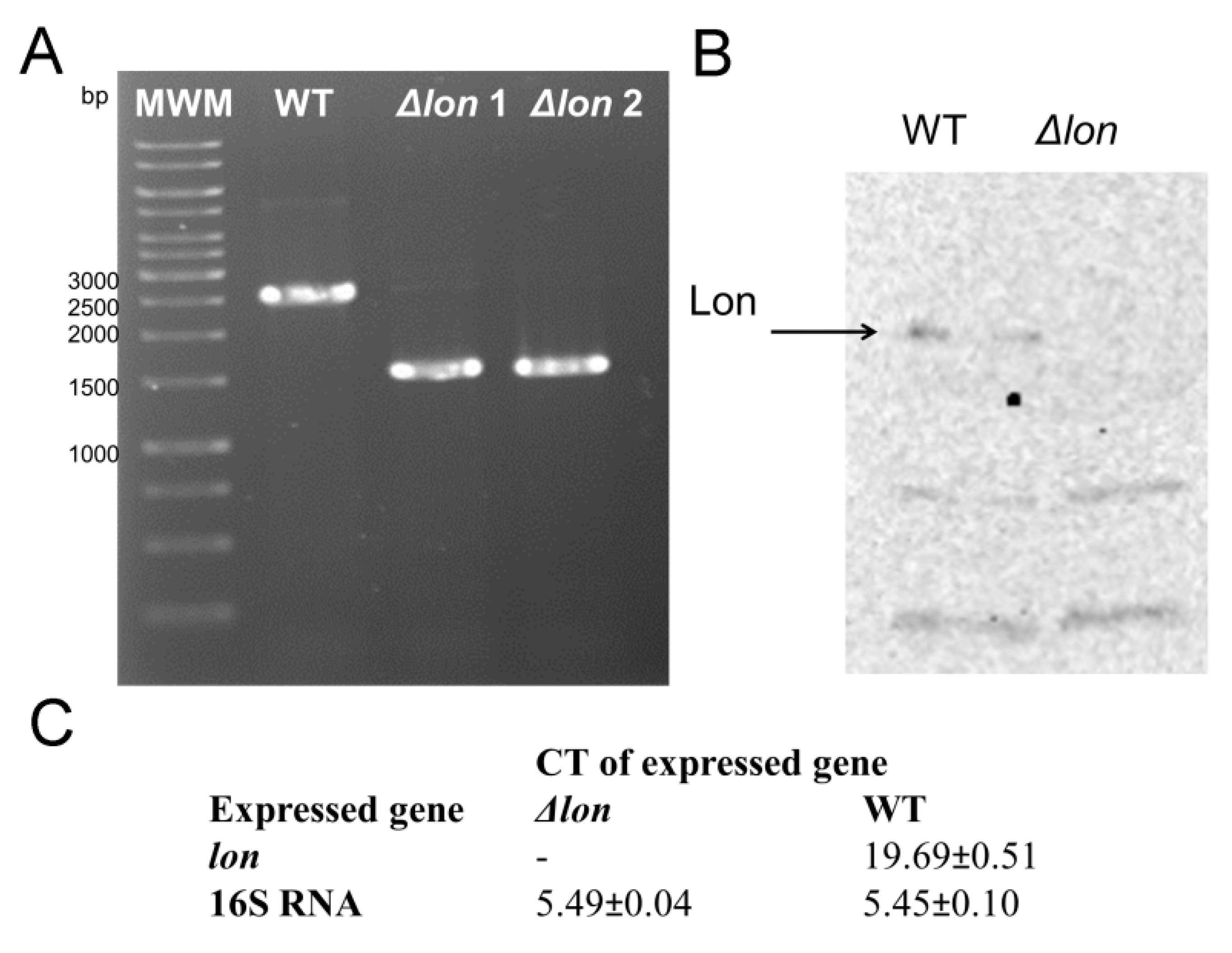
2.1. Construction of the D. solani IPO 2222 Δlon and the Complemented D. solani IPO 2222 Δlon/lon Strains
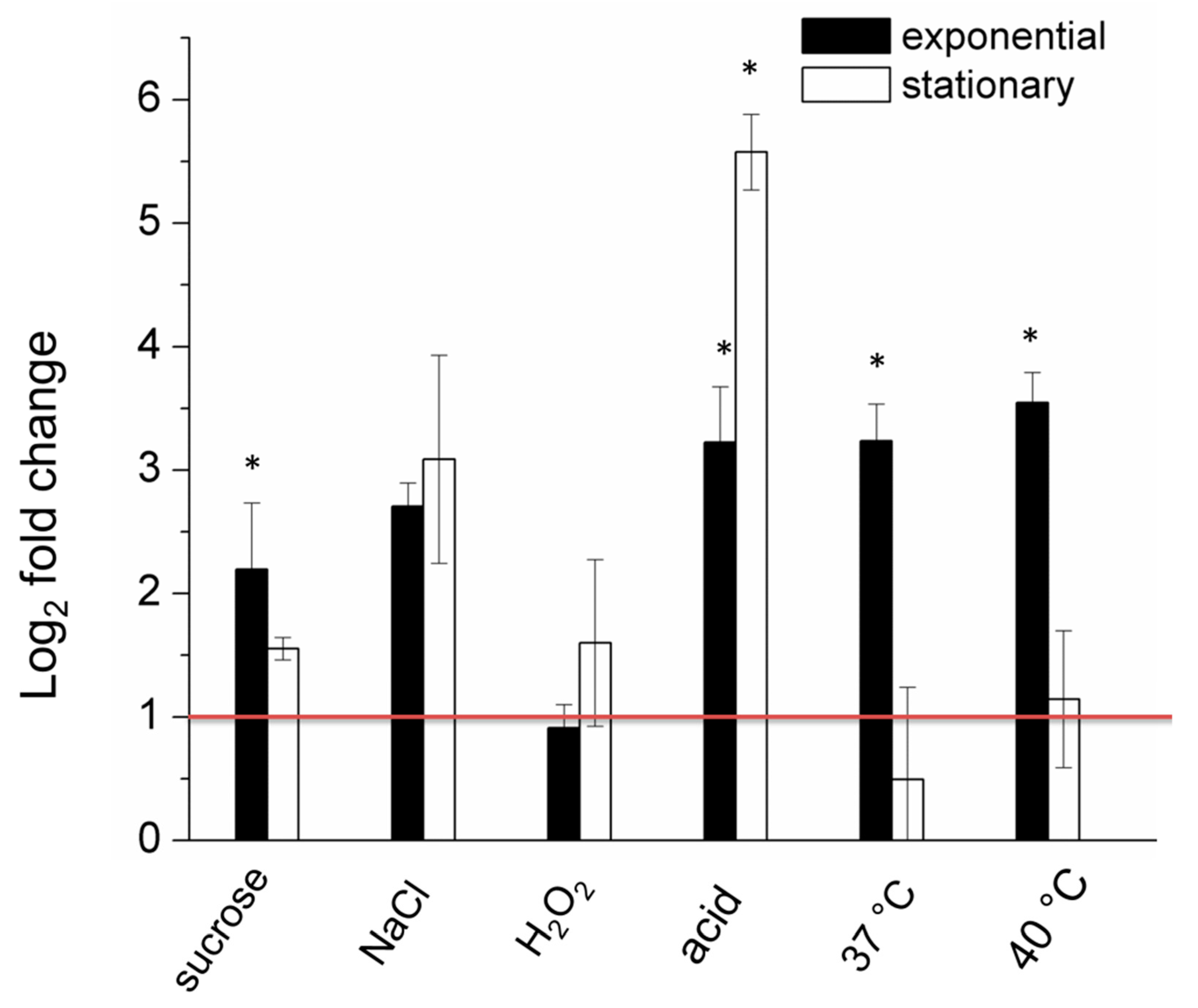
2.2. The Expression of the lon Gene is Upregulated under Certain Stressful Conditions
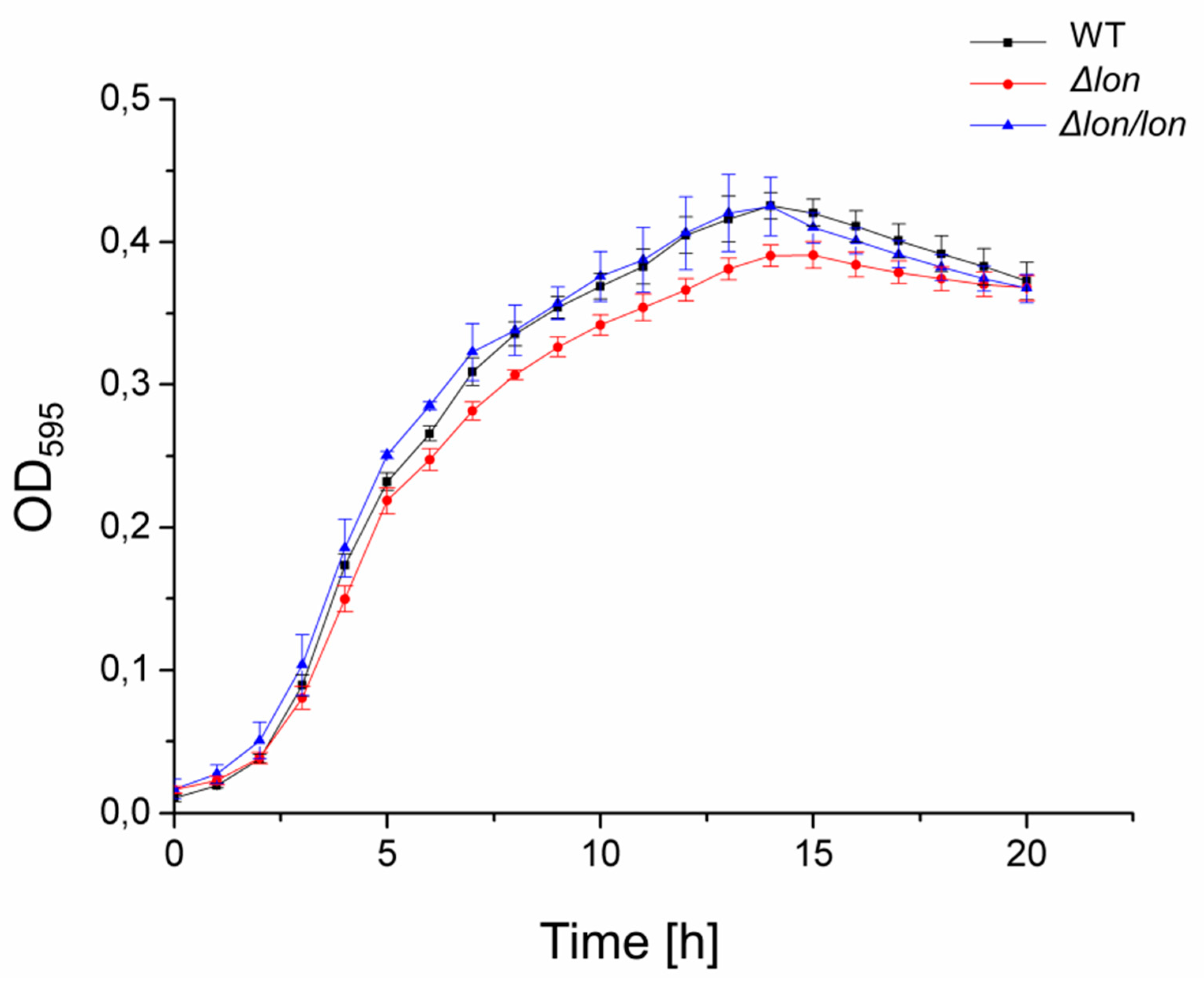
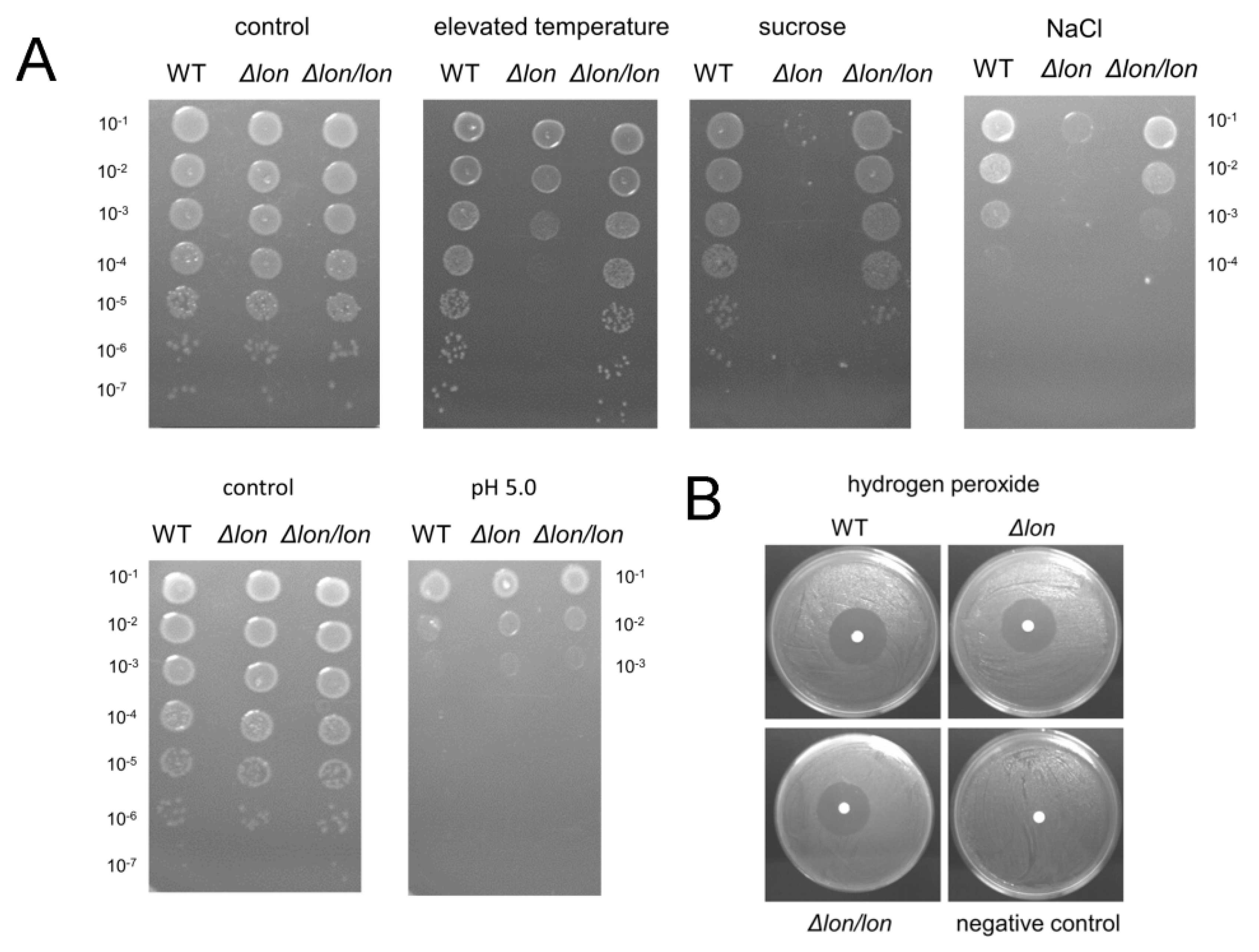
2.3. Lon Protease Plays a Protective Role under Ionic and High-Temperature Stresses
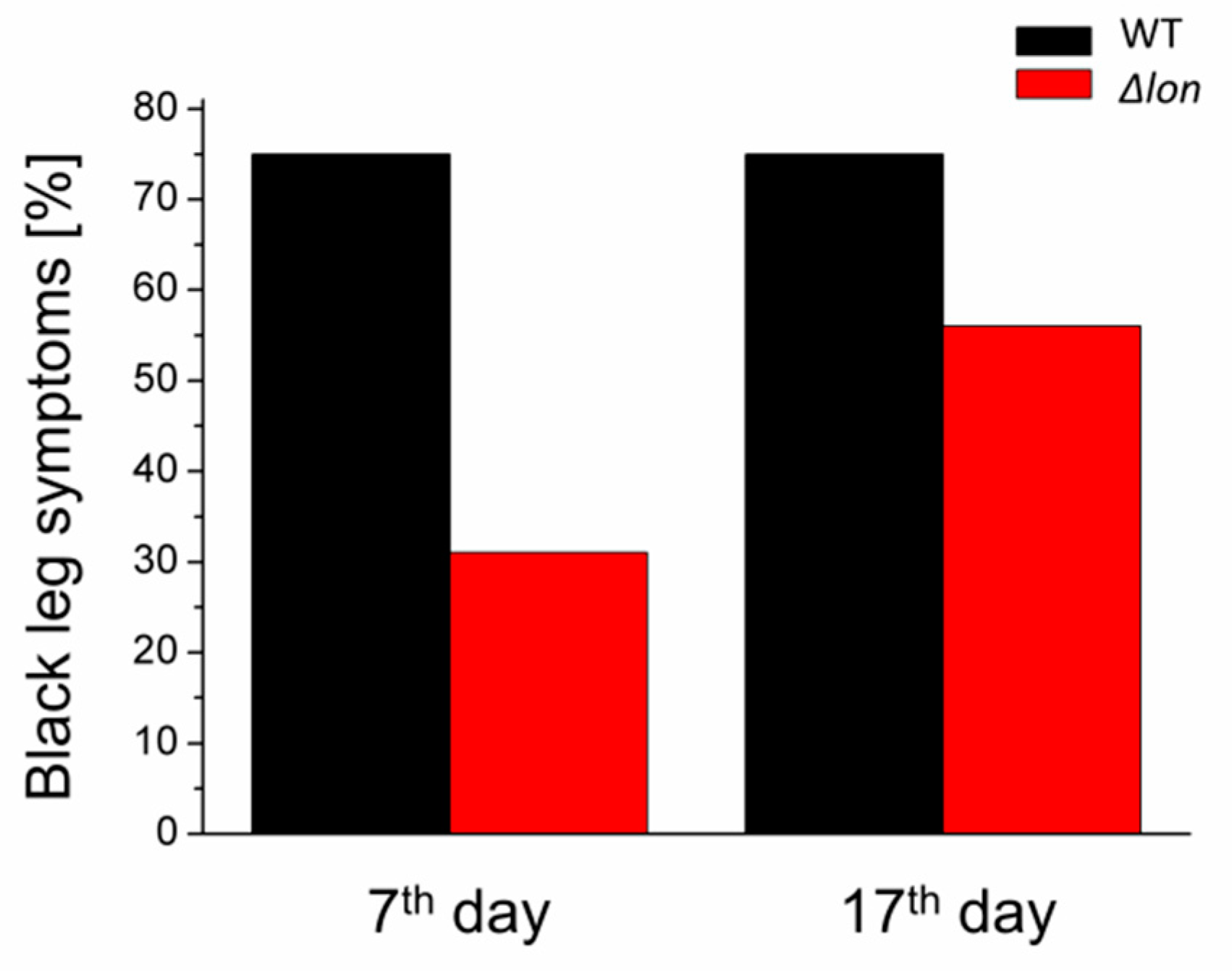
2.4. Deletion of the lon Gene Delays the Onset of the Infection Symptoms
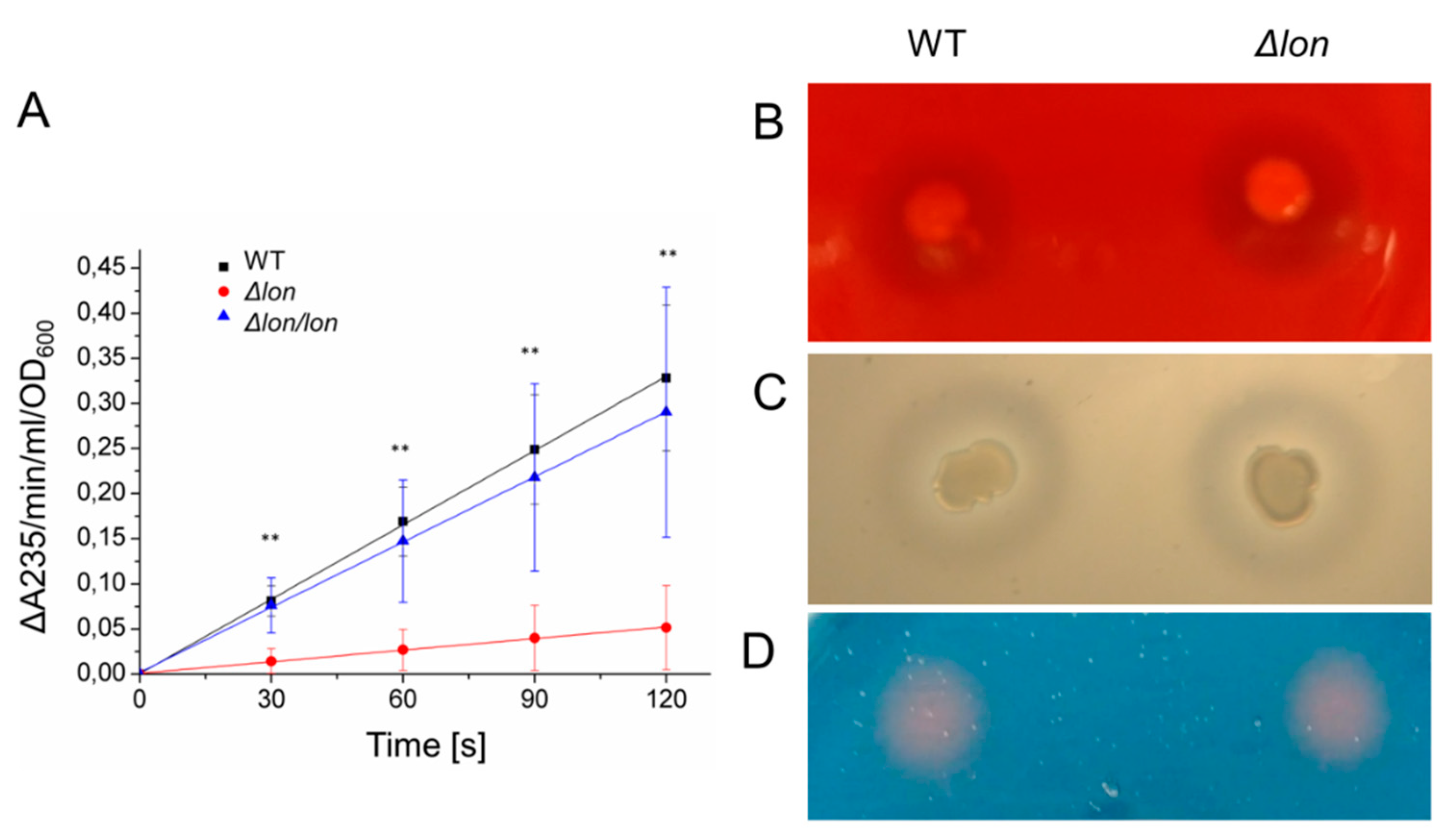
2.5. The Deletion of lon Affects the Activity of Secreted Pectate Lyases
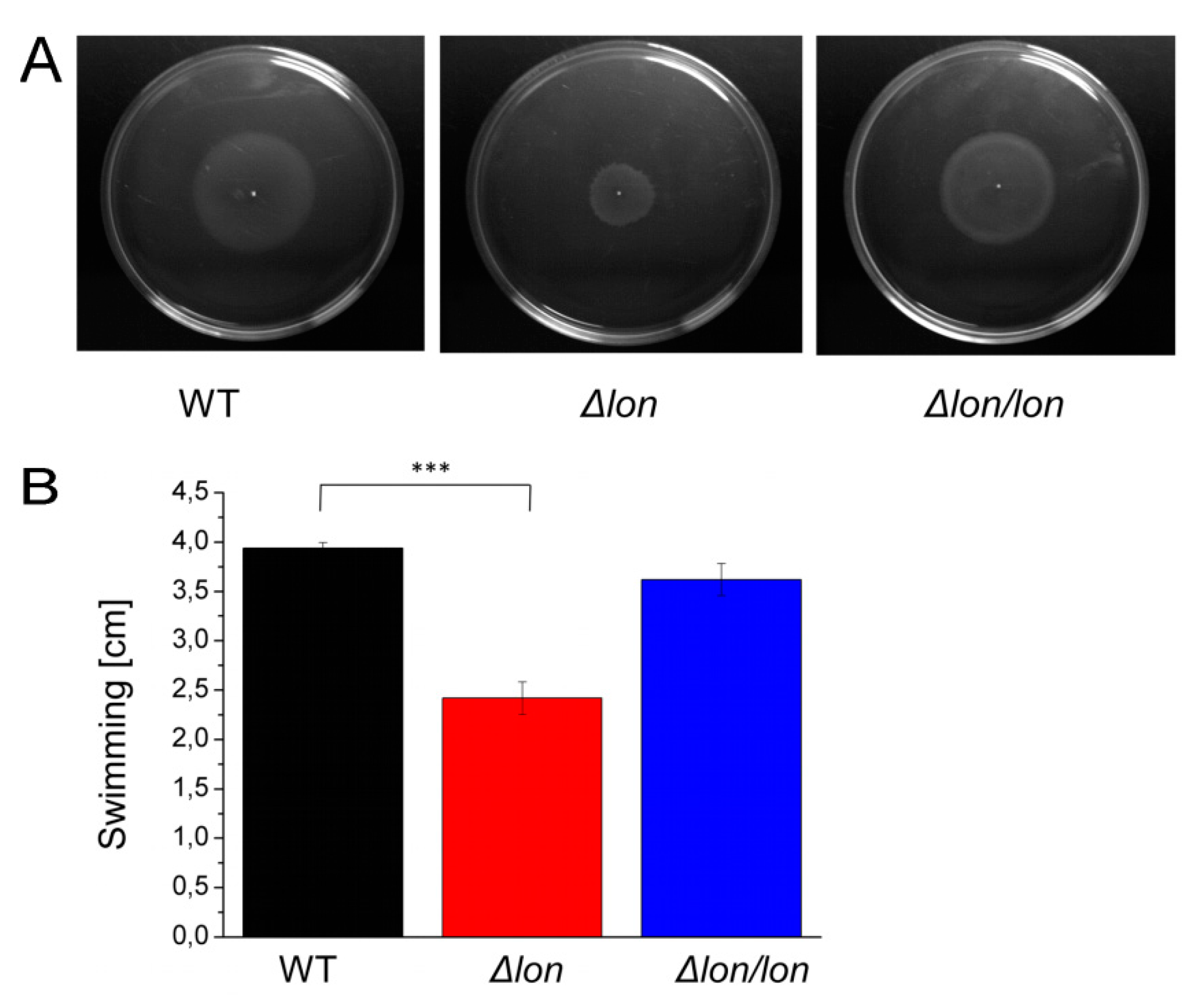
2.6. Lon Protease is Essential for Efficient Motility
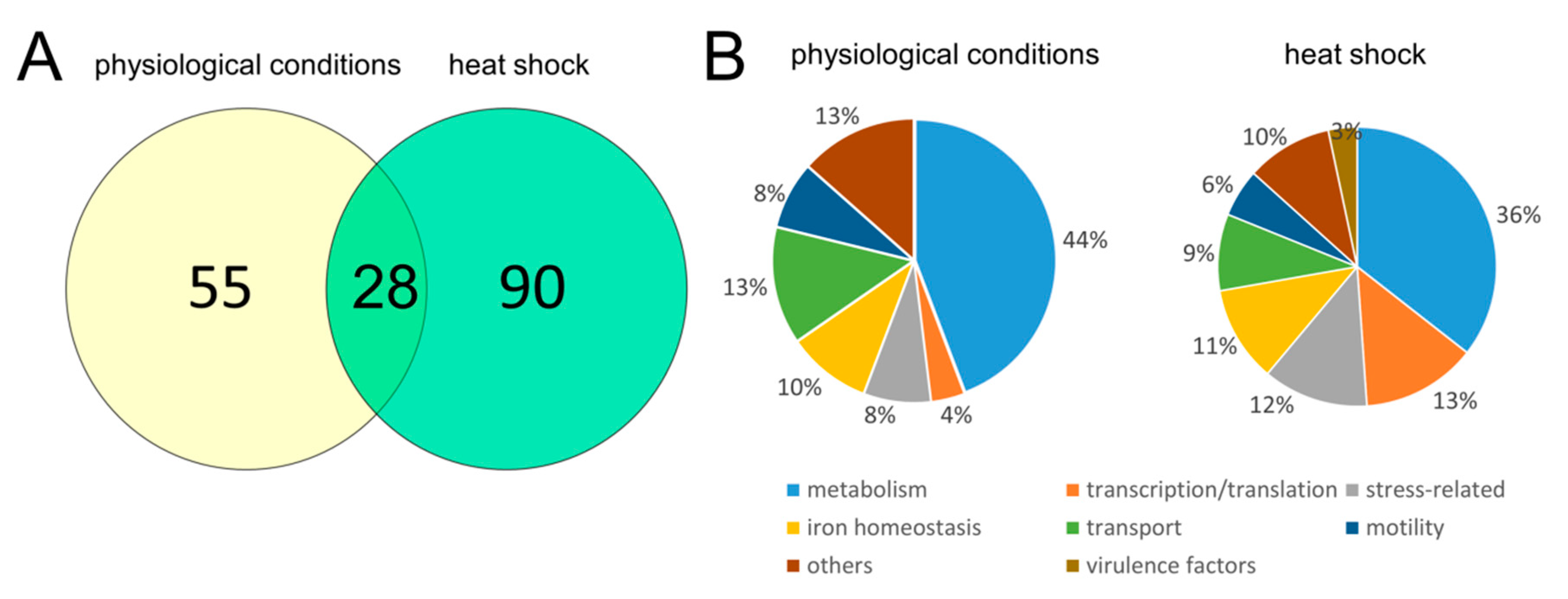
2.7. Comparison of Proteomic Profiles of the D. solani Δlon and WT Cells under Physiological and Stressful Conditions
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Bacterial Growth Conditions
4.3. Construction of the lon Deletion Strain
4.4. Single-Copy Complementation
4.5. Plasmid and Genomic DNA Purification
4.6. Preparation of Electrocompetent Cells and Electroporation
4.7. In Vivo Infection of the Potato Plants
4.8. Pathogenicity on Potato Tubers, Chicory and Chinese Cabbage Leaves.
4.9. Determination of Motility
4.10. Determination of Secreted PCWDE Activity
4.11. Siderophore Activity Assay
4.12. RNA Extraction
4.13. Reverse Transcription
4.14. Quantitative Real-Time PCR (qPCR)
4.15. Protein Electrophoresis and Immunodetection
4.16. Sample Preparation for Mass Spectrometry
4.17. Protein Digestion
4.18. Liquid Chromatography and Mass Spectrometry
4.19. SWATH Mass Spectrometry Experiments
4.20. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAS | casamino acids |
| CFU | colony forming units |
| CMC | carboxymethyl cellulose |
| Cq | quantification cycle |
| DAP | diaminopimelic acid |
| LA | Luria Agar |
| MFD | Mu-free donor |
| OD | optical density |
| PGA | polygalacturonic acid |
| PCWDE | plant cell wall degrading enzymes |
| PQCS | protein quality control system |
| SRP | soft rot Pectobacteriaceae |
| SWATH-MS | Sequential Window Acquisition of All Theoretical Mass Spectra |
| T1SS | type I secretion system |
| T2SS | type II secretion system |
| T3SS | type III secretion system |
| WT | wild-type |
References
- Adeolu, M.; Alnajar, S.; Naushad, S.; S Gupta, R. Genome-based phylogeny and taxonomy of the “Enterobacteriales”: Proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar] [PubMed]
- Toth, I.K.; Saddler, G.; Hélias, V.; Van Der Wolf, J.M.; Lojkowska, E.; Pirhonen, M.; Lahkim, L.T.; Elphinstone, J. Dickeya species: An emerging problem for potato production in Europe. Plant Pathol. 2011, 60, 385–399. [Google Scholar] [CrossRef]
- Mansfield, J.; Genin, S.; Magori, S.; Citovsky, V.; Sriariyanun, M.; Ronald, P.; Dow, M.; Verdier, V.; Beer, S.V.; Machado, M.A.; et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 614–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sławiak, M.; Van Beckhoven, J.R.C.M.; Speksnijder, A.; Czajkowski, R.; Grabe, G.; Van Der Wolf, J.M. Biochemical and genetical analysis reveal a new clade of biovar 3 Dickeya spp. strains isolated from potato in Europe. Eur. J. Plant Pathol. 2009, 125, 245–261. [Google Scholar] [CrossRef]
- Czajkowski, R.; Perombelon, M.C.M.; Van Veen, J.A.; Van Der Wolf, J.M. Control of blackleg and tuber soft rot of potato caused by Pectobacterium and Dickeya species: A review. Plant Pathol. 2011, 60, 999–1013. [Google Scholar] [CrossRef]
- Reverchon, S.; Nasser, W. Dickeyaecology, environment sensing and regulation of virulence programme. Environ. Microbiol. Rep. 2013, 5, 622–636. [Google Scholar] [CrossRef]
- Wagner, S.; Grin, I.; Malmsheimer, S.; Singh, N.; E Torres-Vargas, C.; Westerhausen, S. Bacterial type III secretion systems: A complex device for the delivery of bacterial effector proteins into eukaryotic host cells. FEMS Microbiol. Lett. 2018, 365, 365. [Google Scholar] [CrossRef] [Green Version]
- Figaj, D.; Ambroziak, P.; Przepiora, T.; Skorko-Glonek, J. The Role of Proteases in the Virulence of Plant Pathogenic Bacteria. Int. J. Mol. Sci. 2019, 20, 672. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, J.M.; Bewley, M.C. Protein quality control in bacterial cells: Integrated networks of chaperones and ATP-dependent proteases. Genet. Eng. (N.Y.) 2002, 24, 17–47. [Google Scholar]
- Ventura, S.; Villaverde, A. Protein quality in bacterial inclusion bodies. Trends Biotechnol. 2006, 24, 179–185. [Google Scholar] [CrossRef]
- Maurizi, M.R. Proteases and protein degradation inEscherichia coli. Cell. Mol. Life Sci. 1992, 48, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Schoemaker, J.M.; Gayda, R.C.; Markovitz, A. Regulation of cell division in Escherichia coli: SOS induction and cellular location of the sulA protein, a key to lon-associated filamentation and death. J. Bacteriol. 1984, 158, 551–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Cabassa, A.S.; Gottesman, S. Capsule synthesis in Escherichia coli K-12 is regulated by proteolysis. J. Bacteriol. 1987, 169, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsilibaris, V.; Maenhaut-Michel, G.; Van Melderen, L. Biological roles of the Lon ATP-dependent protease. Res. Microbiol. 2006, 157, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.T.; Kovach, M.E.; Allen, C.A.; Ficht, T.A.; Roop, R.M. The Brucella abortus Lon functions as a generalized stress response protease and is required for wild-type virulence in BALB/c mice. Mol. Microbiol. 2002, 35, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Takaya, A.; Suzuki, M.; Matsui, H.; Tomoyasu, T.; Sashinami, H.; Nakane, A.; Yamamoto, T. Lon, a Stress-Induced ATP-Dependent Protease, Is Critically Important for Systemic Salmonella enterica Serovar Typhimurium Infection of Mice. Infect. Immun. 2003, 71, 690–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breidenstein, E.B.M.; Janot, L.; Strehmel, J.; Fernández, L.; Taylor, P.K.; Kukavica-Ibrulj, I.; Gellatly, S.; Levesque, R.C.; Overhage, J.; Hancock, R.E. The Lon Protease Is Essential for Full Virulence in Pseudomonas aeruginosa. PLoS ONE 2012, 7, e49123. [Google Scholar] [CrossRef] [Green Version]
- Su, S. Lon protease of the -proteobacterium Agrobacterium tumefaciens is required for normal growth, cellular morphology and full virulence. Microbiology 2006, 152, 1197–1207. [Google Scholar] [CrossRef] [Green Version]
- Lan, L.; Deng, X.; Xiao, Y.; Zhou, J.-M.; Tang, X. Mutation of Lon Protease Differentially Affects the Expression ofPseudomonas syringaeType III Secretion System Genes in Rich and Minimal Media and Reduces Pathogenicity. Mol. Plant Microbe Interact. 2007, 20, 682–696. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Gross, C.A. Stress-induced remodeling of the bacterial proteome. Curr. Boil. 2014, 24, R424–R434. [Google Scholar] [CrossRef] [Green Version]
- Ron, E.Z. Bacterial Stress Response. In The Prokaryotes: Prokaryotic Physiology and Biochemistry; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 589–603. ISBN 978-3-642-30141-4. [Google Scholar]
- Gottesman, S. Trouble is coming: Signaling pathways that regulate general stress responses in bacteria. J. Boil. Chem. 2019, 294, 11685–11700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przepiora, T.; Figaj, D.; Radzinska, M.; Apanowicz, M.; Sieradzka, M.; Ambroziak, P.; Hugouvieux-Cotte-Pattat, N.; Lojkowska, E.; Skorko-Glonek, J. Effects of stressful physico-chemical factors on the fitness of the plant pathogenic bacterium Dickeya solani. Eur. J. Plant Pathol. 2019, 156, 519–535. [Google Scholar] [CrossRef]
- Kearns, D.B. A field guide to bacterial swarming motility. Nat. Rev. Genet. 2010, 8, 634–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, C.; Gillet, L.C.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH - MS for quantitative proteomics: A tutorial. Mol. Syst. Boil. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Inamine, G.S.; Dubnau, D. ComEA, a Bacillus subtilis integral membrane protein required for genetic transformation, is needed for both DNA binding and transport. J. Bacteriol. 1995, 177, 3045–3051. [Google Scholar] [CrossRef] [Green Version]
- Goff, S.A.; Casson, L.P.; Goldberg, A.L. Heat shock regulatory gene htpR influences rates of protein degradation and expression of the lon gene in Escherichia coli. Proc. Natl. Acad. Sci. USA 1984, 81, 6647–6651. [Google Scholar] [CrossRef] [Green Version]
- Grossman, A.D.; Erickson, J.W.; Gross, C.A. The htpR gene product of E. coli is a sigma factor for heat-shock promoters. Cell 1984, 38, 383–390. [Google Scholar] [CrossRef]
- Grossman, A.D.; Straus, D.B.; A Walter, W.; A Gross, C. Sigma 32 synthesis can regulate the synthesis of heat shock proteins in Escherichia coli. Genes Dev. 1987, 1, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, A.A.; Baneyx, F. Hyperosmotic shock induces the σ32 and σE stress regulons of Escherichia coli. Mol. Microbiol. 1999, 34, 1029–1038. [Google Scholar] [CrossRef] [Green Version]
- Richmond, C.S. Genome-wide expression profiling in Escherichia coli K-12. Nucleic Acids Res. 1999, 27, 3821–3835. [Google Scholar] [CrossRef]
- He, L.; Nair, M.K.M.; Chen, Y.; Liu, X.; Zhang, M.; Hazlett, K.R.O.; Deng, H.; Zhang, J.-R. The Protease Locus of Francisella tularensis LVS Is Required for Stress Tolerance and Infection in the Mammalian Host. Infect. Immun. 2016, 84, 1387–1402. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Zghidi-Abouzid, O.; Oger-Desfeux, C.; Hommais, F.; Greliche, N.; Muskhelishvili, G.; Nasser, W.; Reverchon, S.; Xuejiao, J.; Ouafa, Z.; et al. Global transcriptional response of Dickeya dadantii to environmental stimuli relevant to the plant infection. Environ. Microbiol. 2016, 18, 3651–3672. [Google Scholar] [CrossRef] [PubMed]
- Cebrián, G.; Condón, S.; Mañas, P. Physiology of the Inactivation of Vegetative Bacteria by Thermal Treatments: Mode of Action, Influence of Environmental Factors and Inactivation Kinetics. Foods 2017, 6, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkin, E. RecA protein in the SOS response: Milestones and mysteries. Biochimie 1991, 73, 133–141. [Google Scholar] [CrossRef]
- Sakoh, M.; Ito, K.; Akiyama, Y. Proteolytic Activity of HtpX, a Membrane-bound and Stress-controlled Protease fromEscherichia coli. J. Boil. Chem. 2005, 280, 33305–33310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabala, L.; Bowman, J.P.; Brown, J.; Ross, T.; McMeekin, T.; Shabala, S. Ion transport and osmotic adjustment inEscherichia coliin response to ionic and non-ionic osmotica. Environ. Microbiol. 2009, 11, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Heuveling, J.; Possling, A.; Hengge, R. A role for Lon protease in the control of the acid resistance genes of Escherichia coli. Mol. Microbiol. 2008, 69, 534–547. [Google Scholar] [CrossRef]
- Ingmer, H.; Brøndsted, L. Proteases in bacterial pathogenesis. Res. Microbiol. 2009, 160, 704–710. [Google Scholar] [CrossRef]
- Frees, D.; Brøndsted, L.; Ingmer, H. Bacterial Proteases and Virulence. Membr. Biog. 2013, 66, 161–192. [Google Scholar]
- Hugouvieux-Cotte-Pattat, N.; Condemine, G.; Shevchik, V. Bacterial pectate lyases, structural and functional diversity. Environ. Microbiol. Rep. 2014, 6, 427–440. [Google Scholar] [CrossRef]
- Ciesielski, S.J.; Schilke, B.; Marszalek, J.; Craig, E. Protection of scaffold protein Isu from degradation by the Lon protease Pim1 as a component of Fe–S cluster biogenesis regulation. Mol. Boil. Cell 2016, 27, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.D.; Pettis, G.S.; A McIntosh, M. Promoter and operator determinants for fur-mediated iron regulation in the bidirectional fepA-fes control region of the Escherichia coli enterobactin gene system. J. Bacteriol. 1994, 176, 3944–3955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemmer, K.M.; Rather, P.N. The Lon protease regulates swarming motility and virulence gene expression in Proteus mirabilis. J. Med. Microbiol. 2008, 57, 931–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Bree, A.C.; Liu, J.; Patrick, J.E.; Chien, P.; Kearns, D.B. Adaptor-mediated Lon proteolysis restricts Bacillus subtilis hyperflagellation. Proc. Natl. Acad. Sci. USA 2014, 112, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Ancona, V.; Zhao, Y. Lon protease modulates virulence traits in Erwinia amylovora by direct monitoring of major regulators and indirectly through the Rcs and Gac-Csr regulatory systems. Mol. Plant Pathol. 2017, 19, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Takaya, A.; Tomoyasu, T.; Tokumitsu, A.; Morioka, M.; Yamamoto, T. The ATP-Dependent Lon Protease of Salmonella enterica Serovar Typhimurium Regulates Invasion and Expression of Genes Carried on Salmonella Pathogenicity Island 1. J. Bacteriol. 2002, 184, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.M.; Oliver, M.H.; Nahar, K.; Alam, M.Z.; Hirata, H.; Tsuyumu, S. CytR Homolog of Pectobacterium carotovorum subsp. carotovorum Controls Air-Liquid Biofilm Formation by Regulating Multiple Genes Involved in Cellulose Production, c-di-GMP Signaling, Motility, and Type III Secretion System in Response to Nutritional and Environmental Signals. Front. Microbiol. 2017, 8. [Google Scholar]
- Josenhans, C.; Suerbaum, S. The role of motility as a virulence factor in bacteria. Int. J. Med. Microbiol. 2002, 291, 605–614. [Google Scholar] [CrossRef]
- Jackson, M.W.; Silva-Herzog, E.; Plano, G.V. The ATP-dependent ClpXP and Lon proteases regulate expression of the Yersinia pestis type III secretion system via regulated proteolysis of YmoA, a small histone-like protein. Mol. Microbiol. 2004, 54, 1364–1378. [Google Scholar] [CrossRef]
- Bretz, J.; Losada, L.; Lisboa, K.; Hutcheson, S.W. Lon protease functions as a negative regulator of type III protein secretion in Pseudomonas syringae. Mol. Microbiol. 2002, 45, 397–409. [Google Scholar] [CrossRef]
- Ventrice, C. Functional Characterization of the csp Homologs of E. coli K-12. Electronic Theses and Dissertations. Master’s Thesis, Duquesne University, Pittsburgh, PA, USA, 2004. [Google Scholar]
- Fernández, L.; Breidenstein, E.B.M.; Taylor, P.K.; Bains, M.; De La Fuente-Núñez, C.; Fang, Y.; Foster, L.J.; Hancock, R.E. Interconnection of post-transcriptional regulation: The RNA-binding protein Hfq is a novel target of the Lon protease in Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 26811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golanowska, M. Characterization of Dickeya solani strains and identification of bacterial and plant signals involved in induction of virulence. In Microbiology and Parasitology; INSA de Lyon: Lyon, France, 2015. [Google Scholar]
- Antonova, E.S.; Bernardy, E.; Hammer, B.K. Natural competence in Vibrio cholerae is controlled by a nucleoside scavenging response that requires CytR?dependent anti?activation. Mol. Microbiol. 2012, 86, 1215–1231. [Google Scholar] [CrossRef] [PubMed]
- Porankiewicz, J.; Wang, J.; Clarke, A.K. New insights into the ATP-dependent Clp protease: Escherichia coli and beyond. Mol. Microbiol. 1999, 32, 449–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.H. A Short Course in Bacterial Genetics: Handbook; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1992; ISBN 978-0-87969-349-7. [Google Scholar]
- Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Boil. 1983, 166, 557–580. [Google Scholar] [CrossRef]
- Platt, R.; Drescher, C.; Park, S.-K.; Phillips, G.J. Genetic System for Reversible Integration of DNA Constructs and lacZ Gene Fusions into the Escherichia coli Chromosome. Plasmid 2000, 43, 12–23. [Google Scholar] [CrossRef]
- Ferrières, L.; Hémery, G.; Nham, T.; Guérout, A.-M.; Mazel, D.; Beloin, C.; Ghigo, J.-M. Silent Mischief: Bacteriophage Mu Insertions Contaminate Products of Escherichia coli Random Mutagenesis Performed Using Suicidal Transposon Delivery Plasmids Mobilized by Broad-Host-Range RP4 Conjugative Machinery. J. Bacteriol. 2010, 192, 6418–6427. [Google Scholar] [CrossRef] [Green Version]
- Van Der Wolf, J.M.; Nijhuis, E.H.; Kowalewska, M.J.; Saddler, G.S.; Parkinson, N.; Elphinstone, J.G.; Pritchard, L.; Toth, I.K.; Lojkowska, E.; Potrykus, M.; et al. Dickeya solani sp. nov., a pectinolytic plant-pathogenic bacterium isolated from potato (Solanum tuberosum). Int. J. Syst. Evol. Microbiol. 2014, 64, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.J.; Bingle, L.E.; Heurlier, K.; Pallen, M.J.; Penn, C.W.; Busby, S.; Hobman, J. Gene doctoring: A method for recombineering in laboratory and pathogenic Escherichia coli strains. BMC Microbiol. 2009, 9, 252. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.A.; Keller, L.H.; Schifferli, D.M. Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 1998, 207, 149–157. [Google Scholar] [CrossRef]
- Stukenberg, D.; (Zentrum für Synthetische Mikrobiologie, Marburg, Germany). Personal communication, 2019.
- Isothermal Reaction (Gibson Assembly) Master Mix. Cold Spring Harb. Protoc. 2017, 2017, 090019.
- Tu, Q.; Yin, J.; Fu, J.; Herrmann, J.; Li, Y.-Z.; Yin, Y.; Stewart, A.F.; Müller, R.; Zhang, Y. Room temperature electrocompetent bacterial cells improve DNA transformation and recombineering efficiency. Sci. Rep. 2016, 6, 24648. [Google Scholar] [CrossRef] [PubMed]
- Collmer, A.; Ried, J.L.; Mount, M.S. Assay methods for pectic enzymes. In Methods in Enzymology; Elsevier BV: Amsterdam, The Netherlands, 1988; Volume 161, pp. 329–335. [Google Scholar]
- Tardy, F.; Nasser, W.; Robert-Baudouy, J.; Hugouvieux-Cotte-Pattat, N. Comparative analysis of the five major Erwinia chrysanthemi pectate lyases: Enzyme characteristics and potential inhibitors. J. Bacteriol. 1997, 179, 2503–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andro, T.; Chambost, J.P.; Kotoujansky, A.; Cattaneo, J.; Bertheau, Y.; Barras, F.; Van Gijsegem, F.; Coleno, A. Mutants of Erwinia chrysanthemi defective in secretion of pectinase and cellulase. J. Bacteriol. 1984, 160, 1199–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwyn, B.; Neilands, J. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 1987, 160, 47–56. [Google Scholar] [CrossRef]
- Primer3 Input (Version 0.4.0). Available online: http://bioinfo.ut.ee/primer3-0.4.0/ (accessed on 31 March 2020).
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, 45. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. Relative expression software tool (REST(C)) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, 36. [Google Scholar] [CrossRef]
- REST-BioInformatics in Real-Time PCR. Available online: http://rest.gene-quantification.info/ (accessed on 31 March 2020).
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Oberfelder, R. Detection of proteins on filters by enzymatic method. In Methods in Nonradioactive Detection; Howard, G.C., Ed.; Appleton & Lange: Norwalk, CT, USA, 1993; pp. 83–85. ISBN 978-0-8385-6946-7. [Google Scholar]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Lewandowska, A.E.; Macur, K.; Czaplewska, P.; Liss, J.; Lukaszuk, K.; Ołdziej, S. Human follicular fluid proteomic and peptidomic composition quantitative studies by SWATH-MS methodology. Applicability of high pH RP-HPLC fractionation. J. Proteom. 2019, 191, 131–142. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2018, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | 30 °C | 40 °C | |||
|---|---|---|---|---|---|
| Accession Number | Name | x-Fold | Log2x-Fold | x-Fold | Log2x-Fold |
| Motility | |||||
| • Downregulated | |||||
| A0A2K8VVE7_9GAMM | Flagellin | 0.35 | −0.46 | ||
| A0A2K8VVK5_9GAMM | Protein phosphatase CheZ | 0.41 | −0.39 | ||
| A0A2K8W5V2_9GAMM | Methyl-accepting chemotaxis protein I (Serine chemoreceptor protein) | 0.24 | −0.63 | 0.21 | −0.67 |
| A0A2K8VVH9_9GAMM | Signal transduction histidine kinase CheA | 0.34 | −0.46 | ||
| A0A2K8VXS1_9GAMM | Methyl-accepting chemotaxis protein I (Serine chemoreceptor protein) | 0.43 | −0.37 | ||
| A0A2K8W5V2_9GAMM | Methyl-accepting chemotaxis protein I (Serine chemoreceptor protein) | 0.24 | −0.63 | ||
| A0A2K8VVG7_9GAMM | Flagellar motor switch protein FliG | 0.46 | −0.34 | ||
| A0A2K8VVJ6_9GAMM | Positive regulator of CheA protein activity (CheW) | 0.35 | −0.45 | ||
| Iron metabolism | |||||
| • Downregulated | |||||
| A0A2K8VW36_9GAMM | Ferrichrome-iron receptor | 0.50 | −0.30 | 0.27 | −0.56 |
| A0A2K8VW52_9GAMM | 2,3-dihydroxybenzoate-AMP ligase enterobactinsiderophore | 0.30 | -0.52 | 0.15 | -0.82 |
| A0A2K8W494_9GAMM | Nonspecific DNA-binding protein Dps/Iron-binding ferritin-like antioxidant protein/Ferroxidase | 0.47 | −0.32 | ||
| A0A2K8VUB7_9GAMM | Ferrous iron transport protein B | 0.45 | −0.34 | ||
| A0A2K8VW26_9GAMM | Isochorismatase enterobactin siderophore/Apo-aryl carrier domain of EntB | 0.4 | −0.34 | ||
| A0A2K8VW34_9GAMM | Isochorismate synthase enterobactin siderophore | 0.48 | −0.32 | ||
| A0A2K8VW22_9GAMM | Enterobactin synthetase component F, serine activating enzyme | 0.36 | −0.44 | ||
| • Upregulated | |||||
| A0A2K8W4W8_9GAMM | Achromobactin biosynthesis protein AcsASiderophoresynthetase superfamily, group B | 7.58 | 0.88 | 3.044 | 0.48 |
| A0A2K8VWX6_9GAMM | Iron-sulfur cluster insertion protein ErpA | 4.90 | 0.69 | ||
| A0A2K8W3W2_9GAMM | Ferric uptake regulation protein | 9.87 | 0.99 | ||
| A0A2K8VWQ7_9GAMM | Iron-sulfur cluster assembly scaffold protein IscU | 3.67 | 0.56 | ||
| A0A2K8W4W2_9GAMM | Achromobactin biosynthesis protein AcsD Siderophore synthetase superfamily, group A | 2.77 | 0.44 | ||
| Stress-Related | |||||
| • Downregulated | |||||
| A0A2K8VUY7_9GAMM | Phage shock protein A | 0.31 | −0.51 | 0.35 | −0.45 |
| A0A2K8VZS1_9GAMM | Small heat shock protein IbpA | 0.45 | −0.35 | ||
| A0A2K8VZU2_9GAMM | Universal stress protein | 0.49 | −0.31 | ||
| A0A2K8VUY1_9GAMM | Phage shock protein B OS=Dickeya solani | 0.38 | −0.41 | ||
| • Upregulated | |||||
| A0A2K8VTF5_9GAMM | Protease II | 4.44 | 0.65 | 2.94 | 0.47 |
| A0A2K8VX21_9GAMM | Protein RecA | 2.65 | 0.42 | ||
| A0A2K8W3I9_9GAMM | ATP-dependent Clp protease proteolytic subunit | 2.00 | 0.30 | ||
| A0A2K8W3U6_9GAMM | Cold shock protein CspE | 2.91 | 0.46 | ||
| A0A2K8VZ71_9GAMM | Cold shock protein CspG | 17.27 | 1.24 | ||
| A0A2K8W1Q0 | Osmotically inducible protein OsmY | 2.50 | 0.40 | ||
| A0A2K8VUA2_9GAMM | Protease HtpX | 2.22 | 0.35 | ||
| • Differentially expressed | |||||
| A0A2K8W260_9GAMM | Periplasmic protein related to spheroplast formation | 0.40 | −0.40 | 2.79 | 0.45 |
| Transport | |||||
| • Downregulated | |||||
| A0A2K8W052_9GAMM | Phosphate-binding protein PstS | 0.48 | −0.32 | ||
| A0A2K8VVW1_9GAMM | Histidine ABC transporter, histidine-binding periplasmic protein HisJ | 0.47 | −0.33 | ||
| A0A2K8VU80_9GAMM | N-acetylneuraminic acid outer membrane channel protein NanC | 0.27 | −0.56 | ||
| A0A2K8VTK7_9GAMM | Oligopeptide ABC transporter, periplasmic oligopeptide-binding protein OppA | 0.46 | −0.34 | ||
| A0A2K8VWQ4_9GAMM | Periplasmic substrate-binding transport protein | 0.49 | −0.31 | ||
| A0A2K8W2K3_9GAMM | Inositol transport system sugar-binding protein | 0.41 | −0.39 | ||
| A0A2K8W3K7_9GAMM | Efflux pump membrane transporter | 0.37 | −0.43 | ||
| A0A2K8VZR0_9GAMM | Dipeptide-binding ABC transporter, periplasmic substrate-binding component | 0.39 | −0.41 | ||
| A0A2K8W417_9GAMM | Cobalt/zinc/cadmium efflux RND transporter, membrane fusion protein, CzcB family | 0.47 | −0.337 | ||
| A0A2K8VSS7_9GAMM | Methionine ABC transporter substrate-binding protein | 0.44 | −0.36 | ||
| • Upregulated | |||||
| A0A2K8W0I4_9GAMM | Xylose ABC transporter, periplasmic xylose-binding protein XylF | 3.06 | 0.49 | 2.85 | 0.45 |
| A0A2K8VXC1_9GAMM | L-proline glycine betaine binding ABC transporter protein ProX | 2.25 | 0.35 | 2.46 | 0.39 |
| A0A2K8W3M0_9GAMM | Lead, cadmium, zinc and mercury transporting ATPase | 3.14 | 0.50 | ||
| Metabolism | |||||
| • Downregulated | |||||
| A0A2K8W021_9GAMM | ATP synthase subunit delta | 0.47 | −0.33 | ||
| A0A2K8W4F2_9GAMM | Glutamate-1-semialdehydeaminotransferase | 0.10 | −1.01 | 0.16 | −0.806 |
| A0A2K8W580_9GAMM | Enoyl-acyl-carrier-protein reductase NADPH | 0.38 | −0.42 | ||
| A0A2K8W1R7_9GAMM | Alkyl hydroperoxide reductase protein C | 0.40 | −0.40 | ||
| A0A2K8VTW6_9GAMM | NAD(P) transhydrogenase subunit alpha | 0.47 | −0.33 | ||
| A0A2K8W040_9GAMM | ATP synthase epsilon chain | 0.23 | −0.64 | ||
| A0A2K8W444_9GAMM | 6-phosphogluconolactonase | 0.40 | −0.40 | ||
| A0A2K8VVU5_9GAMM | NADH-quinone oxidoreductase | 0.46 | −0.33 | ||
| A0A2K8VV27_9GAMM | Endo-1,4-beta-xylanase A | 0.230 | −0.53 | 0.13 | −0.89 |
| A0A2K8VXQ3_9GAMM | PTS system, cellobiose-specific IIB component | 0.45 | −0.35 | ||
| A0A2K8W121_9GAMM | Biotin carboxyl carrier protein of acetyl-CoA carboxylase | 0.41 | −0.39 | ||
| A0A2K8W3J3_9GAMM | Cytochrome O ubiquinol oxidase subunit I | 0.47 | −0.32 | ||
| A0A2K8VTY0_9GAMM | Superoxide dismutase [Cu-Zn] | 0.38 | −0.42 | ||
| A0A2K8VU81_9GAMM | Sugar-binding protein | 0.47 | −0.33 | 0.37 | −0.43 |
| A0A2K8W4J6_9GAMM | Putative l-lactate dehydrogenase, Iron-sulfur cluster-binding subunit YkgF | 0.36 | −0.44 | ||
| A0A2K8VWF7_9GAMM | Peptidyl-prolyl cis-trans isomerase | 2.31 | 0.36 | ||
| A0A2K8VT63_9GAMM | ATP phosphoribosyltransferase | 0.45 | −0.35 | ||
| A0A2K8VVY8_9GAMM | Acetyl-coenzyme A carboxylase carboxyl transferase subunit beta | 0.42 | −0.38 | ||
| A0A2K8VX73_9GAMM | Phosphoheptose isomerase | 0.28 | −0.55 | ||
| A0A2K8W077_9GAMM | Bifunctional polymyxin resistance protein ArnA | 0.20 | −0.71 | ||
| • Upregulated | |||||
| A0A2K8VTR1_9GAMM | 3-hydroxypropionate dehydrogenase | 4.17 | 0.62 | 4.24 | 0.63 |
| A0A2K8VV57_9GAMM | Thioredoxin/glutathione peroxidase BtuE | 3.29 | 0.52 | 5.75 | 0.76 |
| A0A2K8VXQ4_9GAMM | Putative phosphatase/kinase | 6.94 | 0.84 | 10.73 | 1.03 |
| A0A2K8VTR5_9GAMM | SAM-dependent methyltransferase YafE (UbiE-like protein) | 4.13 | 0.62 | 4.38 | 0.64 |
| A0A2K8W193_9GAMM | Glyoxalase | 2.42 | 0.38 | ||
| A0A2K8VYC9_9GAMM | Fructose-bisphosphate aldolase class II | 2.80 | 0.45 | ||
| A0A2K8W3S4_9GAMM | Thiol peroxidase, Bcp-type | 2.57 | 0.41 | ||
| A0A2K8VV22_9GAMM | Glutaredoxin | 2.77 | 0.44 | ||
| A0A2K8W3N7_9GAMM | Stomatin/prohibitin-family membrane protease subunit YbbK | 2.66 | 0.43 | ||
| A0A2K8VXG5_9GAMM | Adenylate cyclase | 2.37 | 0.38 | ||
| A0A2K8W3S1_9GAMM | Glycoprotein/polysaccharide metabolism | 14.75 | 1.17 | 3.187 | 0.507 |
| A0A2K8VVX6_9GAMM | Phosphatase YfbT | 2.04 | 0.31 | ||
| A0A2K8VYD6_9GAMM | Biosynthetic arginine decarboxylase | 3.62 | 0.56 | ||
| A0A2K8VSW1_9GAMM | Soluble aldose sugar dehydrogenase, PQQ-dependent PE = 4 SV = 1 | 2.38 | 0.38 | ||
| A0A2K8VXS7_9GAMM | Aminotransferase | 2.36 | 0.37 | ||
| A0A2K8VXE7_9GAMM | Sulfite reductase [NADPH] flavoprotein alpha-component | 2.03 | 0.31 | ||
| A0A2K8VY35_9GAMM | 3-isopropylmalate dehydratase large subunit | 2.21 | 0.34 | ||
| A0A2K8W254_9GAMM | Phosphopentomutase | 2.60 | 0.41 | ||
| A0A2K8VT52_9GAMM | Phosphoserine aminotransferase | 2.47 | 0.39 | ||
| A0A2K8W0R3_9GAMM | ADP-l-glycero-d-manno-heptose-6-epimerase | 4.45 | 0.65 | 5.20 | 0.72 |
| A0A2K8VY36_9GAMM | 3-isopropylmalate dehydratase small subunit | 2.51 | 0.40 | ||
| A0A2K8VWH1_9GAMM | Methylglyoxal synthase | 2.27 | 0.36 | 2.85 | 0.45 |
| • Differentially expressed | |||||
| A0A2K8W2T8_9GAMM | Phosphotransferase system, phosphocarrier protein HPr | 0.49 | −0.31 | 2.66 | 0.42 |
| A0A2K8VYF1_9GAMM | Epimerase domain-containing protein | 2.34 | 0.37 | 0.18 | −0.75 |
| A0A2K8W293_9GAMM | Thiol:disulfide interchange protein | 0.46 | −0.34 | 2.13 | 0.33 |
| Virulence | |||||
| • Upregulated | |||||
| A0A2K8VU37_9GAMM | Various polyols ABC transporter, permease component 2 | 2.75 | 0.44 | ||
| A0A2K8VUF2_9GAMM | Harpin hrpN (Harpin-Ech) | 2.28 | 0.36 | ||
| A0A2K8VUE5_9GAMM | Hrp pili protein hrpA (TTSS pilin hrpA) | 10.64 | 1.03 | ||
| Transcription/Translation | |||||
| • Downregulated | |||||
| A0A2K8VT33_9GAMM | Serine-tRNA ligase | 0.47 | −0.33 | ||
| A0A2K8VW75_9GAMM | JmjC domain-containing protein | 0.48 | −0.32 | ||
| • Upregulated | |||||
| A0A2K8VZG3_9GAMM | Transcriptional (Co)regulator CytR | 3.47 | 0.54 | ||
| A0A2K8VZY6_9GAMM | DNA-directed RNA polymerase subunit omega | 2.45 | 0.39 | ||
| A0A2K8W0W8_9GAMM | 50S ribosomal protein L7/L12 | 2.11 | 0.32 | ||
| A0A2K8VUA3_9GAMM | Translation initiation factor 3 | 2.69 | 0.43 | ||
| A0A2K8VX20_9GAMM | Ribosome hibernation protein YfiA | 13.75 | 1.14 | ||
| A0A2K8W224_9GAMM | 50S ribosomal protein L27 | 2.03 | 0.31 | ||
| A0A2K8W3J6_9GAMM | 50S ribosomal protein L31 type B | 3.42 | 0.53 | ||
| A0A2K8VWY3_9GAMM | RNA polymerase-binding transcription factor DksA | 2.07 | 0.32 | ||
| A0A2K8VY97_9GAMM | ABC transporter, ATP-binding protein | 2.45 | 0.39 | 2.04 | 0.31 |
| A0A2K8VYE8_9GAMM | RNA-binding protein Hfq | 2.70 | 0.43 | ||
| A0A2K8VYF1_9GAMM | Epimerase domain-containing protein | 2.34 | 0.37 | 0.18 | −0.75 |
| Others | |||||
| • Downregulated | |||||
| A0A2K8W376_9GAMM | UPF0325 protein D083_3591 | 0.36 | −0.45 | ||
| A0A2K8W2W2_9GAMM | IncI1 plasmid conjugative transfer protein TraF | 0.30 | −0.53 | 0.26 | −0.58 |
| A0A2K8W4R1_9GAMM | Uncharacterized protein | 0.45 | −0.34 | 0.33 | −0.48 |
| A0A2K8VTN2_9GAMM | Uncharacterized protein | 0.41 | −0.38 | ||
| A0A2K8VV11_9GAMM | Major outer membrane lipoprotein | 0.50 | −0.30 | ||
| • Upregulated | |||||
| A0A2K8W3L1_9GAMM | Putative membrane protein OS=Dickeya solani | 175.34 | 2.24 | 117.58 | 2.07 |
| A0A2K8VWZ4_9GAMM | S-ribosylhomocysteinelyase | 2.03 | 0.31 | ||
| A0A2K8VZT1_9GAMM | Putative membrane protein | 7.12 | 0.85 | ||
| A0A2K8VTR0_9GAMM | Putative secreted protein | 5.63 | 0.75 | ||
| A0A2K8VT84_9GAMM | Uncharacterized protein | 5.23 | 0.72 | 6.61 | 0.82 |
| • Differentially expressed | |||||
| A0A2K8W4I6_9GAMM | Uncharacterized protein | 4.19 | 0.62 | 0.33 | −0.48 |
| Strain | Genotype | Reference or Source |
| Escherichia coli DH5α | F– φ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK–, mK+) phoA supE44 λ– thi-1 gyrA96 relA1 | [58] |
| Escherichia coli DH5α pir | sup E44 ΔlacU169 (ΦlacZΔM15) recA1 endA1 hsdR17 thi-1 gyrA96, relA1 λpir phage lysogen | [59] |
| Escherichia coli MFD pir | MG1655 RP4-2-Tc::[ΔMu1::aac(3)IV-ΔaphA-Δnic35-ΔMu2::zeo] ΔdapA::(erm-pir) ΔrecA | [60] |
| Dickeya solani IPO 2222 | WT | [61] |
| D. solani IPO 2222 Δlon | Δlon | This work |
| D. solani IPO 2222 Δlon/lon | Δlon/lon | This work |
| Plasmids | Feature | Reference or Source |
| pDOC-C | pEX100T, Sce1 -Sce1 sacB AmpR | [62] |
| pDOC-K | pEX100T, Sce1-KanR -Sce1 sacB AmpR | [62] |
| pACBSCE | I-Sce1 λ-Red CmR | [62] |
| pDFDOC-C-lon | pDOC-C Sce1-KanR-Sce1 | This work |
| pRE112 | pRE107 cmR sacB | [63] |
| pmScarlet | pMB1 ori mScarlet AmpR | [64] |
| pLonScar | pRE112 lon mScarlet | This work |
| Primer | Primer Sequence 5′-3′ | Amplified DNA |
|---|---|---|
| lonkan L | CAGGGTACCTTCCCTTAACCTGGCGGAAACGAAACTAAGAGAGAGCTCTGACCGGTCAATTGGCTGGAG | kanamycin resistance gene with added sequences flanking the D. solani lon gene amplified from pDOC-K |
| lonkan R | GCACACTCGAGCCAGCCTTTTT TTCTCAGTGGTTTTTGCGATAGGTCACTAATATCCTCCTTAGTTCC | |
| lonsolani L | CGATTACCTATAGGCGAAACC | lon and kanamycin resistance gene amplified from D. solani and D. solani Δ lon gDNA, respectively |
| lonsolani R | CAGGCTCAACAGTGCTCTAAC | |
| 1 L | AGTGAACTGCATGAATTCCCGTTGATCCAGATCTTGCGCGA | 500 bp upstream from the start codon of lon gene amplified from D. solani gDNA |
| 1 R | GTTCGGAACGCTCAGGGTTCATAGAGCTCTCTCTTAGTTTCGTTTCC | |
| 2 lon L | ATGAACCCTGAGCGTTCCGAA | lon gene amplified from D. solani gDNA |
| 2 lon R | CACGTTTCACTTTCCGGGTTCCTATTTTTTGGCTACCGACTTCAC | |
| 3 scarlet L | GAGACCCGGAAAGTGAAAACGTG | mScarlet gene amplified from pmScarlet |
| 3 scarlet R | TTACCGCCTTTGAGTGAGCTG | |
| 4 L | CAGCTCACTCAAAGGCGGTAATGACCTATCGCAAAAACCAC | 500 bp downstream from the stop codon of lon gene amplified from D. solani gDNA |
| 4 R | ATGCGATATCGAGCTCTCCCAAAACCGTCCCACCTCAGATT |
| FWD Primer Sequence (5′-3′) | REV Primer Sequence (5′-3′) | Amplicon Length [bp] | PCR Efficiency | R2 | Concentration [µM] | |
|---|---|---|---|---|---|---|
| lon | TGGTCATTCCGTTGTTTGTTGGTC | CATCCGTTGAGGCTTCTTTCTGTG | 111 | 1.97 | 1.0 | 0.3 |
| 16S rRNA | GCTCGTGTTGTGAAATGTTGGGTT | GCAGTCTCCCTTGAGTTCCCAC | 94 | 1.96 | 1.0 | 0.225 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figaj, D.; Czaplewska, P.; Przepióra, T.; Ambroziak, P.; Potrykus, M.; Skorko-Glonek, J. Lon Protease Is Important for Growth under Stressful Conditions and Pathogenicity of the Phytopathogen, Bacterium Dickeya solani. Int. J. Mol. Sci. 2020, 21, 3687. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103687
Figaj D, Czaplewska P, Przepióra T, Ambroziak P, Potrykus M, Skorko-Glonek J. Lon Protease Is Important for Growth under Stressful Conditions and Pathogenicity of the Phytopathogen, Bacterium Dickeya solani. International Journal of Molecular Sciences. 2020; 21(10):3687. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103687
Chicago/Turabian StyleFigaj, Donata, Paulina Czaplewska, Tomasz Przepióra, Patrycja Ambroziak, Marta Potrykus, and Joanna Skorko-Glonek. 2020. "Lon Protease Is Important for Growth under Stressful Conditions and Pathogenicity of the Phytopathogen, Bacterium Dickeya solani" International Journal of Molecular Sciences 21, no. 10: 3687. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103687