MnTBAP Reverses Pulmonary Vascular Remodeling and Improves Cardiac Function in Experimentally Induced Pulmonary Arterial Hypertension

 , , , and
, , , and 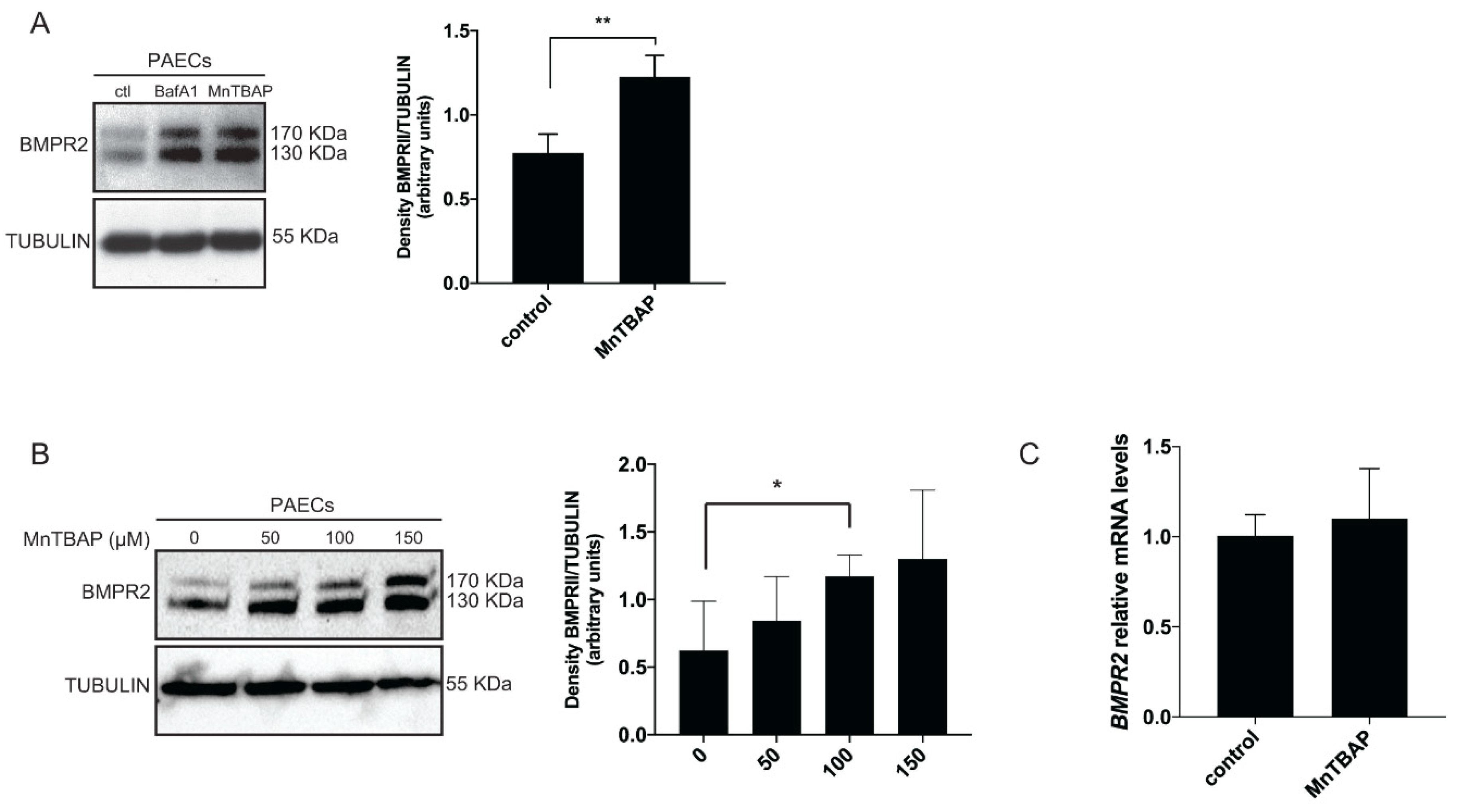{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
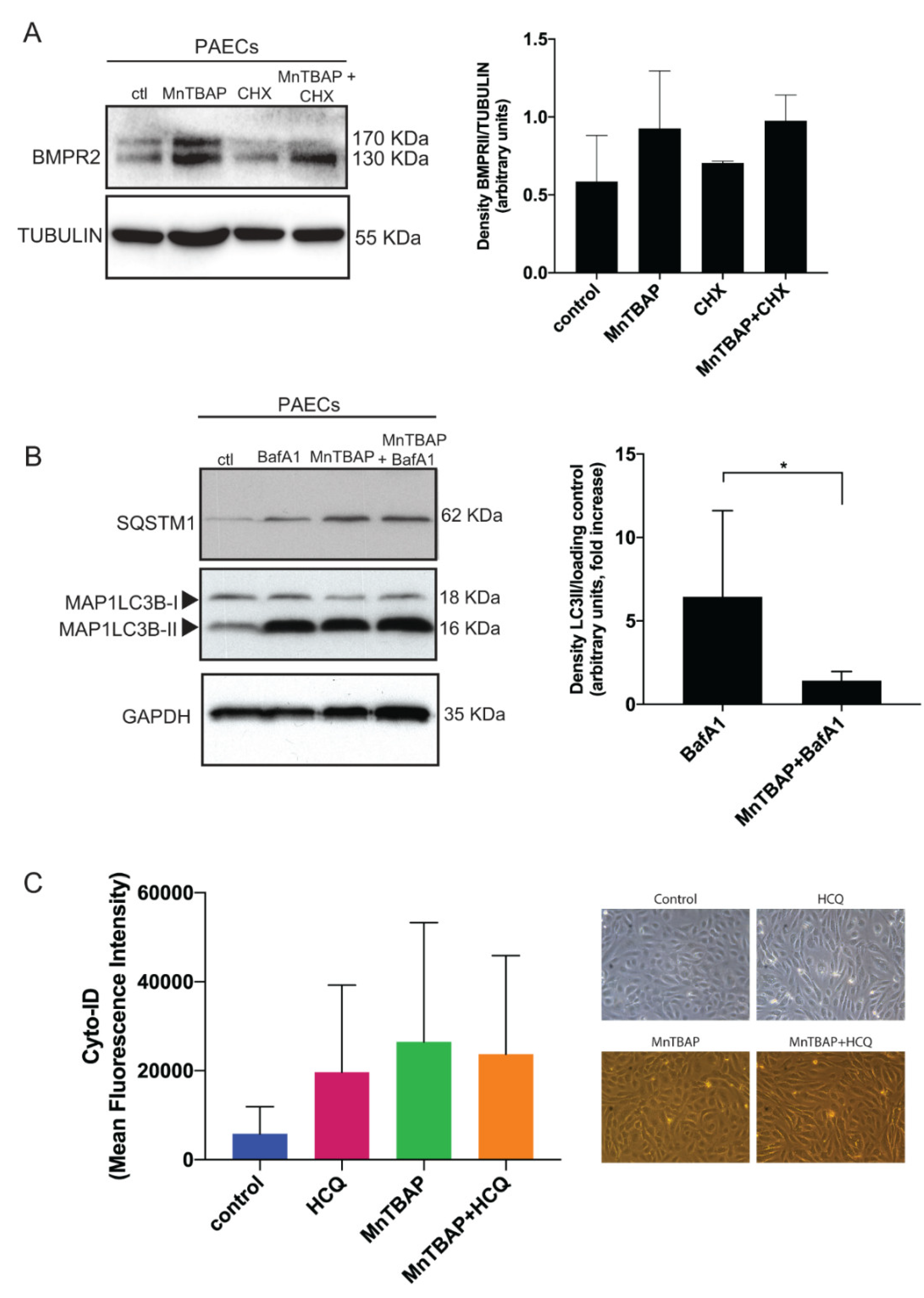
2.1. MnTBAP Increases BMPR2 Levels In Vitro by Inhibiting Autophagy
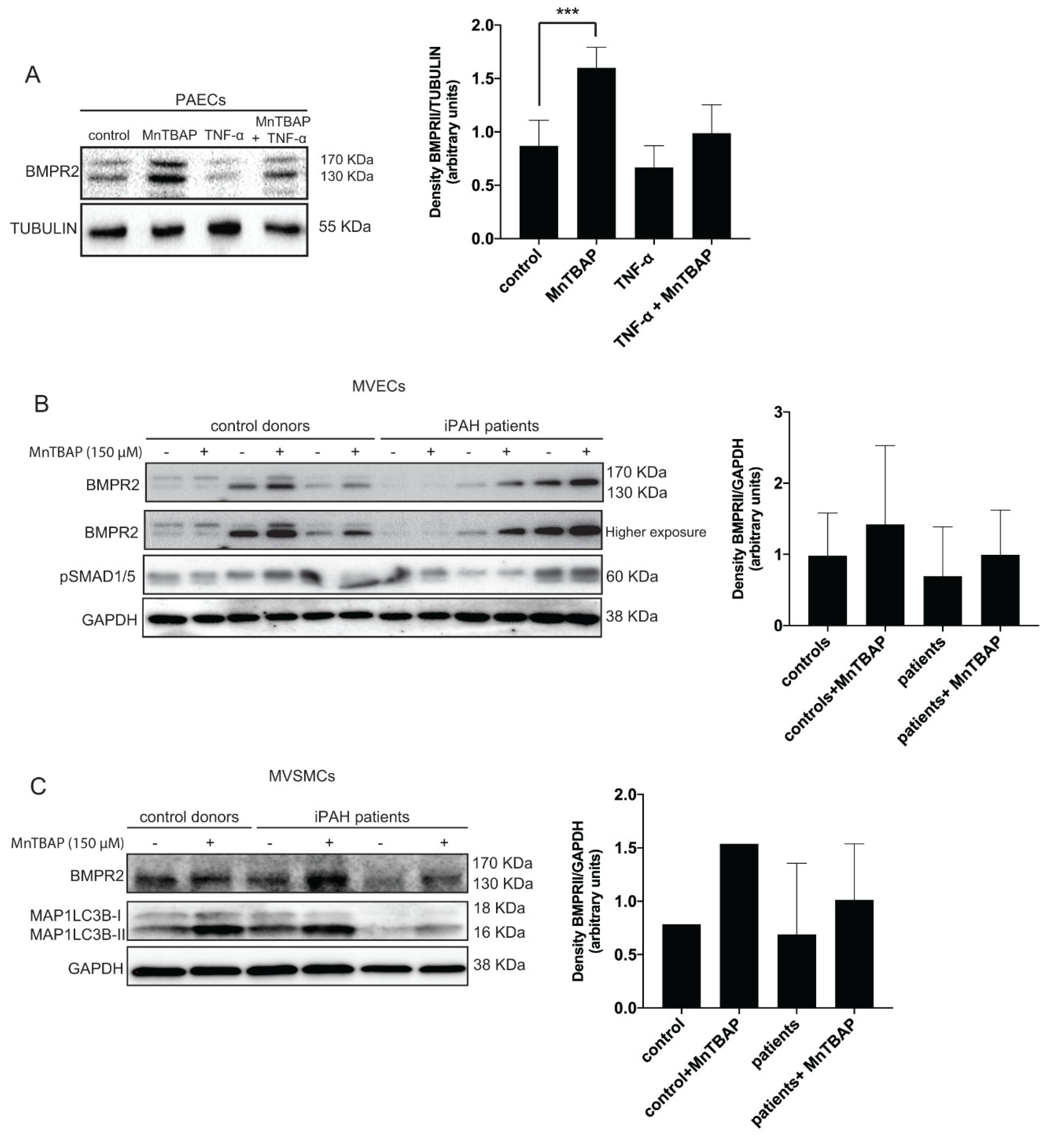
2.2. MnTBAP Increases the Levels of BMPR2 in Pulmonary MVECs and MVSMCs Isolated from iPAH Patients
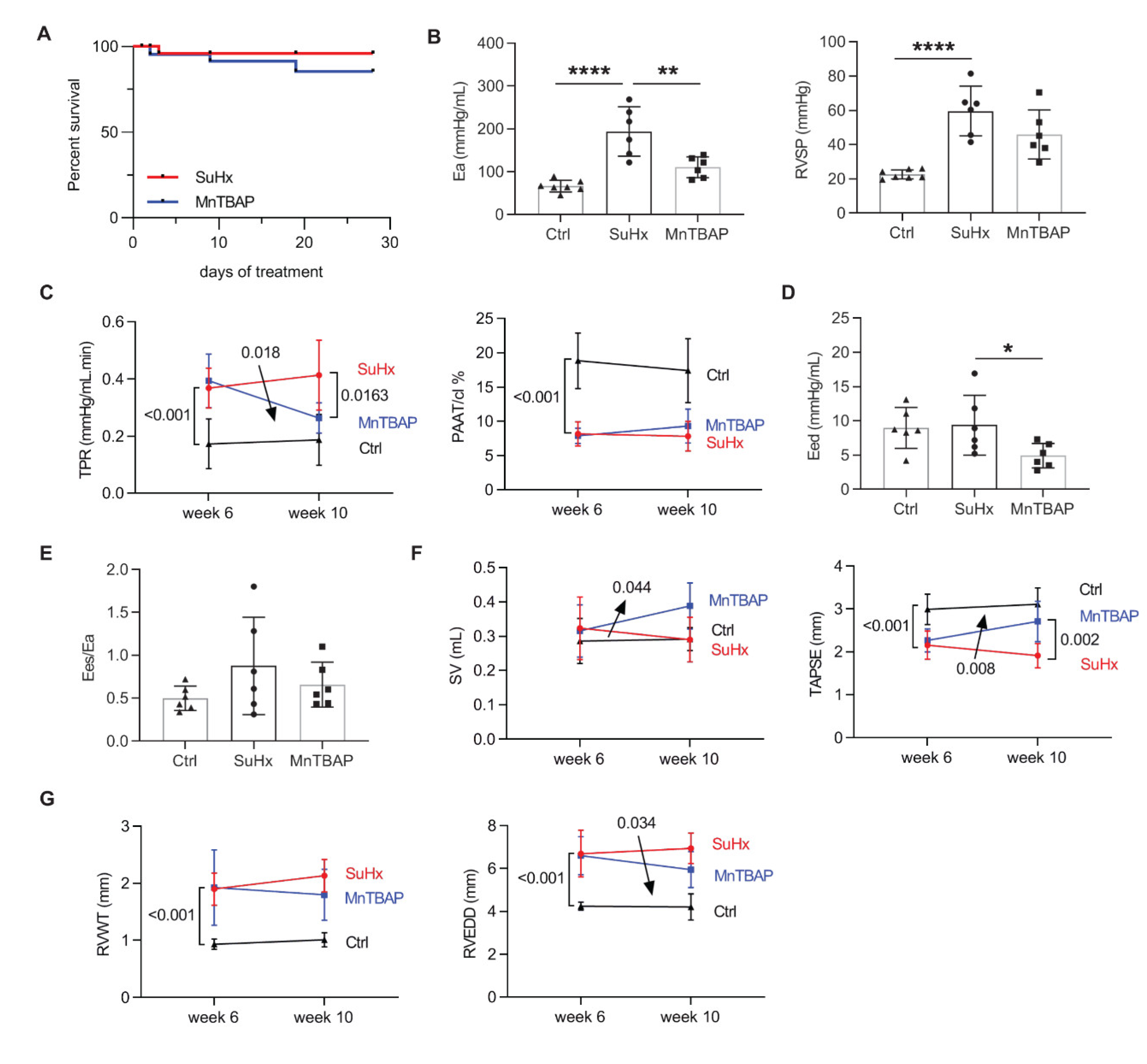
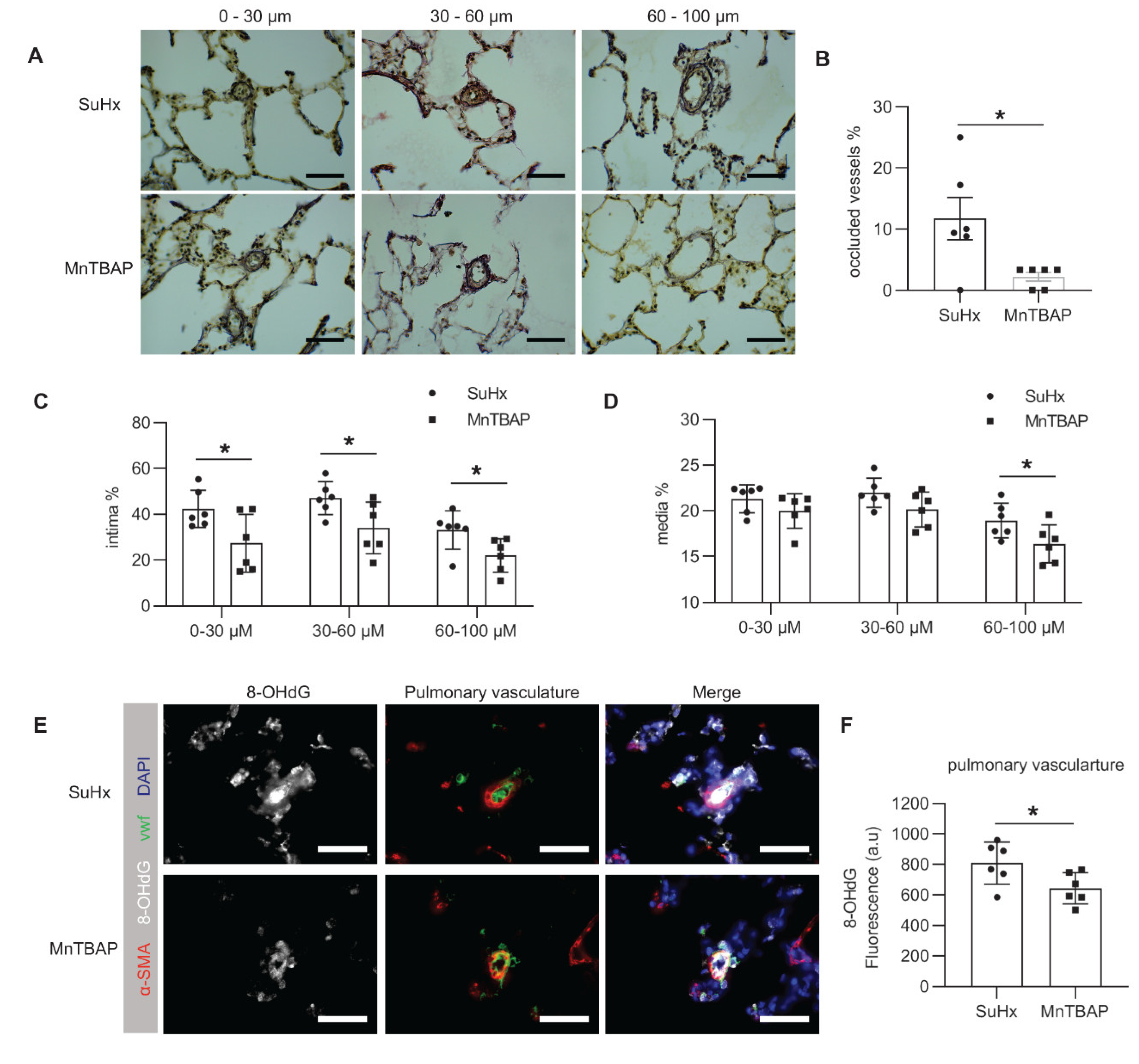
2.3. MnTBAP Reduced RV Afterload in Experimental PAH
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Western Blot Analysis
4.4. Quantitative Real-Time PCR
4.5. Flow Cytometry Analysis
4.6. Experimental Pulmonary Arterial Hypertension
4.7. SuHx Rat Model of Pulmonary Hypertension and MnTBAP Treatment
4.8. Hemodynamic Measurements
4.9. Morphometry of the Pulmonary Vasculatures
4.9.1. Immunofluorescence Staining
4.9.2. Immunofluorescence Quantification
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guignabert, C.; Tu, L.; Le Hiress, M.; Ricard, N.; Sattler, C.; Seferian, A.; Huertas, A.; Humbert, M.; Montani, D. Pathogenesis of pulmonary arterial hypertension: Lessons from cancer. Eur. Respir. Rev. 2013, 22, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenkranz, S. Pulmonary hypertension 2015: Current definitions, terminology, and novel treatment options. Clin. Res. Cardiol. 2015, 104, 197–207. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Aman, J.; Harbaum, L.; Ulrich, A.; Wharton, J.; Rhodes, C.J. Recent advances in pulmonary arterial hypertension. F1000Research 2018, 7, F1000. [Google Scholar] [CrossRef] [Green Version]
- McGoon, M.D.; Benza, R.L.; Escribano-Subias, P.; Jiang, X.; Miller, D.P.; Peacock, A.J.; Pepke-Zaba, J.; Pulido, T.; Rich, S.; Rosenkranz, S.; et al. Pulmonary arterial hypertension: Epidemiology and registries. J. Am. Coll. Cardiol. 2013, 62, D51–D59. [Google Scholar] [CrossRef] [Green Version]
- Provencher, S.; Archer, S.L.; Ramirez, F.D.; Hibbert, B.; Paulin, R.; Boucherat, O.; Lacasse, Y.; Bonnet, S. Standards and Methodological Rigor in Pulmonary Arterial Hypertension Preclinical and Translational Research. Circ. Res. 2018, 122, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Ivy, D. Pulmonary Hypertension in Children. Cardiol. Clin. 2016, 34, 451–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.J. Estrogen: Friend or Foe in Pulmonary Hypertension? Am. J. Respir Crit. Care Med. 2016, 193, 1084–1086. [Google Scholar] [CrossRef] [PubMed]
- Lan, N.S.H.; Massam, B.D.; Kulkarni, S.S.; Lang, C.C. Pulmonary Arterial Hypertension: Pathophysiology and Treatment. Diseases 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, J.R.; Ormiston, M.L.; Perez-Iratxeta, C.; Courtman, D.W.; Jiang, B.; Ferrer, E.; Caruso, P.; Southwood, M.; Foster, W.S.; Morrell, N.W.; et al. Proteomic analysis implicates translationally controlled tumor protein as a novel mediator of occlusive vascular remodeling in pulmonary arterial hypertension. Circulation 2014, 129, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Andruska, A.; Spiekerkoetter, E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and Beyond: Contributions to Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Puerto, M.C.; van Zuijen, I.; Huang, C.J.; Szulcek, R.; Pan, X.; van Dinther, M.A.; Kurakula, K.; Wiesmeijer, C.C.; Goumans, M.J.; Bogaard, H.J.; et al. Autophagy contributes to BMP type 2 receptor degradation and development of pulmonary arterial hypertension. J. Pathol. 2019, 249, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFalpha drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Eickelberg, O.; Elliott, C.G.; Geraci, M.W.; Hanaoka, M.; Loyd, J.E.; Newman, J.H.; Phillips, J.A., 3rd; Soubrier, F.; Trembath, R.C.; et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S32–S42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.; Vrugt, B.; Brock, M.; Speich, R.; Ulrich, S.; Huber, L.C. Inflammatory cytokines in pulmonary hypertension. Respir. Res. 2014, 15, 47. [Google Scholar] [CrossRef] [Green Version]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Orriols, M.; Gomez-Puerto, M.C.; ten Dijke, P. Erratum To: BMP Type II Receptor as a Therapeutic Target in Pulmonary Arterial Hypertension. Cell Mol. Life Sci. 2017, 74, 2997. [Google Scholar] [CrossRef] [Green Version]
- Dunmore, B.J.; Drake, K.M.; Upton, P.D.; Toshner, M.R.; Aldred, M.A.; Morrell, N.W. The lysosomal inhibitor, chloroquine, increases cell surface BMPR-II levels and restores BMP9 signalling in endothelial cells harbouring BMPR-II mutations. Hum. Mol. Genet. 2013, 22, 3667–3679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Invest. 2013, 123, 3600–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumurkhuu, G.; Koide, N.; Dagvadorj, J.; Hassan, F.; Islam, S.; Naiki, Y.; Mori, I.; Yoshida, T.; Yokochi, T. MnTBAP, a synthetic metalloporphyrin, inhibits production of tumor necrosis factor-alpha in lipopolysaccharide-stimulated RAW 264.7 macrophages cells via inhibiting oxidative stress-mediating p38 and SAPK/JNK signaling. FEMS Immunol. Med. Microbiol. 2007, 49, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, M.; Cai, C.L.; Valencia, G.B.; Aranda, J.V.; Beharry, K.D. MnTBAP or Catalase Is More Protective against Oxidative Stress in Human Retinal Endothelial Cells Exposed to Intermittent Hypoxia than Their Co-Administration (EUK-134). React. Oxyg. Species (Apex). 2017, 3, 47–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batinic-Haberle, I.; Cuzzocrea, S.; Reboucas, J.S.; Ferrer-Sueta, G.; Mazzon, E.; Di Paola, R.; Radi, R.; Spasojevic, I.; Benov, L.; Salvemini, D. Pure MnTBAP selectively scavenges peroxynitrite over superoxide: Comparison of pure and commercial MnTBAP samples to MnTE-2-PyP in two models of oxidative stress injury, an SOD-specific Escherichia coli model and carrageenan-induced pleurisy. Free Radic. Biol. Med. 2009, 46, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, X.; Wang, J.; Liu, Y.; Wang, Y.; Ding, W. MnTBAP treatment ameliorates aldosterone-induced renal injury by regulating mitochondrial dysfunction and NLRP3 inflammasome signalling. Am. J. Transl. Res. 2018, 10, 3504–3513. [Google Scholar] [PubMed]
- Zhou, Q.; Einert, M.; Schmitt, H.; Wang, Z.; Pankratz, F.; Olivier, C.B.; Bode, C.; Liao, J.K.; Moser, M. MnTBAP increases BMPR-II expression in endothelial cells and attenuates vascular inflammation. Vascul. Pharmacol. 2016, 84, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suresh, M.V.; Yu, B.; Lakshminrusimha, S.; Machado-Aranda, D.; Talarico, N.; Zeng, L.; Davidson, B.A.; Pennathur, S.; Raghavendran, K. The protective role of MnTBAP in oxidant-mediated injury and inflammation in a rat model of lung contusion. Surgery 2013, 154, 980–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatadri, R.; Iyer, A.K.; Ramesh, V.; Wright, C.; Castro, C.A.; Yakisich, J.S.; Azad, N. MnTBAP Inhibits Bleomycin-Induced Pulmonary Fibrosis by Regulating VEGF and Wnt Signaling. J. Cell Physiol. 2017, 232, 506–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuzzocrea, S.; Zingarelli, B.; Costantino, G.; Caputi, A.P. Beneficial effects of Mn(III)tetrakis (4-benzoic acid) porphyrin (MnTBAP), a superoxide dismutase mimetic, in carrageenan-induced pleurisy. Free Radic. Biol. Med. 1999, 26, 25–33. [Google Scholar] [CrossRef]
- Yu, J.; Mao, S.; Zhang, Y.; Gong, W.; Jia, Z.; Huang, S.; Zhang, A. MnTBAP Therapy Attenuates Renal Fibrosis in Mice with 5/6 Nephrectomy. Oxid Med. Cell Longev. 2016, 2016, 7496930. [Google Scholar] [CrossRef] [Green Version]
- Zahmatkesh, M.; Kadkhodaee, M.; Moosavi, S.M.; Jorjani, M.; Kajbafzadeh, A.; Golestani, A.; Ghaznavi, R. Beneficial effects of MnTBAP, a broad-spectrum reactive species scavenger, in rat renal ischemia/reperfusion injury. Clin. Exp. Nephrol. 2005, 9, 212–218. [Google Scholar] [CrossRef]
- Long, L.; Yang, X.; Southwood, M.; Lu, J.; Marciniak, S.J.; Dunmore, B.J.; Morrell, N.W. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ. Res. 2013, 112, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Durrington, H.J.; Upton, P.D.; Hoer, S.; Boname, J.; Dunmore, B.J.; Yang, J.; Crilley, T.K.; Butler, L.M.; Blackbourn, D.J.; Nash, G.B.; et al. Identification of a lysosomal pathway regulating degradation of the bone morphogenetic protein receptor type II. J. Biol. Chem. 2010, 285, 37641–37649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, J.W.; Amich, J.M.; Andonian, A.; Rosen, V. N-linked glycosylation of the bone morphogenetic protein receptor type 2 (BMPR2) enhances ligand binding. Cell Mol. Life Sci. 2014, 71, 3165–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, A.; Kizhakkedath, P.; Al-Gazali, L.; Ali, B.R. Defective cellular trafficking of the bone morphogenetic protein receptor type II by mutations underlying familial pulmonary arterial hypertension. Gene 2015, 561, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Lilly, B. We Have Contact: Endothelial Cell-Smooth Muscle Cell Interactions. Physiology 2014, 29, 234–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christman, B.W.; Mcpherson, C.D.; Newman, J.H.; King, G.A.; Bernard, G.R.; Groves, B.M.; Loyd, J.E. An Imbalance between the Excretion of Thromboxane and Prostacyclin Metabolites in Pulmonary-Hypertension. New Engl. J. Med. 1992, 327, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Goncalves Bos, D.; Van Der Bruggen, C.E.E.; Kurakula, K.; Sun, X.Q.; Casali, K.R.; Casali, A.G.; Rol, N.; Szulcek, R.; Dos Remedios, C.; Guignabert, C.; et al. Contribution of Impaired Parasympathetic Activity to Right Ventricular Dysfunction and Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. Circulation 2018, 137, 910–924. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.M.; Xia, W.; Holmes, M.D.; Hodge, S.J.; Danilov, S.; Curiel, D.T.; Morrell, N.W.; Reynolds, P.N. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1182–L1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertin, S.; Samson, M.; Pons, C.; Guigonis, J.M.; Gavelli, A.; Baque, P.; Brossette, N.; Pagnotta, S.; Ricci, J.E.; Pierrefite-Carle, V. Comparative Proteomics Study Reveals That Bacterial CpG Motifs Induce Tumor Cell Autophagy in Vitro and in Vivo. Mol. Cell Proteom. 2008, 7, 2311–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thebaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Webb, S.; Tucker, A.; Rabinovitch, M.; O’Brien, R.F.; McMurtry, I.F.; Stelzner, T.J. Factors influencing the idiopathic development of pulmonary hypertension in the fawn hooded rat. Am. Rev. Respir. Dis. 1992, 145, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Provencher, S.; Guignabert, C.; Perros, F.; Boucherat, O.; Schermuly, R.T.; Hassoun, P.M.; Rabinovitch, M.; Nicolls, M.R.; Humbert, M. Translating Research into Improved Patient Care in Pulmonary Arterial Hypertension. Am. J. Resp. Crit. Care 2017, 195, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Mauritz, G.J.; Kind, T.; Marcus, J.T.; Bogaard, H.J.; van de Veerdonk, M.; Postmus, P.E.; Boonstra, A.; Westerhof, N.; Vonk-Noordegraaf, A. Progressive changes in right ventricular geometric shortening and long-term survival in pulmonary arterial hypertension. Chest 2012, 141, 935–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, J.; Bauerle, O.; Palomar, A.; Gomez, A.; Martinez-Guerra, M.L.; Beltran, M.; Guerrero, M.L. Survival in primary pulmonary hypertension. Validation of a prognostic equation. Circulation 1994, 89, 1733–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nojiri, H.; Shimizu, T.; Funakoshi, M.; Yamaguchi, O.; Zhou, H.; Kawakami, S.; Ohta, Y.; Sami, M.; Tachibana, T.; Ishikawa, H.; et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. J. Biol. Chem. 2006, 281, 33789–33801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009, 120, 1951–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szulcek, R.; Happe, C.M.; Rol, N.; Fontijn, R.D.; Dickhoff, C.; Hartemink, K.J.; Grunberg, K.; Tu, L.; Timens, W.; Nossent, G.D.; et al. Delayed Microvascular Shear Adaptation in Pulmonary Arterial Hypertension. Role of Platelet Endothelial Cell Adhesion Molecule-1 Cleavage. Am. J. Respir. Crit. Care Med. 2016, 193, 1410–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Raaf, M.A.; Kroeze, Y.; Middelman, A.; de Man, F.S.; de Jong, H.; Vonk-Noordegraaf, A.; de Korte, C.; Voelkel, N.F.; Homberg, J.; Bogaard, H.J. Serotonin transporter is not required for the development of severe pulmonary hypertension in the Sugen hypoxia rat model. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1164–L1173. [Google Scholar] [CrossRef]
- Stiedl, O.; Spiess, J. Effect of tone-dependent fear conditioning on heart rate and behavior of C57BL/6N mice. Behav. Neurosci. 1997, 111, 703–711. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez-Puerto, M.C.; Sun, X.-Q.; Schalij, I.; Orriols, M.; Pan, X.; Szulcek, R.; Goumans, M.-J.; Bogaard, H.-J.; Zhou, Q.; ten Dijke, P. MnTBAP Reverses Pulmonary Vascular Remodeling and Improves Cardiac Function in Experimentally Induced Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2020, 21, 4130. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114130
Gomez-Puerto MC, Sun X-Q, Schalij I, Orriols M, Pan X, Szulcek R, Goumans M-J, Bogaard H-J, Zhou Q, ten Dijke P. MnTBAP Reverses Pulmonary Vascular Remodeling and Improves Cardiac Function in Experimentally Induced Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2020; 21(11):4130. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114130
Chicago/Turabian StyleGomez-Puerto, Maria Catalina, Xiao-Qing Sun, Ingrid Schalij, Mar Orriols, Xiaoke Pan, Robert Szulcek, Marie-José Goumans, Harm-Jan Bogaard, Qian Zhou, and Peter ten Dijke. 2020. "MnTBAP Reverses Pulmonary Vascular Remodeling and Improves Cardiac Function in Experimentally Induced Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 21, no. 11: 4130. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114130