Genomic Analysis of Intrinsically Disordered Proteins in the Genus Camelus



Abstract
:1. Introduction
2. Results
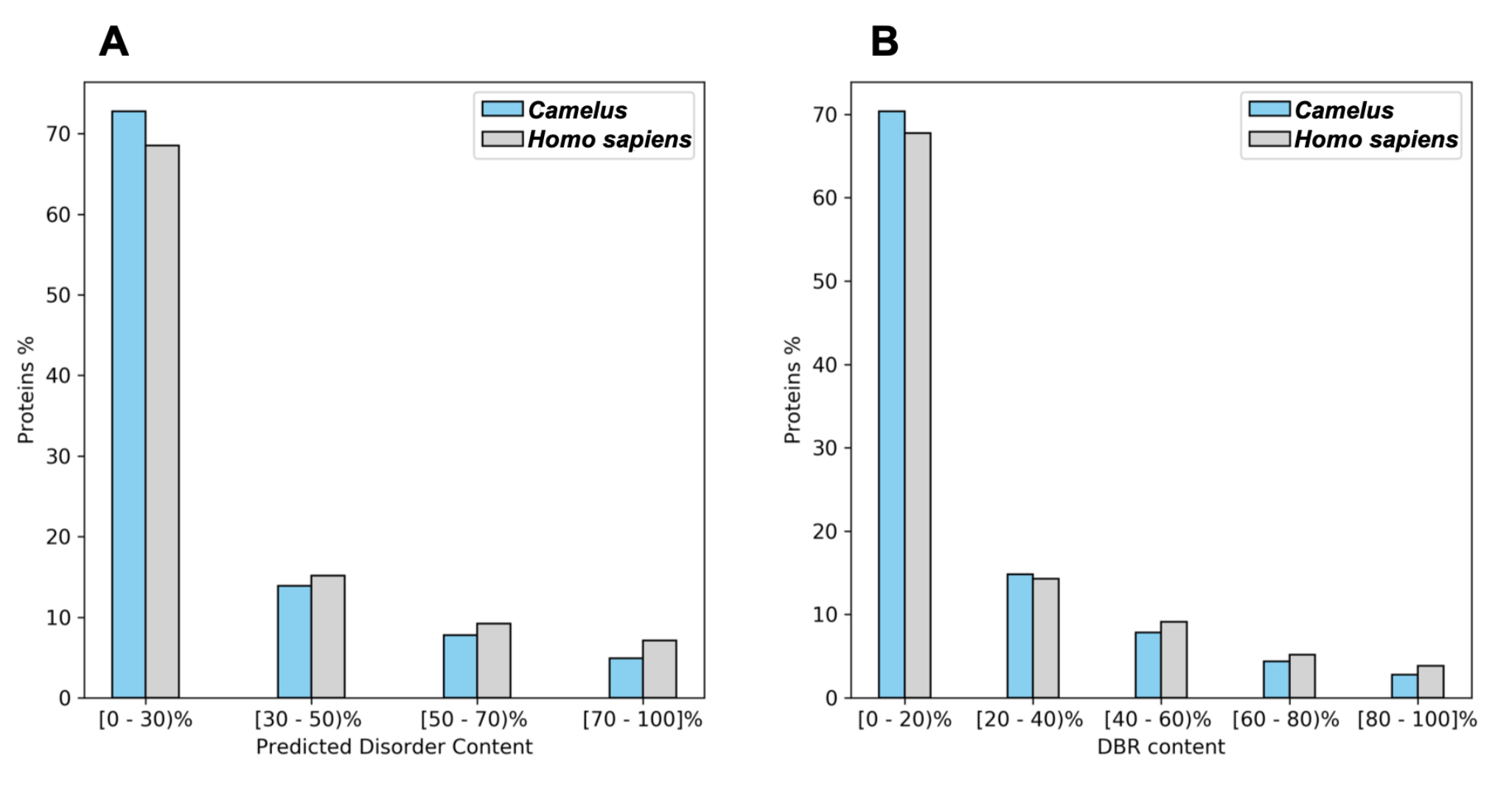
2.1. Disorder and Conserved Regions
2.2. Functional Annotation
2.3. Gene Ontology Enrichment Analysis
2.4. Functional Categories Significantly Disordered in Camelus
2.5. Comparison of GO Functional Categories between Camelus and Homo sapiens
3. Discussion
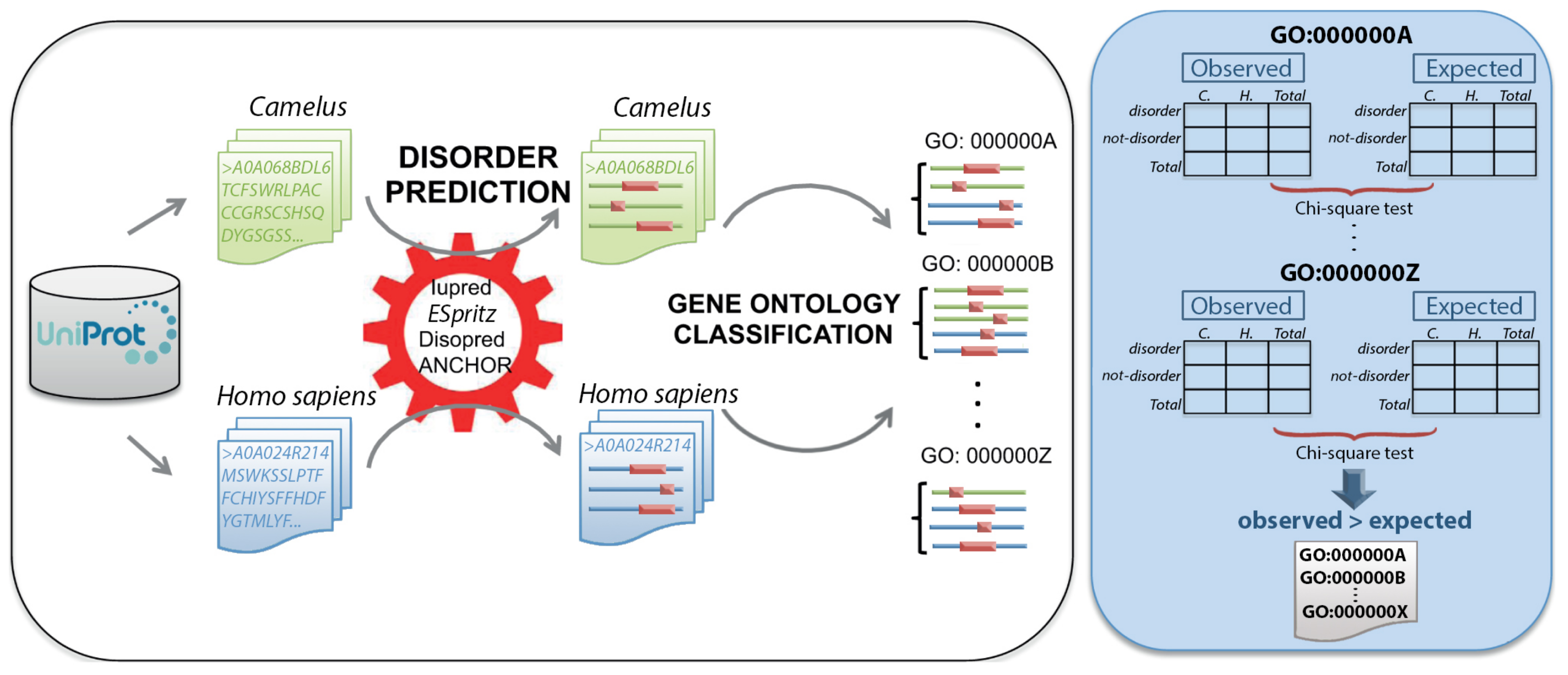
4. Materials and Methods
4.1. Protein Dataset
4.2. Protein Disorder Prediction
4.3. Multiple Sequence Alignment
4.4. Functional Annotation
4.5. Gene Ontology Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernado, P.; Blanchard, L.; Timmins, P.; Marion, D.; Ruigrok, R.W.; Blackledge, M. A structural model for unfolded proteins from residual dipolar couplings and small-angle X-ray scattering. Proc. Natl. Acad. Sci. USA 2005, 102, 17002–17007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Intrinsically disordered proteins and their “mysterious” (meta) physics. Front. Phys. 2019, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Romero, P.R.; Zaidi, S.; Fang, Y.Y.; Uversky, V.N.; Radivojac, P.; Oldfield, C.J.; Cortese, M.S.; Sickmeier, M.; LeGall, T.; Obradovic, Z.; et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl. Acad. Sci. USA 2006, 103, 8390–8395. [Google Scholar] [CrossRef] [Green Version]
- Tantos, A.; Friedrich, P.; Tompa, P. Cold stability of intrinsically disordered proteins. FEBS Lett. 2009, 2, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Dunker, A.K.; Romero, P.; Obradovic, Z.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. 2000, 11, 161–171. [Google Scholar]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef]
- Galea, C.A.; High, A.A.; Obenauer, J.C.; Mishra, A.; Park, C.G.; Punta, M.; Schlessinger, A.; Ma, J.; Rost, B.; Slaughter, C.A.; et al. Large-scale analysis of thermostable, mammalian proteins provides insights into the intrinsically disordered proteome. J. Proteome Res. 2009, 8, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaya, I.E.; Ibrikci, T.; Ersoy, O.K. Prediction of disorder with new computational tool: BVDEA. Expert Syst. Appl. 2011, 38, 14451–14459. [Google Scholar] [CrossRef] [Green Version]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Dunker, A.K. Natively disordered proteins. Appl. Bioinform. 2004, 3, 105–113. [Google Scholar] [CrossRef]
- Ishida, T.; Kinoshita, K. Prediction of disordered regions in proteins based on the meta approach. Bioinformatics 2008, 24, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Mizianty, M.J.; Stach, W.; Chen, K.; Kedarisetti, K.D.; Disfani, F.M.; Kurgan, L. Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics 2010, 26, i489–i496. [Google Scholar] [CrossRef] [Green Version]
- Hatos, A.; Hajdu-Soltész, B.; Monzon, A.M.; Palopoli, N.; Álvarez, L.; Aykac-Fas, B.; Bassot, C.; Benítez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2019, 48, D269–D276. [Google Scholar] [CrossRef] [Green Version]
- Fukuchi, S.; Amemiya, T.; Sakamoto, S.; Nobe, Y.; Hosoda, K.; Kado, Y.; Murakami, S.D.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Res. 2014, 42, D320–D325. [Google Scholar] [CrossRef]
- Pietrosemoli, N.; García-Martín, J.A.; Solano, R.; Pazos, F. Genome-wide analysis of protein disorder in Arabidopsis thaliana: Implications for plant environmental adaptation. PLoS ONE 2013, 8, e55524. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 2013, 22, 693–724. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. Interdiscip. J. 2005, 18, 343–384. [Google Scholar] [CrossRef] [PubMed]
- Redington, J.M.; Breydo, L.; Al-Mehdar, H.A.; Redwan, E.M.; Uversky, V.N. α-Lactalbumin: Of camels and cows. Protein Pept. Lett. 2016, 23, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, S.N.; Alduhaymi, I.S.; Alqahtani, L.; Altammaami, M.A.; Alhoshani, F.M.; Alrabiah, D.K.; Alyemni, S.O.; Alsulami, K.A.; Alghamdi, W.M.; Fallatah, M. Molecular Characterization, Bioinformatic Analysis, and Expression Profile of Lin-28 Gene and Its Protein from Arabian Camel (Camelus dromedarius). Int. J. Mol. Sci. 2019, 20, 2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdos, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. ESpritz: accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Dosztányi, Z.; Mészáros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef]
- Falda, M.; Toppo, S.; Pescarolo, A.; Lavezzo, E.; Di Camillo, B.; Facchinetti, A.; Cilia, E.; Velasco, R.; Fontana, P. Argot2: A large scale function prediction tool relying on semantic similarity of weighted Gene Ontology terms. BMC Bioinform. 2012, 13, S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choura, M.; Ebel, C.; Hanin, M. Genomic analysis of intrinsically disordered proteins in cereals: From mining to meaning. Gene 2019, 714, 143984. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Camelus | Homo sapiens | |
|---|---|---|
| Mean content of disordered residues | 28.16% | 34.04% |
| Proteins with at least one LDR | 47.16% | 52.56% |
| Mean number of residues belonging to LDR | 17.9% | 22.88% |
| Mean number of LDRs | 0.95 | 1.14 |
| Proteins with at least one DBR | 37.75% | 39.58% |
| Mean DBRs per protein | 0.40 | 0.47 |
| Mean residues belonging to DBRs | 16.51% | 19.69% |
| GO Terms Significantly Enriched in Camelus Disordered Proteins | GO Terms More Enriched in Camelus |
|---|---|
| Than Homo sapiens Disordered Proteins | |
| - glutathione catabolism | |
| - microtubule-based process | |
| - negative regulation of canonical Wnt signaling | - detection of chemical stimulus |
| pathway involved in osteoblast differentiation | - involved in sensory perception of smell |
| - protein localization to mitotic spindle | - oxygen transport |
| - protein K27linked deubiquitination | - proteasome assembly |
| - lactose biosynthesis | - protein peptidylprolyl isomerization |
| - hydrogen peroxide biosynthesis | - N-acetylglucosamine metabolism |
| - psychomotor behavior | - antigen processing and presentation of peptide |
| - 5methylcytosine metabolism | or polysaccharide antigen via MHC class II |
| - pigmentation | |
| - demethylation | |
| DNA 3prime dephosphorylation | |
| Camelus | Homo sapiens | Total | |
|---|---|---|---|
| Disordered | 111 | 2 | 113 |
| Not-disordered | 211 | 209 | 420 |
| Total | 322 | 211 | 533 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshehri, M.A.; Manee, M.M.; Al-Fageeh, M.B.; Al-Shomrani, B.M. Genomic Analysis of Intrinsically Disordered Proteins in the Genus Camelus. Int. J. Mol. Sci. 2020, 21, 4010. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114010
Alshehri MA, Manee MM, Al-Fageeh MB, Al-Shomrani BM. Genomic Analysis of Intrinsically Disordered Proteins in the Genus Camelus. International Journal of Molecular Sciences. 2020; 21(11):4010. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114010
Chicago/Turabian StyleAlshehri, Manal A., Manee M. Manee, Mohamed B. Al-Fageeh, and Badr M. Al-Shomrani. 2020. "Genomic Analysis of Intrinsically Disordered Proteins in the Genus Camelus" International Journal of Molecular Sciences 21, no. 11: 4010. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114010