Identification of Substrates of Cytoplasmic Peptidyl-Prolyl Cis/Trans Isomerases and Their Collective Essentiality in Escherichia Coli

 ,
,
Abstract
:1. Introduction
2. Results
2.1. The Peptidyl-Prolyl Cis/Trans Isomerase Activity Is Required for Optimal Growth
2.2. Protein Folding Defects—Accumulation of Various Proteins in Aggregation Fractions in Δ6ppi Bacteria
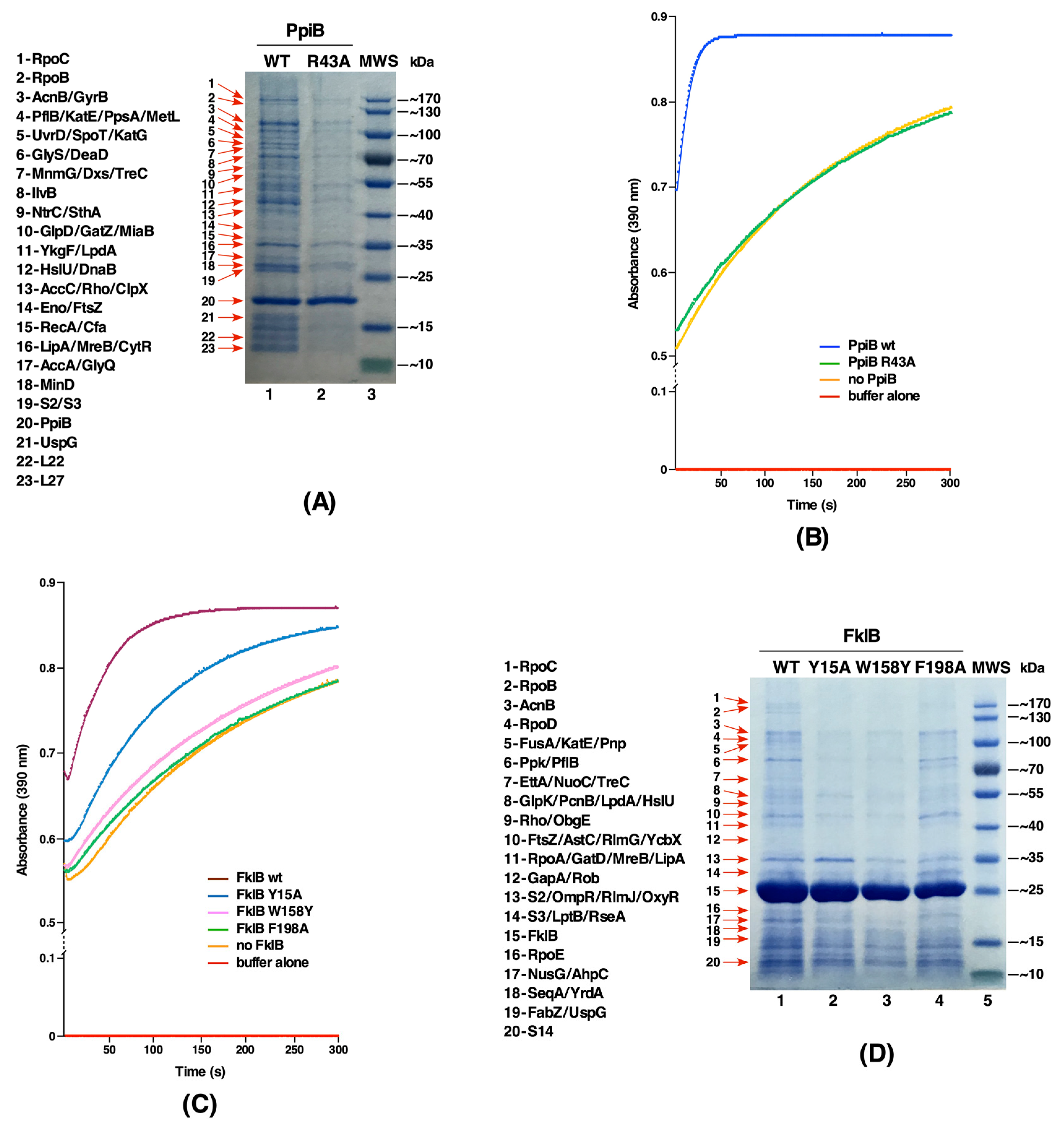
2.3. The PpiB Protein Is the Major Contributor of PPIase Activity in the Cytoplasm
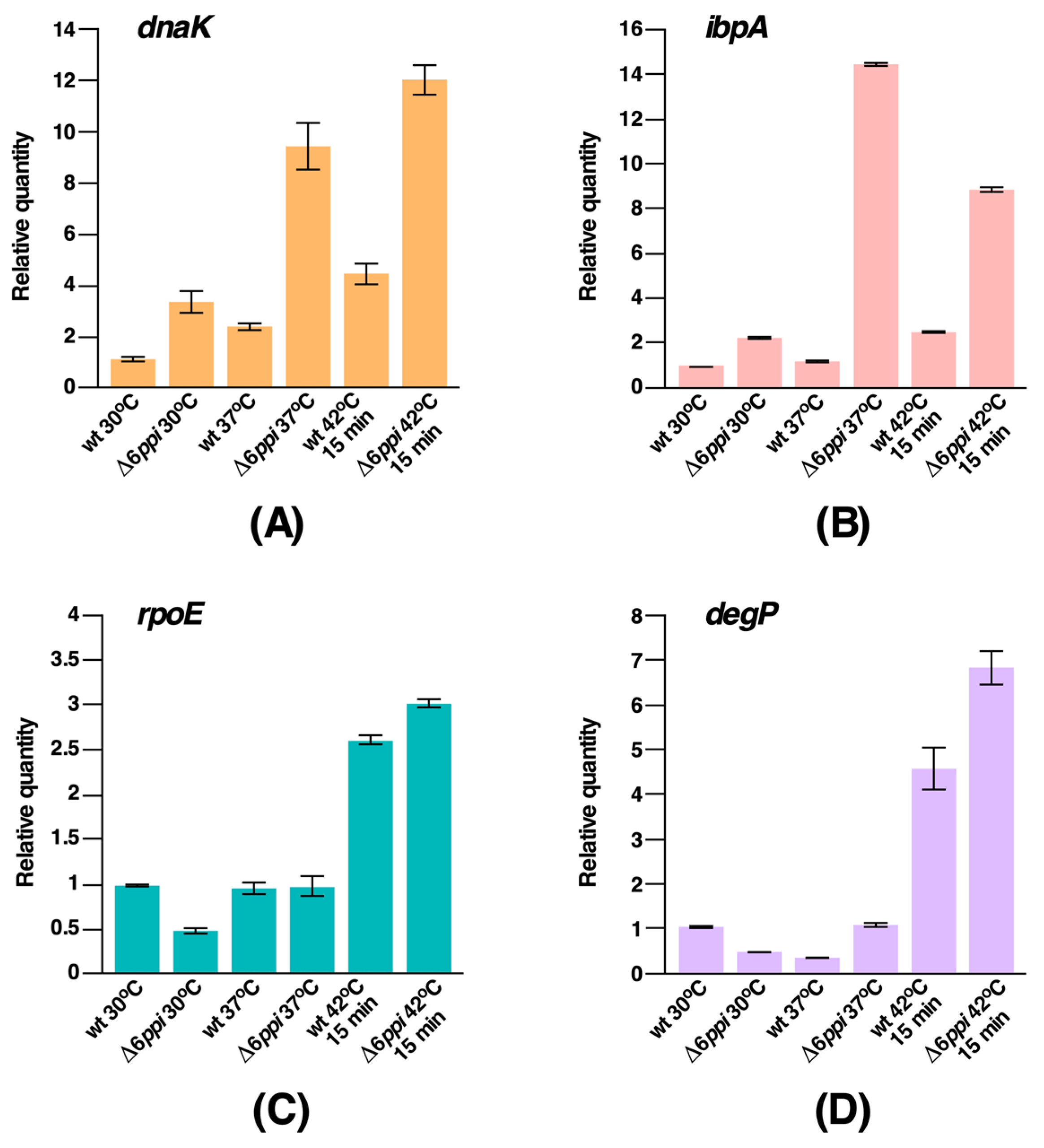
2.4. Δ6ppi Bacteria Exhibit the Constitutive Induction of RpoH-Regulated Heat Shock Response
2.5. Identification of Substrates of the PpiC Protein Reveals a Role in Oxidative Stress, Transcriptional and Essential Metabolic Processes
2.6. Substrates of FkpB Include the RpoE Sigma Factor, Proteins Related to Cell Shape/Division and Lipopolysaccharide (LPS) Transport
2.7. The PpiB Protein Has Larger Number of Substrates That Require Its PPIase Activity
2.8. Identification of Active Site Residues of the FklB Protein and Identification of its Substrates Reveal Certain Unique and Some with an Overlap with Other PPIs
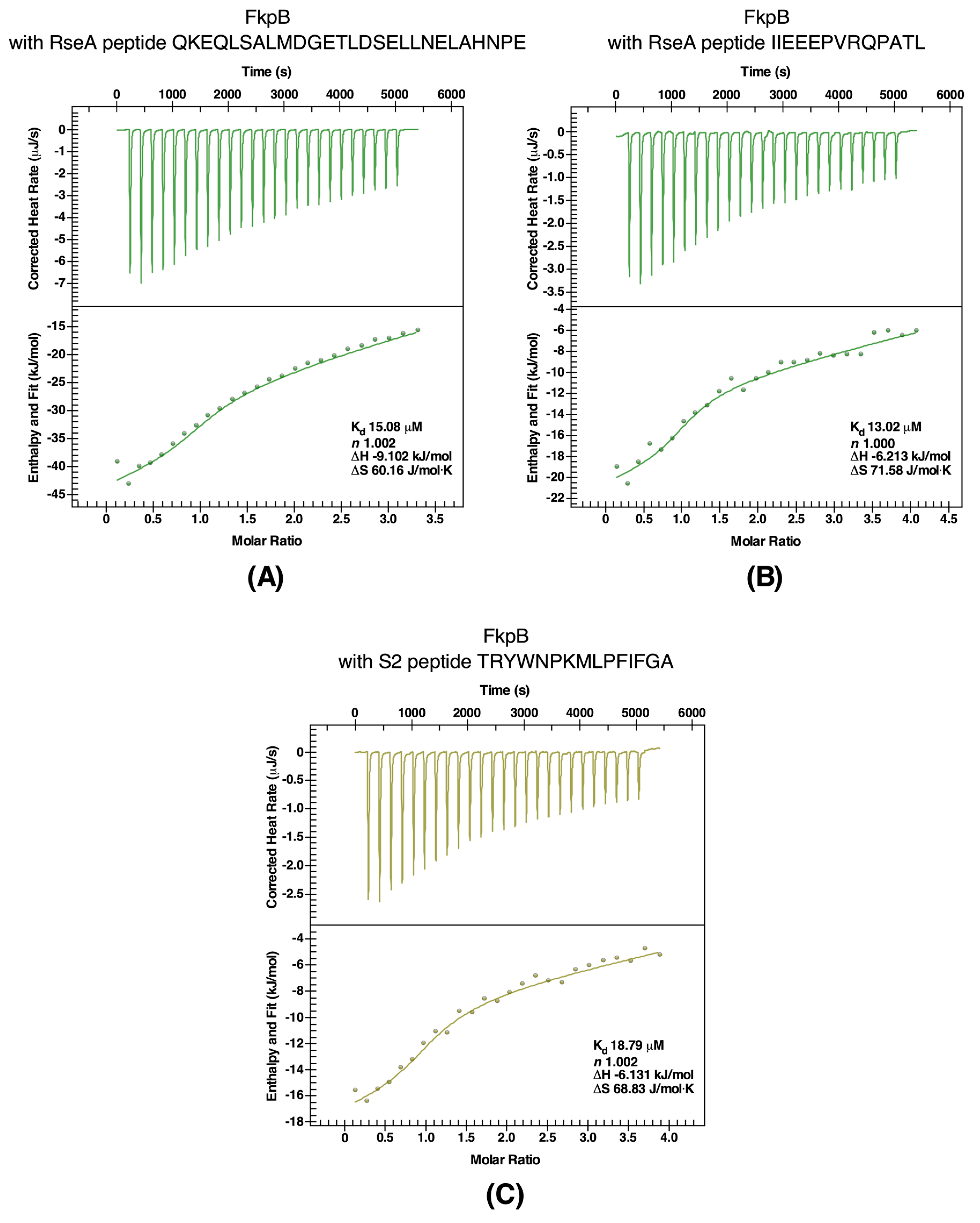
2.9. Validation of RpoE, RseA, and S2 Proteins as Client Proteins of FkpB
2.10. AhpC as a Client Protein of PpiC and FkpB
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids and Media
4.2. Generation of Null Mutations and the Construction of Δ6ppi Derivatives
4.3. Construction of Chromosomal Wild-Type and Single-Copy Mutants of ppiC FLAG Derivatives
4.4. The Isolation of Aggregated Proteins
4.5. Co-immunoprecipitation with 3xFLAG-tagged PpiC Protein
4.6. Protein Purification of Wild-Type and Ppi Mutants
4.7. PPIase Assay
4.8. RNA Purification and qRT-PCR Analysis
4.9. Isothermal Titration Calorimetry (ITC) Measurements
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PPI | peptidyl-prolyl cis/trans isomerase |
| FKBP | FK506-binding protein |
| OMP | outer membrane protein |
| ITC | isothermal titration calorimetry |
References
- Ellis, R.J. Protein misassembly: Macromolecular crowding and molecular chaperones. Adv. Exp. Med. Biol. 2007, 594, 1–13. [Google Scholar] [CrossRef]
- Grathwohl, C.; Wüthrich, K. NMR studies of the rates of proline cis-trans isomerization in oligopeptides. Biopolymers 1981, 20, 2623–2633. [Google Scholar] [CrossRef]
- Nicholson, L.K.; Lu, K.P. Prolyl cis-trans isomerization as a molecular timer in Crk signaling. Mol. Cell 2007, 25, 483–485. [Google Scholar] [CrossRef]
- Fischer, G.; Aumüller, T. Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Rev. Physiol. Biochem. Pharmacol. 2003, 148, 105–150. [Google Scholar] [CrossRef]
- Rahfeld, J.U.; Schierhorn, A.; Mann, K.; Fischer, G. A novel peptidyl-prolyl cis/trans isomerase from Escherichia coli. FEBS Lett. 1994, 343, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Ferbitz, L.; Maier, T.; Patzelt, H.; Bukau, B.; Deuerling, E.; Ban, N. Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins. Nature 2004, 431, 590–596. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.; Becker, A.H.; Sandikci, A.; Huber, D.; Chaba, R.; Gloge, F.; Nichols, R.J.; Typas, A.; Gross, C.A.; Kramer, G.; et al. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell 2011, 147, 1295–1308. [Google Scholar] [CrossRef] [Green Version]
- Hottenrott, S.; Schumann, T.; Plückthun, A.; Fischer, G.; Rahfeld, J.U. The Escherichia coli SlyD is a metal ion-regulated peptidyl-prolyl cis/trans-isomerase. J. Biol. Chem. 1997, 272, 15697–15701. [Google Scholar] [CrossRef] [Green Version]
- Knappe, T.A.; Eckert, B.; Schaarschmidt, P.; Scholz, C.; Schmid, F.X. Insertion of a chaperone domain converts FKBP12 into a powerful catalyst of protein folding. J. Mol. Biol. 2007, 368, 1458–1468. [Google Scholar] [CrossRef]
- Zhang, J.W.; Leach, M.R.; Zamble, D.B. The peptidyl-prolyl isomerase activity of SlyD is not required for maturation of Escherichia coli hydrogenase. J. Bacteriol. 2007, 189, 7942–7944. [Google Scholar] [CrossRef] [Green Version]
- Malešević, M.; Poehlmann, A.; Hernandez Alvarez, B.; Diessner, A.; Träger, M.; Rahfeld, J.U.; Jahreis, G.; Liebscher, S.; Bordusa, F.; Fischer, G.; et al. The protein-free IANUS peptide array uncovers interaction sites between Escherichia coli parvulin 10 and alkyl hydroperoxide reductase. Biochemistry 2010, 49, 8626–8635. [Google Scholar] [CrossRef] [PubMed]
- Quistgaard, E.M.; Nordlund, P.; Löw, C. High-resolution insights into binding of unfolded polypeptides by the PPIase chaperone SlpA. FASEB J. 2012, 26, 4003–4013. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.; Yang, X.; Boulanger, M.J.; Ishiguro, E.E. Occurrence of homologs of the Escherichia coli lytB gene in gram-negative bacterial species. J. Bacteriol. 1998, 180, 1959–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, G.; Kobylak, N.; Lindner, B.; Stupak, A.; Raina, S. Assembly of lipopolysaccharide in Escherichia coli requires the essential LapB heat shock protein. J. Biol. Chem. 2014, 289, 14829–14853. [Google Scholar] [CrossRef] [Green Version]
- Raina, S.; Missiakas, D.; Georgopoulos, C. The rpoE gene encoding the σE (σ24) heat shock sigma factor of Escherichia coli. EMBO J. 1995, 14, 1043–1055. [Google Scholar] [CrossRef]
- Compton, L.A.; Davis, J.M.; Macdonald, R.; Bächinger, H.P. Structural and functional characterization of Escherichia coli peptidyl-prolyl cis-trans isomerases. Eur. J. Biochem. 1992, 206, 927–934. [Google Scholar] [CrossRef]
- Hayano, T.; Takahashi, N.; Kato, S.; Maki, N.; Suzuki, M. Two distinct forms of peptidylprolyl-cis-trans-isomerase are expressed separately in periplasmic and cytoplasmic compartments of Escherichia coli cells. Biochemistry 1991, 30, 3041–3046. [Google Scholar] [CrossRef]
- Zydowsky, L.D.; Etzkorn, F.A.; Chang, H.Y.; Ferguson, S.B.; Stolz, L.A.; Ho, S.I.; Walsh, C.T. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1992, 1, 1092–1099. [Google Scholar] [CrossRef] [Green Version]
- Rahfeld, J.U.; Rücknagel, K.P.; Stoller, G.; Horne, S.M.; Schierhorn, A.; Young, K.D.; Fischer, G. Isolation and amino acid sequence of a new 22-kDa FKBP-like peptidyl-prolyl cis/trans-isomerase of Escherichia coli. Similarity to Mip-like proteins of pathogenic bacteria. J. Biol. Chem. 1996, 271, 22130–22138. [Google Scholar] [CrossRef] [Green Version]
- Budiman, C.; Tadokoro, T.; Angkawidjaja, C.; Koga, Y.; Kanaya, S. Role of polar and nonpolar residues at the active site for PPIase activity of FKBP22 from Shewanella sp. SIB1. FEBS J. 2012, 279, 976–986. [Google Scholar] [CrossRef]
- Polley, S.; Chakravarty, D.; Chakrabarti, G.; Chattopadhyaya, R.; Sau, S. Proline substitutions in a Mip-like peptidyl-prolyl cis-trans isomerase severely affect its structure, stability, shape and activity. Biochim. Open 2015, 1, 28–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiakas, D.; Schwager, F.; Betton, J.M.; Georgopoulos, C.; Raina, S. Identification and characterization of HslVHslU (ClpQ ClpY) proteins involved in overall proteolysis of misfolded proteins in Escherichia coli. EMBO J. 1996, 15, 6899–6909. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.G.; Rosner, J.L. Genomics of the marA/soxS/rob regulon of Escherichia coli: Identification of directly activated promoters by application of molecular genetics and informatics to microarray data. Mol. Microbiol. 2002, 44, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Raina, S. Regulated assembly of LPS, its structural alterations and cellular response to LPS defects. Int. J. Mol. Sci. 2019, 20, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiakas, D.; Mayer, M.P.; Lemaire, M.; Georgopoulos, C.; Raina, S. Modulation of the Escherichia coli σE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Mol. Microbiol. 1997, 24, 355–371. [Google Scholar] [CrossRef] [PubMed]
- De Las Peñas, A.; Connolly, L.; Gross, C.A. The σE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of σE. Mol. Microbiol. 1997, 24, 373–385. [Google Scholar] [CrossRef]
- Tam, C.; Collinet, B.; Lau, G.; Raina, S.; Missiakas, D. Interaction of the conserved region 4.2 of σE with RseA anti-sigma factor. J. Biol. Chem. 2002, 277, 27282–27287. [Google Scholar] [CrossRef] [Green Version]
- Fanghänel, J.; Fischer, G. Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front. Biosci. 2004, 9, 3453–3478. [Google Scholar] [CrossRef] [Green Version]
- Deuerling, E.; Patzelt, H.; Vorderwülbecke, S.; Rauch, T.; Kramer, G.; Schaffitzel, E.; Mogk, A.; Schulze-Specking, A.; Langen, H.; Bukau, B. Trigger Factor and DnaK possess overlapping substrate pools and binding specificities. Mol. Microbiol. 2003, 47, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Vecchi, G.; Vendruscolo, M.; Körner, R.; Hayer-Hartl, M.; Hartl, F.U. The Hsp70 chaperone system stabilizes a thermo-sensitive subproteome in E. coli. Cell Rep. 2019, 28, 1335–1345.e6. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into protein-ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Boël, G.; Hashem, Y.; Ning, W.; Fei, J.; Wang, C.; Gonzalez, R.L., Jr.; Hunt, J.F.; Frank, J. EttA regulates translation by binding the ribosomal E site and restricting ribosome-tRNA dynamics. Nat. Struct. Mol. Biol. 2014, 21, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Dewachter, L.; Deckers, B.; Martin, E.; Herpels, P.; Gkekas, S.; Versées, W.; Verstraeten, N.; Fauvart, M.; Michiels, J. GTP binding is necessary for the activation of a toxic mutant isoform of the essential GTPase ObgE. Int. J. Mol. Sci. 2019, 21, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.; Jeon, H.; Oh, J.I.; Hwang, J. Overexpressed L20 rescues 50S ribosomal subunit assembly defects of bipA-deltion in Escherichia coli. Front. Microbiol. 2020, 10, 2982. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, A. Functional modulation of Escherichia coli RNA polymerase. Annu. Rev. Microbiol. 2000, 54, 499–518. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Stupak, A.; Biernacka, D.; Wojtkiewicz, P.; Lindner, B.; Raina, S. Multiple transcriptional factors regulate transcription of the rpoE gene in Escherichia coli under different growth conditions and when the lipopolysaccharide biosynthesis is defective. J. Biol. Chem. 2016, 291, 22999–23019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dartigalongue, C.; Missiakas, D.; Raina, S. Characterization of the Escherichia coli σE regulon. J. Biol. Chem. 2001, 276, 20866–20875. [Google Scholar] [CrossRef] [Green Version]
- Ades, S.E. Regulation by destruction: Design of the σE envelope stress response. Curr. Opin. Microbiol. 2008, 11, 535–540. [Google Scholar] [CrossRef]
- Campbell, E.A.; Tupy, J.L.; Gruber, T.M.; Wang, S.; Sharp, M.M.; Gross, C.A.; Darst, S.A. Crystal structure of Escherichia coli σE with the cytoplasmic domain of its anti-sigma RseA. Mol. Cell 2003, 11, 1067–1078. [Google Scholar] [CrossRef]
- Daum, S.; Schumann, M.; Mathea, S.; Aumüller, T.; Balsley, M.A.; Constant, S.L.; de Lacroix, B.F.; Kruska, F.; Braun, M.; Schiene-Fischer, C. Isoform-specific inhibition of cyclophilins. Biochemistry 2009, 48, 6268–6277. [Google Scholar] [CrossRef] [Green Version]
- Skagia, A.; Zografou, C.; Vezyri, E.; Venieraki, A.; Katinakis, P.; Dimou, M. Cyclophilin PpiB is involved in motility and biofilm formation via its functional association with certain proteins. Genes Cells 2016, 21, 833–851. [Google Scholar] [CrossRef] [Green Version]
- Rao, N.N.; Kornberg, A. Inorganic polyphosphate supports resistance and survival of stationary-phase Escherichia coli. J. Bacteriol. 1996, 178, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Bochkareva, E.S.; Girshovich, A.S.; Bibi, E. Identification and characterization of the Escherichia coli stress protein UP12, a putative in vivo substrate of GroEL. Eur. J. Biochem. 2002, 269, 3032–3040. [Google Scholar] [CrossRef]
- Klein, G.; Lindner, B.; Brade, H.; Raina, S. Molecular basis of lipopolysaccharide heterogeneity in Escherichia coli: Envelope stress-responsive regulators control the incorporation of glycoforms with a third 3-deoxy-α-D-manno-oct-2-ulosonic acid and rhamnose. J. Biol. Chem. 2011, 286, 42787–42807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzzau, S.; Figueroa-Bossi, N.; Rubino, S.; Bossi, L. Epitope tagging of chromosomal genes in Salmonella. Proc. Natl. Acad. Sci. USA 2001, 98, 15264–15269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomoyasu, T.; Mogk, A.; Langen, H.; Goloubinoff, P.; Bukau, B. Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol. Microbiol. 2001, 40, 397–413. [Google Scholar] [CrossRef]
- Fischer, G.; Wittmann-Liebold, B.; Lang, K.; Kiefhaber, T.; Schmid, F.X. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 1989, 337, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Kofron, J.L.; Kuzmič, P.; Kishore, V.; Colón-Bonilla, E.; Rich, D.H. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 1991, 30, 6127–6134. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 23 °C | 30 °C | 37 °C | 43 °C | |||||
|---|---|---|---|---|---|---|---|---|
| M9 | LA | M9 | LA | M9 | LA | M9 | LA | |
| Δ(fklB slyD fkpB tig ppiB) + vector x ΔppiC | 434 | 8 | 2369 | 27 | 2460 | 47 | − | − |
| Δ(fklB slyD fkpB tig ppiB) + pppiC+ x ΔppiC | 1740 | 1630 | 2280 | 1973 | 2710 | 2432 | 838 | 1140 |
| Δ(fklB slyD tig ppiB ppiC) + vector x ΔfkpB | 342 | 10 | 1732 | 37 | 1562 | 35 | − | − |
| Δ(fklB slyD tig ppiB ppiC) + pfkpB+ x ΔfkpB | 2314 | 2640 | 1948 | 2100 | 1980 | 2013 | 954 | 901 |
| Δ(fklB slyD tig ppiC fkpB) + vector x ΔppiB | 425 | 5 | 1420 | 24 | 1830 | 48 | − | − |
| Δ(fklB slyD tig ppiC fkpB) + pppiB+ x ΔppiB | 1451 | 1620 | 1830 | 1723 | 2620 | 2534 | 1031 | 977 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, G.; Wojtkiewicz, P.; Biernacka, D.; Stupak, A.; Gorzelak, P.; Raina, S. Identification of Substrates of Cytoplasmic Peptidyl-Prolyl Cis/Trans Isomerases and Their Collective Essentiality in Escherichia Coli. Int. J. Mol. Sci. 2020, 21, 4212. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124212
Klein G, Wojtkiewicz P, Biernacka D, Stupak A, Gorzelak P, Raina S. Identification of Substrates of Cytoplasmic Peptidyl-Prolyl Cis/Trans Isomerases and Their Collective Essentiality in Escherichia Coli. International Journal of Molecular Sciences. 2020; 21(12):4212. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124212
Chicago/Turabian StyleKlein, Gracjana, Pawel Wojtkiewicz, Daria Biernacka, Anna Stupak, Patrycja Gorzelak, and Satish Raina. 2020. "Identification of Substrates of Cytoplasmic Peptidyl-Prolyl Cis/Trans Isomerases and Their Collective Essentiality in Escherichia Coli" International Journal of Molecular Sciences 21, no. 12: 4212. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124212