Modulation of DNA Damage Response by Sphingolipid Signaling: An Interplay that Shapes Cell Fate

Abstract
:1. Introduction
2. Overview of the DNA Damage Response
2.1. Types of DNA Damage
2.1.1. Double Strand Breaks
2.1.2. Single Strand Breaks
2.1.3. Base Damage
2.2. Induction of the DNA Damage Response
2.2.1. Detection of Double Strands Breaks (DSBs) and Initiation of the DNA Damage Response (DDR)
2.2.2. Role of Ataxia-Telangiectasia Mutated Protein (ATM) in DSBs Repair
2.2.3. ATM Nuclear Shuttling
2.3. DNA Damage-Induced Cell Death and Senescence
2.3.1. DNA Damage-Induced Senescence
2.3.2. DNA Damage-Induced Apoptosis
2.3.3. DNA Damage-Induced Mitotic Catastrophe
2.3.4. DNA Damage-Induced Necrosis
3. Overview of Sphingolipids
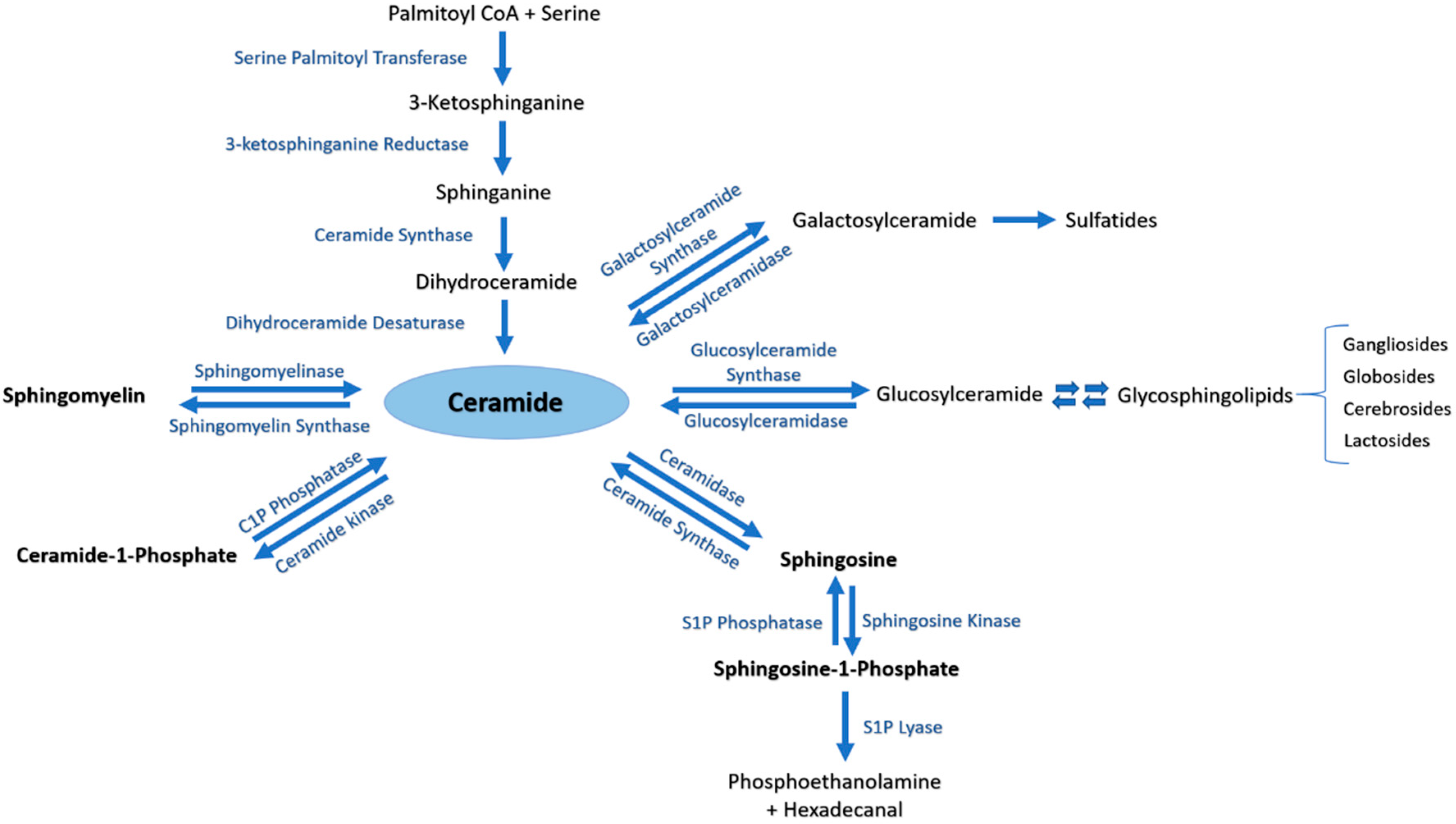
3.1. Sphingolipids Metabolic Pathway
3.2. Sphingolipids in the Nucleus
3.2.1. Nuclear Sphingomyelin and Metabolizing Enzymes
3.2.2. Nuclear Ceramide, Ceramide-1-Phosphate and Metabolizing Enzymes
3.2.3. Nuclear Sphingosine, Sphingosine-1-Phosphate and Metabolizing Enzymes
4. Role of Sphingolipids in the DNA Damage Response
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| Apaf1 | Apoptotic protease activating factor 1 |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad-3 related |
| BD | Base damage |
| Cer | Ceramide |
| CERK | Ceramide kinase |
| CHO | Cholesterol |
| C1P | Ceramide-1-phosphate |
| DDR | DNA damage response |
| DNAPK | DNA-dependent protein kinase |
| DSB | Double strand break |
| Gy | Gray (absorbed dose) |
| HR | Homologous recombination |
| IR | Ionizing radiation |
| MDM2 | Mouse double minute 2 |
| NHEJ | Non-homologous end joining |
| PC | Phosphatidylcholine |
| PUMA | p53 upregulated modulator of apoptosis |
| p53AIP1 | p53 regulated apoptosis inducing protein 1 |
| ROS | Reactive oxygen species |
| SK | Sphingosine kinase |
| SM | Sphingomyelin |
| SMPDL3b | Sphingomyelin phosphodiesterase acid like-3b |
| S1P | Sphingosine-1-phosphate |
| SPP | S1P phosphatase |
| SSB | Single strand break |
| TRAIL | TNF-related apoptosis-inducing ligand |
References
- Carroll, B.; Donaldson, J.C.; Obeid, L. Sphingolipids in the DNA damage response. Adv. Biol. Regul. 2015, 58, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presa, N.; Gomez-Larrauri, A.; Dominguez-Herrera, A.; Trueba, M.; Gomez-Muñoz, A. Novel signaling aspects of ceramide 1-phosphate. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158630. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Okazaki, T. The role of sphingomyelin and sphingomyelin synthases in cell death, proliferation and migration—From cell and animal models to human disorders. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071. [Google Scholar] [CrossRef] [Green Version]
- Saha, G.B. Physics and Radiobiology of Nuclear Medicine; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006; Volume 6. [Google Scholar]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Negritto, C. Repairing double-strand DNA breaks. Nat. Educ. 2010, 3, 26. [Google Scholar]
- Cornforth, M.N.; Bedford, J.S. A quantitative comparison of potentially lethal damage repair and the rejoining of interphase chromosome breaks in low passage normal human fibroblasts. Radiat. Res. 1987, 111, 385–405. [Google Scholar] [CrossRef]
- Bauer, N.C.; Corbett, A.H.; Doetsch, P.W. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015, 43, 10083–10101. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Collins, L.B.; Chen, T.-h.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to nhej mediated mutations. Oncotarget 2016, 7, 25377. [Google Scholar] [CrossRef] [PubMed]
- Bodgi, L.; Foray, N. The nucleo-shuttling of the atm protein as a basis for a novel theory of radiation response: Resolution of the linear-quadratic model. Int. J. Radiat. Biol. 2016, 92, 117–131. [Google Scholar] [CrossRef]
- Isaksson, M.; Raaf, C.L. Environmental Radioactivity and Emergency Preparedness; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res./Rev. Mutat. Res. 2012, 751, 158–246. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and uv radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef] [PubMed]
- Bokhari, B.; Sharma, S. Stress marks on the genome: Use or lose? Int. J. Mol. Sci. 2019, 20, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Pathol. 2010, 38, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Rouse, J.; Jackson, S.P. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002, 297, 547–551. [Google Scholar] [CrossRef]
- Jeggo, P.; Löbrich, M. DNA double-strand breaks: Their cellular and clinical impact? Oncogene 2007, 26, 7717. [Google Scholar] [CrossRef]
- Woodbine, L.; Gennery, A.R.; Jeggo, P.A. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair 2014, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Kühne, M.; Rief, N.; Doherty, A.; Smith, G.C.; Recio, M.a.-J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A. A pathway of double-strand break rejoining dependent upon atm, artemis, and proteins locating to γ-h2ax foci. Mol. Cell 2004, 16, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, E.; Hochegger, H.; Saberi, A.; Taniguchi, Y.; Takeda, S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair 2006, 5, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. Atm and artemis promote homologous recombination of radiation-induced DNA double-strand breaks in g2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [Green Version]
- Abbotts, R.; Wilson III, D.M. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef]
- Khoronenkova, S.V.; Dianov, G.L. Atm prevents dsb formation by coordinating ssb repair and cell cycle progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3997–4002. [Google Scholar] [CrossRef] [Green Version]
- Fortini, P.; Pascucci, B.; Parlanti, E.; D’errico, M.; Simonelli, V.; Dogliotti, E. The base excision repair: Mechanisms and its relevance for cancer susceptibility. Biochimie 2003, 85, 1053–1071. [Google Scholar] [CrossRef]
- Czornak, K.; Chughtai, S.; Chrzanowska, K.H. Mystery of DNA repair: The role of the mrn complex and atm kinase in DNA damage repair. J. Appl. Genet. 2008, 49, 383–396. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. Atr: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y. Atm and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [Green Version]
- Goto, H.; Izawa, I.; Li, P.; Inagaki, M. Novel regulation of checkpoint kinase 1: Is checkpoint kinase 1 a good candidate for anti-cancer therapy? Cancer Sci. 2012, 103, 1195–1200. [Google Scholar] [CrossRef]
- Dasika, G.K.; Lin, S.-C.J.; Zhao, S.; Sung, P.; Tomkinson, A.; Lee, E.Y.P. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene 1999, 18, 7883–7899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone h2ax in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Rappold, I.; Iwabuchi, K.; Date, T.; Chen, J. Tumor suppressor p53 binding protein 1 (53bp1) is involved in DNA damage–signaling pathways. J. Cell Biol. 2001, 153, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. Mechanisms of atm activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. Atm phosphorylates histone h2ax in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [Green Version]
- Ward, I.M.; Chen, J. Histone h2ax is phosphorylated in an atr-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [Green Version]
- Ward, I.M.; Minn, K.; Chen, J. Uv-induced ataxia-telangiectasia-mutated and rad3-related (atr) activation requires replication stress. J. Biol. Chem. 2004, 279, 9677–9680. [Google Scholar] [CrossRef] [Green Version]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.-Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. Atm-dependent and-independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010, 3, rs3. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates atm through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. Atm activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canman, C.E.; Lim, D.-S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the atm kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Bodgi, L.; Granzotto, A.; Devic, C.; Vogin, G.; Lesne, A.; Bottollier-Depois, J.-F.; Victor, J.-M.; Maalouf, M.; Fares, G.; Foray, N. A single formula to describe radiation-induced protein relocalization: Towards a mathematical definition of individual radiosensitivity. J. Theor. Biol. 2013, 333, 135–145. [Google Scholar] [CrossRef]
- Ouenzar, F.; Hendzel, M.J.; Weinfeld, M. Shuttling towards a predictive assay for radiotherapy. Transl. Cancer Res. 2016, 5, S742–S746. [Google Scholar] [CrossRef]
- Ferlazzo, M.L.; Sonzogni, L.; Granzotto, A.; Bodgi, L.; Lartin, O.; Devic, C.; Vogin, G.; Pereira, S.; Foray, N. Mutations of the huntington’s disease protein impact on the atm-dependent signaling and repair pathways of the radiation-induced DNA double-strand breaks: Corrective effect of statins and bisphosphonates. Mol. Neurobiol. 2014, 49, 1200–1211. [Google Scholar] [CrossRef]
- Pereira, S.; Bodgi, L.; Duclos, M.; Canet, A.; Ferlazzo, M.L.; Devic, C.; Granzotto, A.; Deneuve, S.; Vogin, G.; Foray, N. Fast and binary assay for predicting radiosensitivity based on the theory of atm nucleo-shuttling: Development, validation, and performance. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 353–360. [Google Scholar] [CrossRef]
- Ferlazzo, M.L.; Bach-Tobdji, M.K.E.; Djerad, A.; Sonzogni, L.; Devic, C.; Granzotto, A.; Bodgi, L.; Bachelet, J.-T.; Djefal-Kerrar, A.; Hennequin, C. Radiobiological characterization of tuberous sclerosis: A delay in the nucleo-shuttling of atm may be responsible for radiosensitivity. Mol. Neurobiol. 2018, 55, 4973–4983. [Google Scholar] [CrossRef]
- Surova, O.; Zhivotovsky, B. Various modes of cell death induced by DNA damage. Oncogene 2013, 32, 3789–3797. [Google Scholar] [CrossRef] [Green Version]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasty, P.; Campisi, J.; Hoeijmakers, J.; Van Steeg, H.; Vijg, J. Aging and genome maintenance: Lessons from the mouse? Science 2003, 299, 1355–1359. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; Van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med Sci. 2012, 9, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumor Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soussi, T.; Béroud, C. Assessing tp53 status in human tumours to evaluate clinical outcome. Nat. Rev. Cancer 2001, 1, 233. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Lozano, G. P53 mutation heterogeneity in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 834–842. [Google Scholar] [CrossRef]
- Helton, E.S.; Chen, X. P53 modulation of the DNA damage response. J. Cell. Biochem. 2007, 100, 883–896. [Google Scholar] [CrossRef]
- Wang, Z.; Li, B. Mdm2 links genotoxic stress and metabolism to p53. Protein Cell 2010, 1, 1063–1072. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Ma, Q.; Chen, L.; Li, P.; Zhang, M.; Ramamoorthy, S.; Nawaz, Z.; Shimojima, T.; Wang, H.; Yang, Y. Mdm2 acts downstream of p53 as an e3 ligase to promote foxo ubiquitination and degradation. J. Biol. Chem. 2009, 284, 13987–14000. [Google Scholar] [CrossRef] [Green Version]
- Barak, Y.; Gottlieb, E.; Juven-Gershon, T.; Oren, M. Regulation of mdm2 expression by p53: Alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 1994, 8, 1739–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakuma, T.; Lozano, G. Mdm2, an introduction. Mol. Cancer Res. 2003, 1, 993–1000. [Google Scholar] [PubMed]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A.; Holt, S.E.; Elmore, L.W. Accelerated senescence: An emerging role in tumor cell response to chemotherapy and radiation. Biochem. Pharmacol. 2008, 76, 947–957. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Batalni, J.P.; Belloir, C.; Mazabraud, A.; Pilleron, J.P.; Cartigny, A.; Jaulerry, C.; Ghossein, N.A. Desmoid tumors in adults: The role of radiotherapy in their management. Am. J. Surg. 1988, 155, 754–760. [Google Scholar] [CrossRef]
- Cox, J.D.; Kline, R.W. Do prostatic biopsies 12 months or more after external irradiation for adenocarcinoma, stage iii, predict long-term survival? Int. J. Radiat. Oncol. Biol. Phys. 1983, 9, 299–303. [Google Scholar] [CrossRef]
- Shay, J.W.; Roninson, I.B. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 2004, 23, 2919. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Mori, I.; Nakayama, Y.; Miyakoda, M.; Kodama, S.; Watanabe, M. Radiation-induced senescence-like growth arrest requires tp53 function but not telomere shortening. Radiat. Res. 2001, 155, 248–253. [Google Scholar] [CrossRef]
- Ninomiya, Y.; Cui, X.; Yasuda, T.; Wang, B.; Yu, D.; Sekine-Suzuki, E.; Nenoi, M. Arsenite induces premature senescence via p53/p21 pathway as a result of DNA damage in human malignant glioblastoma cells. BMB Rep. 2014, 47, 575. [Google Scholar] [CrossRef] [Green Version]
- Pawlowska, E.; Szczepanska, J.; Szatkowska, M.; Blasiak, J. An interplay between senescence, apoptosis and autophagy in glioblastoma multiforme—Role in pathogenesis and therapeutic perspective. Int. J. Mol. Sci. 2018, 19, 889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hock, A.K.; Vousden, K.H. Tumor suppression by p53: Fall of the triumvirate? Cell 2012, 149, 1183–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Lee, J.M.; Lee, G.; Bhin, J.; Oh, S.K.; Kim, K.; Pyo, K.E.; Lee, J.S.; Yim, H.Y.; Kim, K.I. DNA damage-induced rorα is crucial for p53 stabilization and increased apoptosis. Mol. Cell 2011, 44, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Baliga, B.C.; Colussi, P.A.; Read, S.H.; Dias, M.M.; Jans, D.A.; Kumar, S. Role of prodomain in importin-mediated nuclear localization and activation of caspase-2. J. Biol. Chem. 2003, 278, 4899–4905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinel, A.; Tschopp, J. The piddosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004, 304, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, C.A. Senescence, apoptosis and therapy—Cutting the lifelines of cancer. Nat. Rev. Cancer 2003, 3, 286. [Google Scholar] [CrossRef]
- Galluzzi, L.; Maiuri, M.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell Death Modalities: Classification and Pathophysiological Implications. Cell Death Differ. 2007, 14, 1237–1243. [Google Scholar] [CrossRef]
- Eriksson, D.; Joniani, H.M.; Sheikholvaezin, A.; Löfroth, P.-O.; Johansson, L.; Åhlström, K.R.; Stigbrand, T. Combined low dose radio-and radioimmunotherapy of experimental hela hep 2 tumours. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 895–906. [Google Scholar] [CrossRef]
- Eriksson, D.; Löfroth, P.-O.; Johansson, L.; Riklund, K.Å.; Stigbrand, T. Cell cycle disturbances and mitotic catastrophes in hela hep2 cells following 2.5 to 10 gy of ionizing radiation. Clin. Cancer Res. 2007, 13, 5501s–5508s. [Google Scholar] [CrossRef] [Green Version]
- Castedo, M.; Kroemer, G. Mitotic catastrophe: A special case of apoptosis. J. Soc. Biol. 2004, 198, 97–103. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Kalejs, M.; Ianzini, F.; Kosmacek, E.A.; Mackey, M.; Emzinsh, D.; Cragg, M.S.; Ivanov, A.; Illidge, T.M. Segregation of genomes in polyploid tumour cells following mitotic catastrophe. Cell Biol. Int. 2005, 29, 1005–1011. [Google Scholar] [CrossRef]
- Roninson, I.B.; Broude, E.V.; Chang, B.-D. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist. Updates 2001, 4, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Ianzini, F.; Bertoldo, A.; Kosmacek, E.A.; Phillips, S.L.; Mackey, M.A. Lack of p53 function promotes radiation-induced mitotic catastrophe in mouse embryonic fibroblast cells. Cancer Cell Int. 2006, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourke, E.; Dodson, H.; Merdes, A.; Cuffe, L.; Zachos, G.; Walker, M.; Gillespie, D.; Morrison, C.G. DNA damage induces chk1-dependent centrosome amplification. EMBO Rep. 2007, 8, 603–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, H.; Wheatley, S.P.; Morrison, C.G. Involvement of centrosome amplification in radiation-induced mitotic catastrophe. Cell Cycle 2007, 6, 364–370. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, K.; Fujikawa-Yamamoto, K.; Ozaki, M.; Iwabuchi, K.; Nakashima, H.; Domiki, C.; Morita, N.; Inoue, M.; Tokunaga, K.; Shiba, N. Centrosome hyperamplification and chromosomal damage after exposure to radiation. Oncology 2004, 67, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Morita, N.; Domiki, C.; Fujikawa-Yamamoto, K.; Hashimoto, M.; Iwabuchi, K.; Suzuki, K. Induction of centrosome amplification in p53 sirna-treated human fibroblast cells by radiation exposure. Cancer Sci. 2006, 97, 252–258. [Google Scholar] [CrossRef]
- Hanashiro, K.; Kanai, M.; Geng, Y.; Sicinski, P.; Fukasawa, K. Roles of cyclins a and e in induction of centrosome amplification in p53-compromised cells. Oncogene 2008, 27, 5288. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.; Blagosklonny, M.; El-Deiry, W.; Golstein, P.; Green, D. Classification of cell death: Recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 2009, 16, 3. [Google Scholar] [CrossRef]
- Brandsma, D.; Stalpers, L.; Taal, W.; Sminia, P.; van den Bent, M.J. Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet Oncol. 2008, 9, 453–461. [Google Scholar] [CrossRef]
- Krysko, O.; Aaes, T.L.; Bachert, C.; Vandenabeele, P.; Krysko, D. Many faces of damps in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef] [Green Version]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell Res. 2003, 283, 1–16. [Google Scholar] [CrossRef]
- Cohen–Jonathan, E.; Bernhard, E.J.; McKenna, W.G. How does radiation kill cells? Curr. Opin. Chem. Biol. 1999, 3, 77–83. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. In Sphingolipids as Signaling and Regulatory Molecules; Springer: Berlin, Germany, 2010; pp. 1–23. [Google Scholar]
- Abou Daher, A.; El Jalkh, T.; Eid, A.; Fornoni, A.; Marples, B.; Zeidan, Y. Translational aspects of sphingolipid metabolism in renal disorders. Int. J. Mol. Sci. 2017, 18, 2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidan, Y.H.; Hannun, Y.A. Translational aspects of sphingolipid metabolism. Trends Mol. Med. 2007, 13, 327–336. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139. [Google Scholar] [CrossRef]
- Fröhlich, F.; Petit, C.; Kory, N.; Christiano, R.; Hannibal-Bach, H.-K.; Graham, M.; Liu, X.; Ejsing, C.S.; Farese, R.V., Jr.; Walther, T.C. The garp complex is required for cellular sphingolipid homeostasis. Elife 2015, 4, e08712. [Google Scholar] [CrossRef]
- te Vruchte, D.; Lloyd-Evans, E.; Veldman, R.J.; Neville, D.C.; Dwek, R.A.; Platt, F.M.; van Blitterswijk, W.J.; Sillence, D.J. Accumulation of glycosphingolipids in niemann-pick c disease disrupts endosomal transport. J. Biol. Chem. 2004, 279, 26167–26175. [Google Scholar] [CrossRef] [Green Version]
- Höglinger, D.; Haberkant, P.; Aguilera-Romero, A.; Riezman, H.; Porter, F.D.; Platt, F.M.; Galione, A.; Schultz, C. Intracellular sphingosine releases calcium from lysosomes. Elife 2015, 4, e10616. [Google Scholar] [CrossRef]
- Schuchman, E. The pathogenesis and treatment of acid sphingomyelinase-deficient niemann–pick disease. J. Inherit. Metab. Dis. 2007, 30, 654. [Google Scholar] [CrossRef] [PubMed]
- Merscher, S.; Fornoni, A. Podocyte pathology and nephropathy–sphingolipids in glomerular diseases. Front. Endocrinol. 2014, 5, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boath, A.; Graf, C.; Lidome, E.; Ullrich, T.; Nussbaumer, P.; Bornancin, F. Regulation and traffic of ceramide 1-phosphate produced by ceramide kinase comparative analysis to glucosylceramide and sphingomyelin. J. Biol. Chem. 2008, 283, 8517–8526. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, C.; Kihara, A.; Gokoh, M.; Igarashi, Y. Identification and characterization of a novel human sphingosine-1-phosphate phosphohydrolase, hspp2. J. Biol. Chem. 2003, 278, 1268–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandala, S.M.; Thornton, R.; Galve-Roperh, I.; Poulton, S.; Peterson, C.; Olivera, A.; Bergstrom, J.; Kurtz, M.B.; Spiegel, S. Molecular cloning and characterization of a lipid phosphohydrolase that degrades sphingosine-1-phosphate and induces cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 7859–7864. [Google Scholar] [CrossRef] [Green Version]
- Pyne, S.; Long, J.; Ktistakis, N.; Pyne, N. Lipid phosphate phosphatases and lipid phosphate signalling. Biochem. Soc. Trans. 2005, 33, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Kihara, A.; Igarashi, Y. Sphingosine-1-phosphate lyase spl is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem. Biophys. Res. Commun. 2004, 325, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Albi, E. Role of intranuclear lipids in health and disease. Clin. Lipidol. 2011, 6, 59–69. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. Thematic review series: Sphingolipids. Nuclear sphingolipids: Metabolism and signaling. J. Lipid Res. 2008, 49, 1176–1186. [Google Scholar] [CrossRef] [Green Version]
- Bernardini, I.; Bartoccini, E.; Viola Magni, M. Nuclear lipids and cell fate. Dyn. Cell Biol. 2017, 1, 42–47. [Google Scholar]
- Divecha, N.; Banfic, H.; Irvine, R.F. Inositides and the nucleus and inositides in the nucleus. Cell 1993, 74, 405–407. [Google Scholar] [CrossRef]
- Cocco, L.; Martelli, A.M.; Gilmour, R.S.; Rhee, S.G.; Manzoli, F.A. Nuclear phospholipase c and signaling. Biochim. Biophys. Acta 2001, 1530, 1. [Google Scholar] [CrossRef]
- Maraldi, N.; Cocco, L.; Capitani, S.; Mazzotti, G.; Barnabei, O.; Manzoli, F. Lipid-dependent nuclear signalling: Morphological and functional features. Adv. Enzym. Regul. 1994, 34, 129–143. [Google Scholar] [CrossRef]
- Kleuser, B.; Maceyka, M.; Milstien, S.; Spiegel, S. Stimulation of nuclear sphingosine kinase activity by platelet-derived growth factor. FEBS Lett. 2001, 503, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Ledeen, R.W.; Wu, G. Sphingolipids of the nucleus and their role in nuclear signaling. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2006, 1761, 588–598. [Google Scholar] [CrossRef]
- Neitcheva, T.; Peeva, D. Phospholipid composition, phospholipase a2 and sphingomyelinase activities in rat liver nuclear membrane and matrix. Int. J. Biochem. Cell Biol. 1995, 27, 995–1001. [Google Scholar] [CrossRef]
- Pliss, A.; Kuzmin, A.N.; Kachynski, A.V.; Prasad, P.N. Nonlinear optical imaging and raman microspectrometry of the cell nucleus throughout the cell cycle. Biophys. J. 2010, 99, 3483–3491. [Google Scholar] [CrossRef] [Green Version]
- Albi, E.; Mersel, M.; Leray, C.; Tomassoni, M.; Viola-Magni, M. Rat liver chromatin phospholipids. Lipids 1994, 29, 715–719. [Google Scholar] [CrossRef]
- Cave, C.F.; Gahan, P. A cytochemical and autoradiographic investigation of nucleolar phospholipids. Caryologia 1970, 23, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Fu, P.; Ebenezer, D.L.; Ha, A.W.; Suryadevara, V.; Harijith, A.; Natarajan, V. Nuclear lipid mediators: Role of nuclear sphingolipids and sphingosine-1-phosphate signaling in epigenetic regulation of inflammation and gene expression. J. Cell. Biochem. 2018, 119, 6337–6353. [Google Scholar] [CrossRef]
- Tomassoni, M.-L.; Amori, D.; Magni, M.V. Changes of nuclear membrane lipid composition affect rna nucleocytoplasmic transport. Biochem. Biophys. Res. Commun. 1999, 258, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Albi, E.; Tomassoni, M.L.; Viola-Magni, M. Effect of lipid composition on rat liver nuclear membrane fluidity. Cell Biochem. Funct. Cell. Biochem. Modul. Act. Agents Dis. 1997, 15, 181–190. [Google Scholar] [CrossRef]
- Xie, X.; Wu, G.; Lu, Z.H.; Ledeen, R.W. Potentiation of a sodium–calcium exchanger in the nuclear envelope by nuclear gm1 ganglioside. J. Neurochem. 2002, 81, 1185–1195. [Google Scholar] [CrossRef]
- Albi, E.; Magni, M.V. Chromatin-associated sphingomyelin: Metabolism in relation to cell function. Cell Biochem. Funct. Cell. Biochem. Modul. Act. Agents Dis. 2003, 21, 211–215. [Google Scholar] [CrossRef]
- Lucki, N.C.; Sewer, M.B. Nuclear sphingolipid metabolism. Annu. Rev. Physiol. 2012, 74, 131–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessenko, A.; Chatterjee, S. Neutral sphingomyelinase: Localization in rat liver nuclei and involvement in regeneration/proliferation. Mol. Cell. Biochem. 1995, 143, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lu, Z.H.; Ledeen, R.W. Gm1 ganglioside in the nuclear membrane modulates nuclear calcium homeostasis during neurite outgrowth. J. Neurochem. 1995, 65, 1419–1422. [Google Scholar] [CrossRef] [PubMed]
- Micheli, M.; Albi, E.; Leray, C.; Magni, M.V. Nuclear sphingomyelin protects rna from rnase action. FEBS Lett. 1998, 431, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Tsugane, K.; Tamiya-Koizumi, K.; Nagino, M.; Nimura, Y.; Yoshida, S. A possible role of nuclear ceramide and sphingosine in hepatocyte apoptosis in rat liver. J. Hepatol. 1999, 31, 8–17. [Google Scholar] [CrossRef]
- Rossi, G.; Magni, M.V.; Albi, E. Sphingomyelin-cholesterol and double stranded rna relationship in the intranuclear complex. Arch. Biochem. Biophys. 2007, 459, 27–32. [Google Scholar] [CrossRef]
- Albi, E.; Cataldi, S.; Rossi, G.; Magni, M.V.; Toller, M.; Casani, S.; Perrella, G. The nuclear ceramide/diacylglycerol balance depends on the physiological state of thyroid cells and changes during uv-c radiation-induced apoptosis. Arch. Biochem. Biophys. 2008, 478, 52–58. [Google Scholar] [CrossRef]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Maurya, M.R.; Merrill, A.H., Jr.; Glass, C.K.; Subramaniam, S. Integration of lipidomics and transcriptomics data towards a systems biology model of sphingolipid metabolism. BMC Syst. Biol. 2011, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Albi, E.; Viola Magni, M. Sphingomyelin: A small-big molecule in the nucleus. Recent Res. Dev. Biophys. Biochem. 2006, 37, 211–227. [Google Scholar]
- Exton, J. Signaling through phosphatidylcholine breakdown. J. Biol. Chem. 1990, 265, 1–4. [Google Scholar]
- Reszka, A.A.; Halasy-Nagy, J.; Rodan, G.A. Nitrogen-bisphosphonates block retinoblastoma phosphorylation and cell growth by inhibiting the cholesterol biosynthetic pathway in a keratinocyte model for esophageal irritation. Mol. Pharmacol. 2001, 59, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novello, F.; Muchmore, J.; Bonora, B.; Capitani, S.; Manzoli, F. Effect of phospholipids on the activity of DNA polymerase i from e. Coli. Ital. J. Biochem. 1975, 24, 325–334. [Google Scholar]
- Martelli, A.M.; Follo, M.Y.; Evangelisti, C.; Fala, F.; Fiume, R.; Billi, A.M.; Cocco, L. Nuclear inositol lipid metabolism: More than just second messenger generation? J. Cell. Biochem. 2005, 96, 285–292. [Google Scholar] [CrossRef]
- Scassellati, C.; Albi, E.; Cmarko, D.; Tiberi, C.; Cmarkova, J.; Bouchet-Marquis, C.; Verschure, P.J.; Van Driel, R.; Magni, M.V.; Fakan, S. Intranuclear sphingomyelin is associated with transcriptionally active chromatin and plays a role in nuclear integrity. Biol. Cell 2010, 102, 361–375. [Google Scholar] [CrossRef]
- Albi, E.; Lazzarini, R.; Magni, M.V. Reverse sphingomyelin-synthase in rat liver chromatin. FEBS Lett. 2003, 549, 152–156. [Google Scholar] [CrossRef] [Green Version]
- Albi, E.; Magni, M.V. Sphingomyelin synthase in rat liver nuclear membrane and chromatin. FEBS Lett. 1999, 460, 369–372. [Google Scholar] [CrossRef] [Green Version]
- Albi, E.; Magni, M.V. Chromatin neutral sphingomyelinase and its role in hepatic regeneration. Biochem. Biophys. Res. Commun. 1997, 236, 29–33. [Google Scholar] [CrossRef]
- Venkataraman, K.; Riebeling, C.; Bodennec, J.; Riezman, H.; Allegood, J.C.; Sullards, M.C.; Merrill, A.H.; Futerman, A.H. Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (lag1), regulatesn-stearoyl-sphinganine (c18-(dihydro) ceramide) synthesis in a fumonisin b1-independent manner in mammalian cells. J. Biol. Chem. 2002, 277, 35642–35649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riebeling, C.; Allegood, J.C.; Wang, E.; Merrill, A.H.; Futerman, A.H. Two mammalian longevity assurance gene (lag1) family members, trh1 and trh4, regulate dihydroceramide synthesis using different fatty acyl-coa donors. J. Biol. Chem. 2003, 278, 43452–43459. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. Mammalian lass6 and its related family members regulate synthesis of specific ceramides. Biochem. J. 2005, 390, 263–271. [Google Scholar] [CrossRef]
- Min, J.; Mesika, A.; Sivaguru, M.; Van Veldhoven, P.P.; Alexander, H.; Futerman, A.H.; Alexander, S. (dihydro) ceramide synthase 1–regulated sensitivity to cisplatin is associated with the activation of p38 mitogen-activated protein kinase and is abrogated by sphingosine kinase 1. Mol. Cancer Res. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, T.; Imai, S.; Uda, Y. The presence of ceramidase activity in liver nuclear membrane. Biol. Pharm. Bull. 2003, 26, 775–779. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Kitano, T.; Kondo, T.; Yabu, T.; Taguchi, Y.; Tashima, M.; Umehara, H.; Domae, N.; Uchiyama, T.; Okazaki, T. Increase of nuclear ceramide through caspase-3-dependent regulation of the “sphingomyelin cycle” in fas-induced apoptosis. Cancer Res. 2004, 64, 1000–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albi, E.; Cataldi, S.; Bartoccini, E.; Magni, M.V.; Marini, F.; Mazzoni, F.; Rainaldi, G.; Evangelista, M.; Garcia-Gil, M. Nuclear sphingomyelin pathway in serum deprivation-induced apoptosis of embryonic hippocampal cells. J. Cell. Physiol. 2006, 206, 189–195. [Google Scholar] [CrossRef]
- Chocian, G.; Chabowski, A.; Żendzian-Piotrowska, M.; Harasim, E.; Łukaszuk, B.; Górski, J. High fat diet induces ceramide and sphingomyelin formation in rat’s liver nuclei. Mol. Cell. Biochem. 2010, 340, 125–131. [Google Scholar] [CrossRef]
- Schroeder, F.; Petrescu, A.D.; Huang, H.; Atshaves, B.P.; McIntosh, A.L.; Martin, G.G.; Hostetler, H.A.; Vespa, A.; Landrock, D.; Landrock, K.K. Role of fatty acid binding proteins and long chain fatty acids in modulating nuclear receptors and gene transcription. Lipids 2008, 43, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, T.; Kumagai, K.; Tomishige, N.; Hanada, K. Two sphingolipid transfer proteins, cert and fapp2: Their roles in sphingolipid metabolism. Iubmb Life 2008, 60, 511–518. [Google Scholar] [CrossRef]
- Sugiura, M.; Kono, K.; Liu, H.; Shimizugawa, T.; Minekura, H.; Spiegel, S.; Kohama, T. Ceramide kinase, a novel lipid kinase molecular cloning and functional characterization. J. Biol. Chem. 2002, 277, 23294–23300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simanshu, D.K.; Kamlekar, R.K.; Wijesinghe, D.S.; Zou, X.; Zhai, X.; Mishra, S.K.; Molotkovsky, J.G.; Malinina, L.; Hinchcliffe, E.H.; Chalfant, C.E. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature 2013, 500, 463–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovina, P.; Schanzer, A.; Graf, C.; Mechtcheriakova, D.; Jaritz, M.; Bornancin, F. Subcellular localization of ceramide kinase and ceramide kinase-like protein requires interplay of their pleckstrin homology domain-containing n-terminal regions together with c-terminal domains. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2009, 1791, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Urs, A.N.; Dammer, E.; Kelly, S.; Wang, E.; Merrill, A.H., Jr.; Sewer, M.B. Steroidogenic factor-1 is a sphingolipid binding protein. Mol. Cell. Endocrinol. 2007, 265, 174–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urs, A.N.; Dammer, E.; Sewer, M.B. Sphingosine regulates the transcription of cyp17 by binding to steroidogenic factor-1. Endocrinology 2006, 147, 5249–5258. [Google Scholar] [CrossRef]
- Sewer, M.B.; Waterman, M.R. Acth modulation of transcription factors responsible for steroid hydroxylase gene expression in the adrenal cortex. Microsc. Res. Tech. 2003, 61, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. Functions of the multifaceted family of sphingosine kinases and some close relatives. J. Biol. Chem. 2007, 282, 2125–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H. Sphk1 and sphk2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alemany, R.; van Koppen, C.J.; Danneberg, K.; Ter Braak, M.; Zu Heringdorf, D.M. Regulation and functional roles of sphingosine kinases. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2007, 374, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Selvam, S.P.; De Palma, R.M.; Oaks, J.J.; Oleinik, N.; Peterson, Y.K.; Stahelin, R.V.; Skordalakes, E.; Ponnusamy, S.; Garrett-Mayer, E.; Smith, C.D. Binding of the sphingolipid s1p to htert stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 2015, 8, ra58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stunff, H.L.; Milstien, S.; Spiegel, S. Generation and metabolism of bioactive sphingosine-1-phosphate. J. Cell. Biochem. 2004, 92, 882–899. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mao, J.; Redfield, S.; Mo, Y.; Lage, J.M.; Zhou, X. Systemic distribution, subcellular localization and differential expression of sphingosine-1-phosphate receptors in benign and malignant human tissues. Exp. Mol. Pathol. 2014, 97, 259–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lépine, S.; Allegood, J.; Park, M.; Dent, P.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate phosphohydrolase-1 regulates er stress-induced autophagy. Cell Death Differ. 2011, 18, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Schwiebs, A.; Thomas, D.; Kleuser, B.; Pfeilschifter, J.M.; Radeke, H.H. Nuclear translocation of sgpp-1 and decrease of sgpl-1 activity contribute to sphingolipid rheostat regulation of inflammatory dendritic cells. Mediat. Inflamm. 2017, 2017, 5187368. [Google Scholar] [CrossRef] [Green Version]
- Ebenezer, D.; Fu, P.; Berdyshev, E.; Natarajan, V. Nuclear s1p lyase regulates histone acetylation in pseudomonas aeruginosa-induced lung inflammation. FASEB J. 2015, 29, 863.26. [Google Scholar]
- Ebenezer, D.L.; Fu, P.; Mangio, L.A.; Berdyshev, E.; Schumacher, F.; Kleuser, B.; Van Veldhoven, P.P.; Natarajan, V. Δ-2 hexadecenal generated from s1p by nuclear s1p lyase is a regulator of hdac1/2 activity and histone acetylation in lung epithelial cells. FASEB J. 2019, 33, 489.3. [Google Scholar]
- Reynolds, C.P.; Maurer, B.J.; Kolesnick, R.N. Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Lett. 2004, 206, 169–180. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M. Still benched on its way to the bedside: Sphingosine kinase 1 as an emerging target in cancer chemotherapy. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Dbaibo, G.S.; Pushkareva, M.Y.; Rachid, R.A.; Alter, N.; Smyth, M.J.; Obeid, L.M.; Hannun, Y.A. P53-dependent ceramide response to genotoxic stress. J. Clin. Investig. 1998, 102, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Vit, J.-P.; Rosselli, F. Role of the ceramide-signaling pathways in ionizing radiation-induced apoptosis. Oncogene 2003, 22, 8645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, M.; Nakashima, S.; Kiyono, T.; Nakagawa, M.; Yamada, J.; Yamakawa, H.; Banno, Y.; Shinoda, J.; Nishimura, Y.; Nozawa, Y. P53 regulates ceramide formation by neutral sphingomyelinase through reactive oxygen species in human glioma cells. Oncogene 2001, 20, 1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, C.A.; He, Q.; Ponnusamy, S.; Ogretmen, B.; Huang, Y.; Sheikh, M.S. Neutral sphingomyelinase-3 is a DNA damage and nongenotoxic stress-regulated gene that is deregulated in human malignancies. Mol. Cancer Res. 2008, 6, 795–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffrézou, J.-P.; Bruno, A.P.; Moisand, A.; Levade, T.; Laurent, G. Activation of a nuclear sphingomyelinase in radiation-induced apoptosis. FASEB J. 2001, 15, 123–133. [Google Scholar] [CrossRef]
- Ravid, T.; Tsaba, A.; Gee, P.; Rasooly, R.; Medina, E.A.; Goldkorn, T. Ceramide accumulation precedes caspase-3 activation during apoptosis of a549 human lung adenocarcinoma cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L1082–L1092. [Google Scholar] [CrossRef] [Green Version]
- Dbaibo, G.S.; Pushkareva, M.Y.; Jayadev, S.; Schwarz, J.K.; Horowitz, J.M.; Obeid, L.M.; Hannun, Y.A. Retinoblastoma gene product as a downstream target for a ceramide-dependent pathway of growth arrest. Proc. Natl. Acad. Sci. USA 1995, 92, 1347–1351. [Google Scholar] [CrossRef] [Green Version]
- Phillips, D.; Hunt, J.; Moneypenny, C.; Maclean, K.; McKenzie, P.; Harris, L.; Houghton, J. Ceramide-induced g 2 arrest in rhabdomyosarcoma (rms) cells requires p21 cip1/waf1 induction and is prevented by mdm2 overexpression. Cell Death Differ. 2007, 14, 1780. [Google Scholar] [CrossRef]
- Xu, R.; Garcia-Barros, M.; Wen, S.; Li, F.; Lin, C.-L.; Hannun, Y.A.; Obeid, L.M.; Mao, C. Tumor suppressor p53 links ceramide metabolism to DNA damage response through alkaline ceramidase 2. Cell Death Differ. 2017. [Google Scholar] [CrossRef] [Green Version]
- Hoeferlin, L.A.; Fekry, B.; Ogretmen, B.; Krupenko, S.A.; Krupenko, N.I. Folate stress induces apoptosis via p53-dependent de novo ceramide synthesis and up-regulation of ceramide synthase 6. J. Biol. Chem. 2013, 288, 12880–12890. [Google Scholar] [CrossRef] [Green Version]
- Fekry, B.; Jeffries, K.A.; Esmaeilniakooshkghazi, A.; Szulc, Z.M.; Knagge, K.J.; Kirchner, D.R.; Horita, D.A.; Krupenko, S.A.; Krupenko, N.I. C 16-ceramide is a natural regulatory ligand of p53 in cellular stress response. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Taha, T.A.; Osta, W.; Kozhaya, L.; Bielawski, J.; Johnson, K.R.; Gillanders, W.E.; Dbaibo, G.S.; Hannun, Y.A.; Obeid, L.M. Down-regulation of sphingosine kinase-1 by DNA damage dependence on proteases and p53. J. Biol. Chem. 2004, 279, 20546–20554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankala, H.M.; Hait, N.C.; Paugh, S.W.; Shida, D.; Lépine, S.; Elmore, L.W.; Dent, P.; Milstien, S.; Spiegel, S. Involvement of sphingosine kinase 2 in p53-independent induction of p21 by the chemotherapeutic drug doxorubicin. Cancer Res. 2007, 67, 10466–10474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.R.; Johnson, K.Y.; Becker, K.P.; Bielawski, J.; Mao, C.; Obeid, L.M. Role of human sphingosine-1-phosphate phosphatase 1 in the regulation of intra-and extracellular sphingosine-1-phosphate levels and cell viability. J. Biol. Chem. 2003, 278, 34541–34547. [Google Scholar] [CrossRef] [Green Version]
- Oskouian, B.; Sooriyakumaran, P.; Borowsky, A.D.; Crans, A.; Dillard-Telm, L.; Tam, Y.Y.; Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase potentiates apoptosis via p53-and p38-dependent pathways and is down-regulated in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 17384–17389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Oskouian, B.; Fyrst, H.; Zhang, M.; Paris, F.; Saba, J. S1p lyase regulates DNA damage responses through a novel sphingolipid feedback mechanism. Cell Death Dis. 2011, 2, e119. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Mitrofanova, A.; Bielawski, J.; Yang, Y.; Marples, B.; Fornoni, A.; Zeidan, Y.H. Sphingomyelinase-like phosphodiesterase 3b mediates radiation-induced damage of renal podocytes. FASEB J. 2016, 31, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornoni, A.; Sageshima, J.; Wei, C.; Merscher-Gomez, S.; Aguillon-Prada, R.; Jauregui, A.N.; Li, J.; Mattiazzi, A.; Ciancio, G.; Chen, L. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci. Transl. Med. 2011, 3, ra46–ra85. [Google Scholar] [CrossRef] [Green Version]
- Mitrofanova, A.; Mallela, S.; Ducasa, G.; Yoo, T.; Rosenfeld-Gur, E.; Zelnik, I.; Molina, J.; Santos, J.V.; Ge, M.; Sloan, A. Smpdl3b modulates insulin receptor signaling in diabetic kidney disease. Nat. Commun. 2019, 10, 2692. [Google Scholar] [CrossRef] [Green Version]
- Mallela, S.K.; Mitrofanova, A.; Merscher, S.; Fornoni, A. Regulation of the amount of ceramide-1-phosphate synthesized in differentiated human podocytes. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2019, 1864, 158517. [Google Scholar] [CrossRef]
- Beckham, T.H.; Cheng, J.C.; Marrison, S.T.; Norris, J.S.; Liu, X. Interdiction of sphingolipid metabolism to improve standard cancer therapies. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2013; Volume 117, pp. 1–36. [Google Scholar]
- Savić, R.; Schuchman, E.H. Use of acid sphingomyelinase for cancer therapy. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2013; Volume 117, pp. 91–115. [Google Scholar]
- Albi, E.; Cataldi, S.; Ceccarini, M.R.; Conte, C.; Ferri, I.; Fettucciari, K.; Patria, F.F.; Beccari, T.; Codini, M. Gentamicin targets acid sphingomyelinase in cancer: The case of the human gastric cancer nci-n87 cells. Int. J. Mol. Sci. 2019, 20, 4375. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Ji, C.; Zhou, Y.; Huang, W.; Ni, W.; Tong, X.; Wei, J.-F. Sphingosine kinase inhibitors: A patent review. Int. J. Mol. Med. 2018, 41, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.; Rio, E.; Ripoche, N.; Ferchaud-Roucher, V.; Gaugler, M.-H.; Campion, L.; Krempf, M.; Carrie, C.; Mahé, M.; Mirabel, X. Plasma ceramide, a real-time predictive marker of pulmonary and hepatic metastases response to stereotactic body radiation therapy combined with irinotecan. Radiother. Oncol. 2016, 119, 229–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Smith, C.D. Ablation of sphingosine kinase-2 inhibits tumor cell proliferation and migration. Mol. Cancer Res. 2011, 9, 1509–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W. A phase i study of abc294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Nuclear Sphingolipid Metabolites | Nuclear Sphingolipid Producing Enzymes | Nuclear Sphingolipid Degrading or Converting Enzymes | Main Nuclear Functions |
|---|---|---|---|
| Sphingomyelin | Sphingomyelin synthase | Reverse sphingomyelin synthase Neutral sphingomyelinase | Maintenance of NE and nucleoplasm structure Regulation of NE permeability and Fluidity Stabilization of DNA and dsRNA |
| Ceramide | Ceramide synthase Ceramide desaturase Neutral sphingomyelinase | Ceramidase Ceramide kinase | Regulation of Cell cycle arrest, Senescence, and Apoptosis |
| Ceramide-1-phosphate | Ceramide kinase | C1P phosphatase | Regulation of cell growth and survival |
| Sphingosine | Ceramidase | Ceramide synthase Sphingosine kinase 2 | Regulation of gene transcription and apoptosis |
| Sphingosine-1-phosphate | Sphingosine kinase 2 | S1P lyase S1P phosphatase | Epigenetic modulation of gene transcription Regulation of cell cycle progression and apoptosis Stabilization of human telomerase |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francis, M.; Abou Daher, A.; Azzam, P.; Mroueh, M.; Zeidan, Y.H. Modulation of DNA Damage Response by Sphingolipid Signaling: An Interplay that Shapes Cell Fate. Int. J. Mol. Sci. 2020, 21, 4481. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124481
Francis M, Abou Daher A, Azzam P, Mroueh M, Zeidan YH. Modulation of DNA Damage Response by Sphingolipid Signaling: An Interplay that Shapes Cell Fate. International Journal of Molecular Sciences. 2020; 21(12):4481. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124481
Chicago/Turabian StyleFrancis, Marina, Alaa Abou Daher, Patrick Azzam, Manal Mroueh, and Youssef H. Zeidan. 2020. "Modulation of DNA Damage Response by Sphingolipid Signaling: An Interplay that Shapes Cell Fate" International Journal of Molecular Sciences 21, no. 12: 4481. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124481