Inflammasome and Mitophagy Connection in Health and Disease


1
Department of Infection Biology, Chungnam National University School of Medicine, Daejeon 35015, Korea
2
Infection Control Convergence Research Center, Chungnam National University School of Medicine, Daejeon 35015, Korea
3
Department of Medical Science, Chungnam National University School of Medicine, Daejeon 35015, Korea
4
Department of Microbiology, Chungnam National University School of Medicine, Daejeon 35015, Korea
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(13), 4714; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134714
Submission received: 2 June 2020
/
Revised: 26 June 2020
/
Accepted: 28 June 2020
/
Published: 1 July 2020
(This article belongs to the Special Issue Inflammasome)
Abstract
:The inflammasome is a large intracellular protein complex that activates inflammatory caspase-1 and induces the maturation of interleukin (IL)-1β and IL-18. Mitophagy plays an essential role in the maintenance of mitochondrial homeostasis during stress. Previous studies have indicated compelling evidence of the crosstalk between inflammasome and mitophagy. Mitophagy regulation of the inflammasome, or vice versa, is crucial for various biological functions, such as controlling inflammation and metabolism, immune and anti-tumor responses, and pyroptotic cell death. Uncontrolled regulation of the inflammasome often results in pathological inflammation and pyroptosis, and causes a variety of human diseases, including metabolic and inflammatory diseases, infection, and cancer. Here, we discuss how improved understanding of the interactions between inflammasome and mitophagy can lead to novel therapies against various disease pathologies, and how the inflammasome-mitophagy connection is currently being targeted pharmacologically by diverse agents and small molecules. A deeper understanding of the inflammasome-mitophagy connection will provide new insights into human health and disease through the balance between mitochondrial clearance and pathology.
1. Introduction
Inflammasomes are intracellular multiprotein complexes that trigger the activation of the enzymatic activity of caspase-1 to release mature interleukin (IL)-1β and IL-18, and also induce pyroptotic cell death [1,2]. The integrated regulation of inflammasome activation and pyroptosis contributes to promoting the antimicrobial host defense and the clearance of pathogens [3,4,5]. However, aberrant activation of inflammasomes leads to excessive inflammatory and pathological autoimmune responses potentially harmful to the host. The inflammasome-related diseases include metabolic and inflammatory diseases, atherosclerosis, arthritic diseases, gout, tissue injury, infectious diseases, and sepsis [6,7,8].
Autophagy is an intracellular catabolic process for the degradation of large macromolecules and/or damaged organelles. Although autophagy has generally been accepted as a bulk- and nonselective degradation process for intracellular cargoes, numerous studies have highlighted the selective nature of autophagy, targeting various organelles or substances [9]. Mitophagy is a selective autophagy pathway that targets and destroys mitochondria to maintain mitochondrial quality control [10]. Accumulating data suggest that mitophagy processes are mediated through several pathways involving PTEN-induced putative kinase 1 (PINK1)/Parkin and diverse mitophagy receptors under different cell condition contexts [11,12]. Dysregulation of mitophagy leads to mitochondrial dysfunction, increased production of mitochondrial reactive oxygen species (ROS), and translocation of mitochondrial DNA into the cytosol, all of which are involved in the activation of the inflammasome [13,14,15].
Here, we review the crosstalk mechanisms between inflammasomes and mitophagy. Dysregulated inflammasome activation, at least partly due to defective mitophagy, contributes to the pathogenesis of a variety of human diseases [16,17]. The mitophagy pathway has been shown to regulate mitochondrial ROS generation and elimination of damaged mitochondria, thus controlling mitochondrial homeostasis to prevent excessive activation of the inflammasomes [12,18,19]. We also discuss recent progress in identifying various agents and molecules to regulate the mitophagy and inflammasome pathways, and which may provide opportunities for novel therapeutics against a variety of human diseases caused by dysregulation of inflammasome and mitophagy.
2. Overview of Inflammasomes
Innate immune responses are the primary immune defense, which is activated by pattern recognition receptors (PRRs) that recognize and respond to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) [20]. Among PRRs, NOD-like receptors (NLRs) are located in the cytosol and constitute a large family of PRRs that include nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) [20].
Inflammasomes are crucial signaling platforms for the maturation of IL-1β and IL-18, and pyroptosis through caspase-1-dependent processing [21,22]. The mechanisms and functions of the nucleotide-binding domain leucine-rich repeat-containing receptor (NLR) family and, especially, the NLRP3 inflammasome (i.e., the domain containing pyrin (NLRP)) is the most extensively characterized among numerous NLR family members. The NLRP3 inflammasome comprises NLRP3, the adaptor apoptosis-associated speck-like protein containing a CARD (ASC), and caspase-1, a major polyprotein complex. The absent in melanoma 2 (AIM2) inflammasome is formed by AIM2, ASC, and caspase-1, to recognize double-stranded DNA in the cytosol [23]. Also, NLR family, apoptosis inhibitory protein (NAIP) function as specific cytosolic receptors for several bacterial protein ligands, and they assemble with NLR family CARD domain containing 4 (NLRC4) to form inflammasome which activates caspase-1 [24]. The adaptor protein ASC, a protein comprised of a pyrin domain (PYD) and a CARD, is required for the inflammasome activation of AIM2, NLRP3, and pyrin, all of which carry a PYD domain [25,26,27]. In addition, pyrin (TRIM20), encoded by MEFV gene, is a PRR for the pyrin inflammasome in response to bacterial toxins [28,29]. Pyrin inflammasome activation is mediated through effectors that modify RhoA [28].
In this section, we focus on the activation process and roles of the NLRP3, AIM2, and NLRC4 inflammasomes and discuss them in the context of mitophagy, which is summarized in Figure 1.
2.1. NLRP3 Inflammasome
Upon pathogen and/or damage-associated stimulation, NLRP3 inflammasome activation is triggered and assembled [30]. The NLRP3 inflammasome is an intracellular multimeric protein complex that is composed of the NLRP3 as the PRR, the adaptor protein ASC, and pro-caspase-1 [30,31]. Although numerous PAMPs and DAMPs are involved in the activation of NLRP3 inflammasome complex, the exact molecular mechanisms have not been fully elucidated to show how NLRP3 inflammasome responds to various PAMPs and DAMPs. It is generally thought that a two-component process, involving innate and cytokine receptor signaling through the Toll-like receptor (TLR) 4, IL-1 receptor (R), and tumor necrosis factor (TNF)-R (priming), and secondary signals (assembly), are involved in NLRP3 inflammasome activation [22,32,33] (Figure 1).
The first signal for inflammasome priming is essential for the expression of pro-IL-1β and NLRP3 in innate immune cells. Indeed, the inflammasome activation is tightly regulated by a number of factors at the transcriptional level [31]. In addition to NF-κB activation, interferon regulatory factor 1 (IRF1), AP-1, and other transcriptional factors are involved in the transcriptional regulation of the components of NLRP3 inflammasome assembly [31,34,35]. These priming signals also induce post-translational modifications for several inflammasome components, i.e., NLRP3 deubiquitination and the ubiquitination and phosphorylation of ASC, for further activation of inflammasome complex assembly [36]. Although the inflammasome assembly remains fully understood, the second signals are required for the NLRP3 inflammasome assembly. The assembly of NLRP3 inflammasome complexes involves the association of NLRP3 and ASC, and the specks formation of ASC, leading to the recruitment and activation of pro-caspase-1 [36,37]. The second signals include potassium efflux via ATP-dependent P2X purinoreceptor 7 receptor activation, bacterial pore-forming toxins, mitochondrial ROS generation, and lysosomal damage-mediated release of cathepsins such as cathepsin B or L [38]. Pannexin-1, a channel-forming glycoprotein, is an important driver of ATP release during apoptosis, and is also implicated in canonical or non-canonical NLRP3 inflammasome activation during apoptosis [39,40].
Importantly, mitochondrial perturbation and dysfunction are critically involved in NLRP3 activation through enhanced mitochondrial ROS as the second signal [36,41,42,43]. Mitochondrial Ca2+ overload leads to the increased generation of mitochondrial ROS, resulting in the activation of the NLRP3 inflammasome [36] (Figure 1). In this event, the mitochondria-associated membrane acts as a platform for inflammasome assembly [41] and cytosolic translocation of mitochondrial DNA act as danger signals for the activation of the NLRP3 inflammasome [44,45]. The molecular mechanisms of upstream signals for NLRP3 activation are not fully understood. Mitochondrial outer membrane proteins such as mitochondrial antiviral signaling protein (MAVS) participate in the NLRP3 inflammasome activation through interaction with NLPR3 [35,46,47,48]. In addition, serine-threonine kinase NIMA related kinase 7 (NEK7) functions as an upstream regulator of NLRP3 inflammasome activation through interactions with- and facilitation of NLRP3 oligomerization [49,50,51].
Pyroptosis is an inflammatory programmed cell death, which is characterized by the release of proinflammatory cytokines (IL-1β and IL-18). To date, two pyroptotic pathways are known; the canonical (caspase-1 mediated) and noncanonical (caspase-4/5/11-mediated) inflammasome pathways [8]. During pyroptosis, gasdermin D (GSDMD) acts as a key substrate of inflammatory caspases (caspase-1/4/5/11). It should be noted that LPS of Gram-negative bacteria can activate a non-canonical inflammasome to induce pyroptosis and the secretion of IL-1β and IL-18 through a direct interaction with human caspase-4 and mouse caspase-11 [52,53]. The GSDMD N-terminal domain has an intrinsic pyroptosis-inducing ability through the formation of cytotoxic pores on the plasma membrane [54,55]. The appropriate activation of the NLRP3 inflammasome and pyroptotic cell death are required for the induction of the innate and adaptive immune responses and antimicrobial host defense against a variety of infections [56,57,58,59]. However, dysregulation of the NLRP3 inflammasome and pyroptosis results in pathological inflammation during infection and is associated with pathogenesis in multiple disease conditions, including inflammatory autoimmune diseases, metabolic, degenerative, and neuroinflammatory diseases [60,61,62]. Therefore, further studies are warranted to understand clearly the complicated mechanisms of NLRP3 activation, which will aid the development of therapeutic agents against NLRP3 inflammasome-associated pathologies.
2.2. AIM2 Inflammasome
AIM2 is a cytosolic innate receptor for dsDNA derived from microbes and host cells, and activates ASC-dependent caspase-1, leading to maturation of IL-1β [63,64,65]. AIM2 consists of a PYD domain that associates with ASC, as well as a 200-amino acid repeat (HIN-200) domain that binds the DNA in the cytosol. The positively charged surface of the HIN domain binds to the DNA, and the PYD recruits ASC to assemble the inflammasome complex [64,65,66] (Figure 1). AIM2 inflammasome activation is triggered by infections resulting from DNA viruses and intracellular bacteria, which then participates in the orchestration of the host defense [67,68,69,70]. Since the AIM2 inflammasome can sense self-DNA molecules, its activation is associated with several human diseases, including systemic lupus erythematosus, psoriasis, and tumorigenesis of colorectal cancer [71,72,73]. Circulating cell-free mitochondrial DNA derived from type 2 diabetes patients is able to activate the AIM2 inflammasome in macrophages, suggesting a role for the AIM2 inflammasome in the induction of chronic inflammation during type 2 diabetes [74]. Understanding of the regulatory mechanisms of AIM2 inflammasome activation may offer the potential to prevent various human diseases.
2.3. NAIP/NLRC4 Inflammasome
Among many inflammasomes, NLR family, NAIP/NLRC4 inflammasome is critically involved in the recognition and control of intracellular bacterial infections [24] (Figure 1). The NAIP/NLRC4 inflammasome is the protein complex composed of a single NAIP bound to NLRC4 subunit. The ligands for NAIP/NLRC4 inflammasomes include intracellular bacterial flagellin or the components of type-3 secretion system, derived from pathogens [75]. Once it detects intracellular pathogens, it activates a signaling cascade resulting in pyroptosis and cytokine release to inhibit the replication of pathogenic bacteria [24,76]. At the structural basis, NAIP recognition of bacterial pathogens leads to the oligomerization of NLRC4, to activate the NAIP/NLRC4 inflammasomes that are assembled into multisubunit disk-like structures [24,77,78]. In addition, the gain-of-function mutation of NLRC4 is associated with autoinflammatory diseases in humans [24,79]. Substantial evidence now exists to support a protective role for NAIP/NLRC4 against the infection with Salmonella typhimurium in vivo [80], respiratory melioidosis [81]; however, NAIP/NLRC4 inflammasome does not protect against melanoma [82]. Therefore NAIP/NLRC4 inflammasomes are considered to be crucial regulators of infection and immunity.
3. Overview of Mitophagy
Mitophagy plays a key role in mitochondrial quality control and homeostasis. It is known that several different effectors participate in the process of mitophagy. These effectors include PINK1/Parkin with autophagic receptors, and multiple mitophagy receptors, such as NIP3-like protein X (NIX; also known as BNIP3L), BNIP3, FUN14 domain containing 1 (FUNDC1), FK506 Binding Protein 8 (FKBP8), and Syntaxin 17 (STX17) [12,83]. Also, Parkin-independent ubiquitin-mediated mitophagy and lipid-mediated mitophagy activation have been discovered [83,84,85,86] (Figure 2). Here, we briefly discuss several mechanisms underlying mitophagy processes, including Parkin/PINK1-induced and mitophagy receptor-mediated pathways.
3.1. PINK1/Parkin-Mediated Mitophagy
Mitophagy is a selective autophagic process that primes and targets damaged mitochondria to the lysosomes for degradation [87,88]. PINK1/Parkin-dependent mitophagy is a well-known post-translational signaling cascade that recognizes the cargo through the polyubiquitination of mitochondrial proteins and the recruitment of the autophagic machinery [88,89] (Figure 2). Mitochondrial Ser/Thr kinase PINK1 level is very low through constitutive degradation in physiological condition [90]. Once ΔΨM is dissipated, translocase of the inner membrane (TIM)23-mediated import of PINK1 is inhibited, thus being accumulated on the outer membrane (OMM) [90]. A supercomplex containing PINK1 homodimers facilitates PINK1 activation to phosphorylate the ubiquitinated substrates on the OMM [90]. In addition, the activated PINK1 recruits and phosphorylates Parkin, the E3 ubiquitin ligase [89,91]. Once recruited, Parkin induces the ubiquitination of the OMM proteins, which can be recognized by several autophagic receptors, including SQSTM1/p62 (sequestosome 1), NBR1, NDP52, and optineurin (OPTN) [87,92,93,94]. The autophagic receptors, having both a ubiquitin-binding domain and LC3-interacting region (LIR) motifs, are able to bridge ubiquitinated cargos with ubiquitin-like proteins belonging to the LC3/ γ-aminobutyric acid A receptor-associated protein (GABARAP) family, and the components of the core autophagic machinery to further activate selective autophagy [87,94,95,96]. Notably, both OPTN and NDP52 may play essential roles in mitophagy, independently of Parkin [97,98], whereas other receptors (p62, NBR1, and TAX1BP1) showed a lesser contribution [92].
In addition, prohibitin 2 (PHB2), containing a LIR domain, is identified as the inner mitochondrial membrane (IMM) mitophagy receptor that works to prime mitochondria during Parkin-dependent mitophagy [99]. PGAM (phosphoglycerate mutase) family member 5 (PGAM5, mitochondrial serine/threonine protein phosphatase) is also required for the participation of PHB2-mediated stabilization of PINK1, and promoting PINK1/Parkin-mediated mitophagy [100] (Figure 2).
3.2. The Mitophagy Receptor-Mediated Pathway
In mammals, mitophagy is mediated by specific mitophagy receptors, including NIX, BNIP3, FUNDC1, FKBP8, and STX17. Among these receptors, BNIP3, NIX, and FUNDC1 are the mammalian functional counterparts of yeast Atg32 [101,102] and are recruited to the OMM. As mitophagy receptors, they contain LIRs to enable them to interact with LC3, thereby activating selective autophagy against damaged mitochondria (Figure 2). The BCL2 family proteins BNIP3 and BNIP3L/NIX cooperate with the LC3 family proteins to eliminate damaged mitochondria [92,103]. NIX, a protein on the mitochondrial OMM [104], is essential for erythroid maturation through mitophagy activation in the reticulocytes [105,106]. During viral infection, both NIX/BNIP3L and BNIP3 are required for the generation of natural killer (NK) memory and the enhancement of NK cell proliferation through protective mitophagy activation via the Atg3-dependent mechanism [107]. Additionally, during retinal development, the tissue hypoxia signal induces the expression of NIX/BNIP3L and NIX-mediated mitophagy to induce a metabolic shift toward glycolysis and promote M1/proinflammatory macrophage polarization in retinal ganglion cell development [108]. In addition, FUNDC1 is a crucial mitophagy receptor in hypoxia-induced mitophagy through interaction with LC3 and subsequent activation of mitophagy [109]. FUNDC1-mediated mitophagy is regulated by a phosphorylation/dephosphorylation system, i.e., Unc-51-like autophagy activating kinase (ULK1)-mediated phosphorylation of FUNDC1 at Ser-17 and PGAM5-mediated dephosphorylation at Ser-13 [110,111]. Under normoxic conditions, FUNDC1-mediated mitophagy is inhibited by Src kinase and casein kinase 2 (CK2), which phosphorylate FUNDC1 at Tyr-18 and Ser-13, respectively [103,112].
3.3. Lipid-Mediated Mitophagy: Cardiolipin
Cardiolipin, a tetra-acyl anionic phospholipid found at the IMM, is required for the maintenance of mitochondrial membrane structures and normal bioenergetic processes [113,114]. During mitochondrial oxidative stress, cardiolipin is translocated into the external mitochondrial membrane [113,114,115]. Transmembrane redistribution of cardiolipin can lead to the activation of mitophagy through interactions with LC3B or GABARAP [116,117,118] (Figure 2). Mechanistically, the protein kinase C (PKC) pathway [119] and Tafazzin (TAZ), a phospholipid transacylase [120], are involved in the cardiolipin-mediated initiation of mitophagy. However, the regulatory pathway and/or the players involved in the activation of cardiolipin-mediated mitophagy remain largely unknown.
4. Mitophagy Crosstalk with the Inflammasome
Although there is increasing evidence of the link between mitophagy and the inflammasome in human health and disease [1,17,121,122,123], few studies have reported the key components through which mitophagy regulates the inflammasome and vice versa. The interrelationship between inflammasome and mitophagy is summarized in Figure 3. The balanced activation of inflammasome-mitophagy pathway may contribute to protective host immunity and the prevention of harmful inflammatory responses, and thus maintain human health. However, dysregulation between inflammasome and mitophagy pathways may lead to pathological inflammatory responses to aggravate mitochondrial damage further, and pyroptotic cell death (Figure 3). In this section, we briefly discuss the molecular mechanisms by which inflammasomes and mitophagy are communicated.
4.1. Mitochondrial Reactive Oxygen Species (ROS) Generation and Translocation of Mitochondrial DNA into the Cytosol
As discussed above, perturbation of the mitochondria and the generation of mitochondrial ROS trigger signal 2 for inflammasome activation. Also, the cytosolic translocation of mitochondrial DNA acts as a crucial DAMP signal to activate the NLRP3 and AIM2 inflammasomes [36,74,124,125]. At the same time, mitophagy is activated to remove the damaged mitochondria [88,126]. Defective mitophagy increases the production of mitochondrial ROS and the translocation of mitochondrial DNA into the cytosol, amplifying the increased inflammasome activation [17,43,46,127]. Mechanistically, defective mitophagy leads to mitochondrial dysfunction, impairment of membrane integrity, mtDNA leaking out of damaged mitochondria through opening mitochondrial permeability transition pore (mPTP), forming a vicious cycle that ultimately aggravates insufficient mitochondrial quality control and excessive inflammation [128,129,130]. Furthermore, cardiolipin interacts with NLRP3 [45], can be externalized into the surface of the mitochondria through mitochondrial phospholipid scramblase-3 (PLS-3) [131]. The transfer of cardiolipin from inner to outer membrane of mitochondria is mediated by two inner membrane space proteins, the mitochondrial creatine kinase and nucleoside diphosphate kinase (NDPK-D/NM23-H4) through binding and formation of oligomeric complexes with cardiolipin [131,132,133]. After transport into the outer membrane of mitochondria, cardiolipin can trigger mitophagy and activate the NLRP3 inflammasome complex in a context-dependent manner [131]. However, the mechanisms by which cardiolipin detects mitophagic and inflammasome signaling are not fully understood.
4.2. Multiple Regulators That Connect between Inflammasome and Mitophagy
In macrophages, an important innate immune signaling mediator, NF-κB, reportedly adopts the p62-mitophagy pathway as an intrinsic system for tissue repair and anti-inflammatory homeostasis. The NF-κB-p62-mitophagy pathway is required to control excessive activation of the NLRP3 inflammasome and IL-1β-mediated inflammation [134]. IL-10, an anti-inflammatory cytokine, inhibits the mammalian target of rapamycin (mTOR) activity, promotes mitophagy, and eliminates dysfunctional mitochondria in macrophages. IL-10 signaling is protective against colitis through the regulation of the NLRP3 inflammasome and IL-1β secretion. Also, the oxidation of phospholipids, such as oxidized phosphatidylcholine, during cellular stress and damage, triggered the activation of the NLRP3 inflammasome and secretion of IL-1β in macrophages [135]. Moreover, deficiency of the kinase JNK2 leads to defective mitophagy, tissue damage, and hyperactivation of inflammasomes in a mouse sepsis model [136], suggesting a role for JNK2 in the regulation of stress-induced mitophagy. These findings suggest that innate immune players participate in the connection between inflammasome and mitophagy to maintain immunological homeostasis.
In addition, the autophagy protein, GABARAP has been found to regulate NLRP3 inflammasome activation through the clearance of damaged mitochondria in macrophages [137]. Although this study suggests that GABARAP functions in mitochondrial quality control in macrophages [137], the mechanisms by which GABARAP regulates mitophagy remain to be determined. Another study showed that the metabolic component choline transporter CTL1 links mitophagy and the inflammasome. Blockade of choline transporter CTL1 expression or choline phosphorylation inhibits NLRP3 inflammasome activation through the activation of AMPK and the subsequent stimulation of mitophagy [138]. These data strongly suggest that choline kinase inhibitors are beneficial in attenuating acute and chronic inflammation driven by IL-1β.
Interestingly, specific cell types participate in the regulation of the inflammasome activation of other cell types. A previous study has shown that mesenchymal stromal cells have beneficial effects on systemic inflammation during sepsis through mitophagy activation and suppression of mitochondrial ROS and the NLRP3 inflammasome in macrophages [139]. However, it is unclear whether cell-to-cell interaction or soluble factors regulated the inflammasome/mitophagy axis.
5. Crosstalk between Inflammasomes and Mitophagy during Infection
Appropriate regulation of the inflammasome pathways is crucial for the activation of the host defense against a variety of microbial infections. However, dysregulation of NLRP3 inflammasome activation can lead to pathological responses during infection. Emerging data suggest a role for mitophagy in various bacterial, viral, and parasitic infections [140]. We discuss the current understanding of the interaction between inflammasome and mitophagy pathways in the context of various infections.
5.1. The Inflammasome-Mitophagy Axis in Bacterial Infections
Pseudomonas aeruginosa (PA) activates the NLRC4 inflammasome via T3SS-dependent mitochondrial damage and ROS [141]. Mitophagy activation to remove damaged mitochondria markedly attenuated PA-induced NLRC4 inflammasome activation [141]. Several studies have reported the virulence mechanism by which bacteria escape from inflammasome activation. The bacterial Type I CRISPR-Cas system of the PA strain UCBPP-PA14 inhibits TLR4 recognition by targeting the mRNA of LasR, thus diminishing the proinflammatory responses of the host cell and in in vivo models [142]. Mechanistically, the bacterial Type I CRISPR-Cas system of PA activates mitophagy to promote bacterial escape ability from the host immunity through TLR4-mediated inflammation and inhibition of inflammasome activation [143]. MyD88-dependent inflammasome activation and defective autophagy/mitophagy contributed to the pathogenesis of acute liver injury induced by Ehrlichia, a gram-negative bacteria [144].
In addition, Lactobacillus johnsonii L531 ameliorated Salmonella-induced enteritis through suppression of mitochondrial damage and regulation of NLRC4 and NLRP3 inflammasome activation. Although the mechanism is unclear, L. johnsonii L531 played a role in the attenuation of impaired mitophagy in the host cells during Salmonella infantis infection [145]. Through the elimination of damaged mitochondria L. johnsonii L531 ameliorates enteritis in a S. infantis model [145]. Together, these findings suggest that bacterial pathogens modulate the inflammasome/mitophagy axis, which in turn regulates host defensive pathways during bacterial infections.
5.2. The Inflammasome-Mitophagy Axis in Viral Infections
During influenza A virus infection, RIPK2 regulates mitophagy through phosphorylation of ULK1, which is needed to control mitochondrial ROS and the activation of caspase-1. NOD2-RIPK2 signaling has been found to be protective against influenza viral infection and immunopathology through inhibition of the NLRP3 inflammasome via ULK1-dependent mitophagy [146].
In human astrocytes, HIV infection induces mitochondrial injury and inflammasome activation, which contributes to the development of neurodegenerative diseases [147]. In astrocytes that were productively infected with HIV, mitophagy activation was required for cell death resistance through the inhibition of mitochondrial ROS and injury. In bystander cells with abortive infection, impaired mitophagy resulted in excessive mitochondrial damage and inflammasome-induced cell death [147]. These data suggest that crosstalk between mitophagy and the inflammasome determines the cell fate in the context of productive and abortive HIV infection [147]. Additionally, specific GU-rich single-stranded RNA from the HIV long terminal repeat region (ssRNA40) has been shown to be capable of triggering NLRP3 inflammasome activation in glial cells and cause neurotoxicity [148]. Also, ssRNA40 mediated the impairment of autophagy/mitophagy and was not able to control NLRP3 inflammasome activity and the secretion of neurotoxic cytokines [148]. Furthermore, the HIV-1 transactivator of transcription (TAT) protein increased the expression of mitophagy signaling proteins, such as PINK1, PRKN, and DNM1L, but decreased mitophagy flux, leading to the enhancement of proinflammatory cytokines, such as Tnf, Il1b, and Il6 [149]. Other studies showed that either the HIV TAT protein or the HIV-1 viral protein R (Vpr) induces the priming signal (signal 1) to activate the NLRP3 inflammasome complex and the secretion of IL-1β in macrophages and microglia, thus promoting neuroinflammation during HIV infection [150,151].
Parkin appeared to be a critical regulator of the antiviral response through inhibition of antiviral inflammation [152]. Parkin deficiency promotes antiviral immune responses and improves viral clearance and survival by enhancing the mitochondrial ROS-mediated NLRP3 inflammasome pathway [152]. Also, Parkin expression levels are downregulated in peripheral blood mononuclear cells from patients during viral infection [152]. Together, these studies provide new insights into the regulatory functions of mitophagy during the host defense against viral infections.
6. Altered Crosstalk between Inflammasome and Mitophagy in the Context of Human Disease
Defective inflammasome and mitophagy activation contribute to the pathogenic responses in a variety of inflammatory and degenerative diseases. In this session, we will briefly introduce several examples for human diseases related with dysregulated crosstalk between inflammasome and mitophagy, prior to the discussion of pharmacologic modulation in the following session.
6.1. Metabolic and Cardiovascular Diseases
Human type 2 diabetes (T2DM) is one of the best characterized human diseases associated with defective autophagy and NLRP3 inflammasome activation. Palmitate (PA) induces defective mitophagy, increased mitochondrial ROS production, and NLRP3 inflammasome activation, thus aggravating lipotoxicity and leading to insulin resistance [153]. In T2DM mice, the impairment of mitophagy and activation of inflammasomes AIM2 and NLRC4 in cardiomyocytes and cardiac macrophages are associated with myocardial infarction [154]. On the other hand, upregulation of autophagy and dysregulation of inflammation may contribute to detrimental effects in cardiac pathology, resulting in the decline of cardiovascular function and heart failure [155,156].
Acute exercise strongly triggers mitochondrial ROS-mediated NLRP3 inflammasome activation and the induction of mitophagy proteins, including Beclin1, LC3, BNIP3, and NLRP3 in the rat myocardium [157]. Although the relationship between the inflammasome and mitophagy was not examined, the data suggest that elevated mitophagy minimizes inflammation-induced myocardial injury [157]. These data also suggest that the maintenance of mitochondrial homeostasis by controlled activation of the inflammasome/mitophagy axis could provide a new preventive and therapeutic platform against cardiovascular disease, including heart failure and myocardial infarction associated with T2DM.
In the context of cardiovascular diseases, dysregulation of the cholesterol pathway leads to mitochondrial dysfunction and ROS production, which are related to defective autophagy/mitophagy and further activation of the NLRP3 inflammasome [122]. Lipid-activated eukaryotic initiation factor 2 α (eIF2α) signaling suppresses Parkin-mediated mitophagy, and reduces mitochondrial damage and inflammasome activation [158]. Since eIF2α phosphorylation is persistently activated in mouse and human atheroma, blockade of the eIF2α-mediated stress response could prove useful in the treatment of atherosclerosis [158]. In addition, development of nonalcoholic steatohepatitis (NASH) is linked to the impaired mitophagy leading to hepatic NLRP3 inflammasome activation [159]. These data suggest that the pathogenesis of metabolic and cardiovascular diseases is likely to be associated with defective mitophagy/autophagy and promotion of the inflammasomes.
6.2. Neuronal and Nephrotic Inflammation, and Sepsis
Defective mitophagy, mitochondrial dysfunction, and dysregulated activation of inflammasomes are associated with pathogenesis of neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis [160,161]. Systemic inflammation can activate TLR4 and NLRP3 inflammasome in the microglial cells, leading to neuroinflammation, accumulation of fibrillary amyloid-β, and neurodegeneration [162]. In chronic cerebral hypoperfusion models, microglial cells are excessively activated, and ROS are accumulated, in combination with the activated NLRP3 inflammasome and IL-1β production [163]. The inflammasome activation occurs in parallel with mitophagy in another model of retinal neurodegenerative disease, in which pathogenesis was associated with NLRP3 inflammasome activation [164].
Several factors involved in the mitophagy process are shown to regulate nephrotic, intestinal, and systemic inflammation. In a contrast-induced acute kidney injury (CI-AKI), the activation of PINK1-Parkin-mediated mitophagy ameliorates tissue damage and suppress apoptosis in renal tubular epithelial cells (RTECs) through the inhibition of mitochondrial ROS and NLRP3 inflammasome activation [165]. In addition, the role of PHB2 was reported in the context of inflammasome/mitophagy axis in a model of renal tubular injury. Silencing of PHB2 inhibits mitophagy and increases renal cell death, whereas PHB2 overexpression suppresses NLRP3 inflammasome activation in renal proximal tubular cells by attenuating mitochondrial dysfunction [166]. In renal biopsy tissue samples from patients with diabetic nephropathy, decreased renal expression of OPTN is negatively associated with urinary levels of IL-1β and IL-18. Also, the treatment of RTECs leads to a significant decrease in OPTN mRNA and protein levels. However, RTECs overexpressing OPTN enhance mitophagy, which decreased NLRP3 activation and the release of IL-1β and IL-18 [161]. These data collectively suggest that the autophagic receptors PHB2 and OPTN are clinically relevant in renal injury/inflammation through inhibition of the NLRP3 inflammasome by mitophagy activation.
Previous studies showed that pink1-/- and park2-/- mice have an increased susceptibility to polymicrobial sepsis through NLRP3-dependent inflammasome activation [167]. In septic patients, NLRP3 levels are upregulated, whereas PINK1 and PARK2 levels are downregulated, in peripheral blood mononuclear cells [167]. These data indicate that targeting mitophagy factors may provide new therapeutics against neuronal and nephrotic inflammation/injury and sepsis through the inhibition of NLRP3 inflammasome activation.
6.3. Cancers
Mitophagy is induced by a variety of stresses, including hypoxia, nutrient starvation, and mitochondrial and DNA damage, which are all present during tumorigenesis. Thus, emerging evidence suggests roles for mitophagy in the crosstalk with the inflammation and inflammasome pathways in the context of cancer [168]. In a pancreatic cancer mouse model, deficiency of Pink1 and Park2 accelerated pancreatic tumorigenesis through mitochondrial iron accumulation and AIM2 inflammasome activation in tumor cells [169]. Genetic depletion of AIM2 in pink1-/- and park2-/- mice increases the protective effects in pancreatic tumorigenesis [169]. In addition, a low level of PARK2, but increased AIM2 expression, has been associated with a poor prognosis in pancreatic cancer patients [169]. FUNDC1, a mitophagy receptor, was accumulated in human hepatocellular carcinomas (HCCs), and played a protective role in the initiation and development of HCCs [170]. In FUNDC1-depleted hepatocytes, dysfunctional mitochondria were accumulated, leading to activation of the inflammasome, excessive production of IL-1β, and hyperproliferation [170]. Thus, defective mitophagy-mediated dysregulation of mitochondrial homeostasis and the resulting activation of the inflammasome may contribute to tumorigenesis, and could be a potential target for therapeutic intervention.
7. The Pharmacological Regulation of Inflammasome-Mitophagy Axis to Control Human Disease
Emerging studies have revealed multiple agents/small molecules as potential therapeutic strategies based on the connection between inflammasome and mitophagy for the treatment of various human diseases. In this section, we discuss the current knowledge on pharmacological regulation of crosstalk between inflammasomes and mitophagy and the potential mechanisms by which the agents/molecules regulate the pathological responses during disease, which are summarized in Table 1.
7.1. Pharmacological Regulation in Models of Metabolic and Cardiovascular Diseases
In T2DM patients, metformin therapy increases the expression of mRNA and proteins of mitophagy markers and p-AMPKα (T172), and improve mitochondrial morphology in mononuclear cells from a metformin-treated patient group [171]. The expression levels of mitophagy kinase, PINK1, significantly correlate with homeostatic model assessment of β-cell function (HOMA-β) indices, which represent the insulin sensitivity [171]. Another drug, Miltefosine, an FDA-approved drug for the treatment of leishmaniasis, confers beneficial effects on lipid homeostasis through the activation of lipophagy and mitophagy, autophagy-inducing AMPK, and ULK1 in macrophages [172]. Additionally, Miltefosine inhibits TLR signaling and NLRP3 inflammasome assembly/activity, thereby affecting both signals 1 and 2 of NLRP3 inflammasome activation, and IL-1β release [172].
The protective effects in NASH conferred by liraglutide, a glucagon-like peptide-1 (GLP-1) analog, are mediated through the inhibition of the NLRP3 inflammasome and pyroptosis, and activation of mitophagy in hepatocytes. Liraglutide treatment reduces lipid accumulation, inhibits NLRP3 inflammasome and pyroptosis activation, attenuates mitochondrial dysfunction and ROS generation, and augments mitophagy in hepatocytes [175]. Another study using a NASH model reported that a high-fat diet or treatment with palmitic acid leads to impaired mitophagy, contributing to the activation of the hepatic NLRP3 inflammasome [159]. Several pharmacological inhibitors for the NLRP3 inflammasome (MCC950, N-acetyl cysteine or acetyl-L-carnitine) recover mitophagic flux, at least partially, and inhibit inflammasome activation and pyroptosis, thus being helpful in improving the NASH model [159].
7.2. Pharmacological Regulation in Models of Neurological and Nephrotic Diseases
The fatty acid amide hydrolase inhibitor URB597 (URB) [183] reduced the impairment of mitophagy and inhibition of the NLRP3 inflammasome pathway [163]. The beneficial effects of melatonin are reported in a subarachnoid hemorrhage model, by activation of mitophagy and inhibition of the NLRP3 inflammasome [174]. Based on the well-established effect of inflammation and mitophagy in traumatic brain injury, simultaneous activation of mitophagy and blockade of the NLRP3 inflammasome is achieved by combined treatment with MCC950 (an NLRP3 selective inhibitor) and Rapamycin (an mTOR inhibitor) [184]. In addition, ATF4 overexpression reduces cerebral ischemia/reperfusion (I/R) injury through enhancement of Parkin expression, mitophagy activity, and inhibition of NLRP3 inflammasome activation [185]. Importantly, mitophagy inhibition by either mdivi-1, an inhibitor of mitophagy, or Parkin knockdown, significantly counteracts the effects of ATF4 to inhibit NLRP3 inflammasome activation [185]. In Alzheimer’s disease model, 2,3,5,4’-Tetrahydroxystilbene-2-O-β-D-glucoside (TSG), the major active component extracted from Polygonum multiflorum, reduces β-amyloid-mediated inflammation through AMPK/PINK1/Parkin-dependent mitophagy activation [160]. Given these results, autophagy/mitophagy, combined with NLRP3 inflammasome suppression, may contribute to the development of new strategies for neuroprotection in neurodegenerative and neurological inflammation or injury [184].
In addition, auranofin, an inhibitor of the thioredoxin receptor (TrxR), leads to mitochondrial dysfunction (reduced Δψm and ATP), oxidative stress (increased H2O2), and mitophagy activation in a human retinal pigment epithelium (RPE) cell line (APRE-19) [186]. Auranofin increases NLRP3 inflammasome activation, which is improved by the antioxidant N-acetylcysteine, mitochondrial antioxidant, and anti-inflammatory drugs (amlexanox and tranilast) [186]. Moreover, mitochondrial protection by SS-31 (elamipretide) contributes to the reduction of mitochondrial damage and inflammasome activation during acute kidney injury (AKI) and arrests the progression of chronic kidney disease (CKD) [176]. Together, these data suggest that multiple agents/molecules targeting inflammasome-mitophagy/autophagy axis are promising candidates for future therapeutics against neurodegenerative diseases and renal injury/inflammation.
7.3. Pharmacological Regulation in Models of Inflammatory Disease: Sepsis and Colitis
Either a dopamine agonist (e.g., pramipexole) or an NLRP3-inhibitor (e.g., MCC950) suppresses the sepsis-mediated lethality in pink1-/- and park2-/- mice, suggesting that PINK1-PARK2-mediated neuroimmunology circuits are involved in the protection against septic shock [167]. Also, SESN2 (sestrin 2), a stress-inducible protein, functions in the inhibition of NLRP3 inflammasome activation through the induction of mitophagy [177]. Sesn2-deficient mice show defective mitophagy and increase the susceptibility to sepsis [177].
In an ulcerative colitis (UC) model, palmatine, a herb-derived isoquinoline alkaloid, has protective effects on dextran sulfate sodium (DSS)-induced colitis by enhancing mitophagy, which inhibits NLRP3 inflammasome activation [178]. In the colonic tissue of DSS mice and THP-1 macrophages, palmatine treatment enhances the expression of mitophagy-related proteins, including LC3, PINK1, and Parkin [178]. In addition, ginsenoside Rd alleviates DSS-induced colitis pathologies through autophagy/mitophagy-mediated inhibition of the NLRP3 inflammasome [179]. Andrographolide (Andro), a small molecule, protects mice against colitis-induced colon carcinogenesis by activating mitophagy, suppressing the NLRP3 inflammasome, and reducing IL1B secretion [180]. Further studies are warranted to gather evidence of future therapeutic molecules/agents based on increased mitophagy and suppression of inflammasome activation in the context of sepsis, colitis, or colitis-mediated carcinogenesis.
7.4. Pharmacological Regulation Targeting Sirtuins Nuclear Receptor Rev-erbα
Resveratrol, an agent to activate sirtuins 1 and 3, induces autophagy/mitophagy through the activation of AMPK in peritoneal mesothelial cells to ameliorate inflammatory responses triggered by mitochondrial ROS-mediated NLRP3 inflammasome activation [181]. Furthermore, honokiol (HNK), an activator of sirtuin 3 [187], attenuates mitochondrial damage and ROS production, and increases the levels of autophagy/mitophagy markers, including Parkin and PINK1 after surgery/sevoflurane treatment. Importantly, HNK-mediated mitophagy suppresses NLRP3 inflammasome activation and neuroinflammation, which is reversed by the inhibition of an autophagy inhibitor [182]. These data suggest that HNK-induced mitophagy exhibits neuroprotective effects through the regulation of NLRP3 inflammasome activation, thus suggesting its potential use in neurodegenerative diseases.
Melatonin treatment shows a preventive effect on the progression of atherosclerosis by the inhibition of NLRP3 inflammasome activation through scavenging ROS via mitophagy activation in macrophages [173]. Mechanistically, the melatonin-mediated protective effect is counteracted by knockdown of Sirt3 or pharmacologic inhibition of autophagy, suggesting a role for Sirt3-mediated mitophagy [173]. In addition, the nuclear receptor Rev-erbα (NR1D1) controls autophagy and mitochondrial biogenesis [188] and is associated with circadian variation of NLRP3 [189,190]. Rev-erbα activation inhibits fulminant hepatitis, colitis, Myocardial I/R, and lung infection in an NLRP3 dependent manner [189,190,191,192,193]. Because melatonin and sleep are controlled by the circadian clock, targeting Rev-erbα may give us more insights into the regulation of mitophagy/autophagy-NLRP3 inflammasome axis in these contexts. Future studies are warranted to investigate the possibility of linking sirtuins and/or nuclear receptor Rev-erbα in the activation of mitophagy through amelioration of numerous pathological responses induced by the NLRP3 inflammasome.
8. Conclusions
Multiple stimulants involved in the activation of the inflammasome are capable of triggering mitochondrial dysfunction, which displays a depolarized membrane potential, and is therefore targeted for mitophagy and further elimination [12,18,19]. Furthermore, by eliminating the damaged mitochondria, mitophagy controls the activation of the inflammasome to prevent tissue damage [16,17]. Accumulating evidence has increased our knowledge of a variety of biological and pathological events that are induced by crosstalk between inflammasomes and mitophagy. However, the mechanisms underlying this relationship between inflammasomes and mitophagy pathways remain largely unknown. Some additional questions remain to be answered: Are there specific molecules/factors that regulate the balance of inflammasomes and mitophagy to maintain intracellular homeostasis? Can specific types of mitophagy or mitophagy receptors play different roles in regulating inflammasomes and pyroptotic cell death? If specific types of mitophagy and/or mitophagy receptors have a unique role, which molecular targets are required to regulate inflammasome activation?
Dysregulated mitophagy is linked to aberrant activation of the inflammasome pathway and results in a variety of human diseases, including inflammatory, neurodegenerative, and cardiovascular diseases. Several experimental studies have identified numerous pharmacological agents and/or small molecules for control of the multilayered and bidirectional interactions between inflammasomes and mitophagy. However, the clinical significance of pharmacological regulation of the inflammasome/mitophagy connection is largely unknown in the context of human diseases. Future studies on the crosstalk between inflammasomes and mitophagy pathways will move the field forward and aid our understanding of how mitochondrial function interacts with host innate immunity in human health. Understanding the mechanisms underlying the dysregulation of inflammasomes and mitophagy could provide protective and therapeutic targets against numerous human diseases and pathological conditions.
Funding
This work is supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MSIP) (2017R1A5A2015385); by the framework of international cooperation program managed by National Research Foundation of Korea (2015K2A2A6002008); Chungnam National University Hospital Research Fund, 2019.
Acknowledgments
We are indebted to current and past members of our laboratory for discussions and investigations that contributed to this article. We thank H.-W. Suh for critical reading of this paper. We apologize to everyone whose work and publications could not be referenced due to space limitations.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AIM2 | Absent in melanoma 2 |
| AMPK | AMP-activated protein kinase |
| AMPK | 5′-AMP-activated protein kinase |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| Atg | Autophagy-related gene |
| BCCAo | Bilateral common carotid artery occlusion |
| BMDM | Bone marrow-derived macrophages |
| CAC | Colitis-associated cancer |
| CCH | Chronic cerebral hypoperfusion |
| CK2 | Casein kinase 2 |
| CLP | Cecal ligation and puncture |
| DAMP | Damage associated molecular pattern |
| dsDNA | Double-stranded DNA |
| DSS | Dextran sulfate sodium |
| eIF2α | Eukaryotic initiation factor 2 |
| FUNDC1 | FUN14 domain containing 1 |
| GABARAP | Gamma-aminobutyric acid receptor-associated protein |
| GLP-1 | Glucagon like peptide-1 |
| HepG2 | Human hepatocellular carcinoma cell line |
| HMGB1 | High mobility group box 1 |
| IL | Interleukin |
| IMM | Inner mitochondrial membrane |
| LPS | Lipopolysaccharide |
| MFN2 | Mitofusin 2 |
| mtDNA | Mitochondrial DNA |
| mTOR | Mammalian target of rapamycin |
| mtROS | Mitochondrial reactive oxygen species |
| NAIP | NLR family, apoptosis inhibitory protein |
| NEK7 | NIMA related kinase 7 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NIX | NIP3-like protein X |
| NLRC4 | NLR family CARD domain containing 4 |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NR1D1 | Nuclear receptor subfamily 1, group D, member 1 |
| OMM | Outer mitochondrial membrane |
| OPTN | Optineurin |
| ox-LDL | Oxidized low-density lipoprotein |
| P2X7 | P2X purinoceptor 7 |
| PA | Palmitic acid |
| PAMP | Pathogen associated molecular pattern |
| PBMC | Peripheral blood mononuclear cell |
| PGAM5 | PGAM family member 5 |
| PHB2 | Prohibitin 2 |
| PINK1 | PTEN Induced Kinase 1 |
| PKC | Protein kinase C |
| PRR | Pattern recognition receptor |
| RIPK2 | Receptor-interacting serine/threonine-protein kinase 2 |
| ROS | Reactive oxygen species |
| SAH | Subarachnoid hemorrhage |
| SESN2 | Sestrin 2 |
| SQSTM1 | Sequestosome 1 |
| T2DM | Type 2 diabetes mellitus |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| TNFR | Tumor necrosis factor receptor |
| UC | Ulcerative colitis |
| ULK1 | Unc-51 like kinase 1 |
References
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin. Exp. Rheumatol. 2016, 34, 12–16. [Google Scholar]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Krakauer, T. Inflammasomes, Autophagy, and Cell Death: The Trinity of Innate Host Defense against Intracellular Bacteria. Mediat. Inflamm. 2019, 2019, 2471215. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Spel, L.; Martinon, F. Inflammasomes contributing to inflammation in arthritis. Immunol. Rev. 2020, 294, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Zhang, F.; Zhang, L.; Wei, W. The advances in pyroptosis initiated by inflammasome in inflammatory and immune diseases. Inflamm. Res. 2020, 69, 159–166. [Google Scholar] [CrossRef]
- Lu, F.; Lan, Z.; Xin, Z.; He, C.; Guo, Z.; Xia, X.; Hu, T. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J. Cell. Physiol. 2020, 235, 3207–3221. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Gustafsson, A.B.; Dorn, G.W., 2nd. Evolving and Expanding the Roles of Mitophagy as a Homeostatic and Pathogenic Process. Physiol. Rev. 2019, 99, 853–892. [Google Scholar] [CrossRef]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell. Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta 2015, 1853, 2784–2790. [Google Scholar] [CrossRef] [Green Version]
- A, A.M.; Ameenudeen, S.; Kumar, A.; Hemalatha, S.; Ahmed, N.; Ali, N.; AlAsmari, A.F.; Aashique, M.; Waseem, M. Emerging Role of Mitophagy in Inflammatory Diseases: Cellular and Molecular Episodes. Curr. Pharm. Des. 2020, 26, 485–491. [Google Scholar] [CrossRef]
- Fan, P.; Xie, X.H.; Chen, C.H.; Peng, X.; Zhang, P.; Yang, C.; Wang, Y.T. Molecular Regulation Mechanisms and Interactions Between Reactive Oxygen Species and Mitophagy. DNA Cell Biol 2019, 38, 10–22. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, S.; Liu, J.; Wu, X.; Zhou, S.; Dai, K.; Kou, Y. Mitophagy Contributes to the Pathogenesis of Inflammatory Diseases. Inflammation 2018, 41, 1590–1600. [Google Scholar] [CrossRef]
- Harris, J.; Deen, N.; Zamani, S.; Hasnat, M.A. Mitophagy and the release of inflammatory cytokines. Mitochondrion 2018, 41, 2–8. [Google Scholar] [CrossRef]
- Kim, M.J.; Yoon, J.H.; Ryu, J.H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016, 49, 529–535. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.H.; Shin, H.W.; Lee, J.M.; Lee, H.W.; Kim, E.C.; Park, S.H. An Overview of Pathogen Recognition Receptors for Innate Immunity in Dental Pulp. Mediat. Inflamm. 2015, 2015, 794143. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howrylak, J.A.; Nakahira, K. Inflammasomes: Key Mediators of Lung Immunity. Annu. Rev. Physiol. 2017, 79, 471–494. [Google Scholar] [CrossRef]
- Wang, B.; Tian, Y.; Yin, Q. AIM2 Inflammasome Assembly and Signaling. Adv. Exp. Med. Biol. 2019, 1172, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Andrade, W.A.; Zamboni, D.S. NLRC4 biology in immunity and inflammation. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.A.; Mathur, A.; Ngo, C.; Man, S.M. Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action. Microbiol. Mol. Biol. Rev. 2018, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guey, B.; Bodnar, M.; Manie, S.N.; Tardivel, A.; Petrilli, V. Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc. Natl. Acad. Sci. USA 2014, 111, 17254–17259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Opdenbosch, N.; Gurung, P.; Vande Walle, L.; Fossoul, A.; Kanneganti, T.D.; Lamkanfi, M. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat. Commun. 2014, 5, 3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeven, N.A.; Medici, N.P.; Bliska, J.B. The pyrin inflammasome in host-microbe interactions. Curr. Opin. Microbiol. 2020, 54, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, G.; Aminov, R. Update on Pyrin Functions and Mechanisms of Familial Mediterranean Fever. Front. Microbiol. 2016, 7, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Suh, G.Y.; Ryter, S.W.; Choi, A.M. Regulation and Function of the Nucleotide Binding Domain Leucine-Rich Repeat-Containing Receptor, Pyrin Domain-Containing-3 Inflammasome in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.C.; Wu, Y.H.; Wu, C.C.; Lin, H.R.; Stern, A.; Chen, S.H.; Shu, J.C.; Tsun-Yee Chiu, D. Impaired inflammasome activation and bacterial clearance in G6PD deficiency due to defective NOX/p38 MAPK/AP-1 redox signaling. Redox Biol. 2020, 28, 101363. [Google Scholar] [CrossRef]
- Chen, H.; Jiang, Z. The essential adaptors of innate immune signaling. Protein Cell 2013, 4, 27–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Demarco, B.; Broz, P. Pannexin-1 promotes NLRP3 activation during apoptosis but is dispensable for canonical or noncanonical inflammasome activation. Eur. J. Immunol. 2020, 50, 170–177. [Google Scholar] [CrossRef]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Yu, J.W.; Lee, M.S. Mitochondria and the NLRP3 inflammasome: Physiological and pathological relevance. Arch. Pharm. Res. 2016, 39, 1503–1518. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschopp, J. Mitochondria: Sovereign of inflammation? Eur. J. Immunol. 2011, 41, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, A.; Tiwari-Pandey, R.; Pandey, N.R. Mitochondria: The indispensable players in innate immunity and guardians of the inflammatory response. J. Cell Commun. Signal. 2019, 13, 303–318. [Google Scholar] [CrossRef]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.W.; Alnemri, E.S. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef]
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J. Biol. Chem. 2016, 291, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.S. Caspase-11 non-canonical inflammasome: A critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology 2017, 152, 207–217. [Google Scholar] [CrossRef]
- Casson, C.N.; Copenhaver, A.M.; Zwack, E.E.; Nguyen, H.T.; Strowig, T.; Javdan, B.; Bradley, W.P.; Fung, T.C.; Flavell, R.A.; Brodsky, I.E.; et al. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog. 2013, 9, e1003400. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kuriakose, T.; Kanneganti, T.D. Regulation and functions of NLRP3 inflammasome during influenza virus infection. Mol. Immunol. 2017, 86, 56–64. [Google Scholar] [CrossRef]
- Rabes, A.; Suttorp, N.; Opitz, B. Inflammasomes in Pneumococcal Infection: Innate Immune Sensing and Bacterial Evasion Strategies. Curr. Top. Microbiol. Immunol. 2016, 397, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Jo, E.K. NLRP3 inflammasome and host protection against bacterial infection. J. Korean Med. Sci. 2013, 28, 1415–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchi, L.; Warner, N.; Viani, K.; Nunez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christgen, S.; Kanneganti, T.D. Inflammasomes and the fine line between defense and disease. Curr. Opin. Immunol. 2020, 62, 39–44. [Google Scholar] [CrossRef]
- Awad, F.; Assrawi, E.; Louvrier, C.; Jumeau, C.; Georgin-Lavialle, S.; Grateau, G.; Amselem, S.; Giurgea, I.; Karabina, S.A. Inflammasome biology, molecular pathology and therapeutic implications. Pharmacol. Ther. 2018, 187, 133–149. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Kanneganti, T.D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 2017, 29, 201–210. [Google Scholar] [CrossRef]
- Burckstummer, T.; Baumann, C.; Bluml, S.; Dixit, E.; Durnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef] [Green Version]
- Marim, F.M.; Franco, M.M.C.; Gomes, M.T.R.; Miraglia, M.C.; Giambartolomei, G.H.; Oliveira, S.C. The role of NLRP3 and AIM2 in inflammasome activation during Brucella abortus infection. Semin Immunopathol. 2017, 39, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Sauer, J.D.; Witte, C.E.; Zemansky, J.; Hanson, B.; Lauer, P.; Portnoy, D.A. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 2010, 7, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Alnemri, T.; Yu, J.W.; Juliana, C.; Solorzano, L.; Kang, S.; Wu, J.; Datta, P.; McCormick, M.; Huang, L.; McDermott, E.; et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 2010, 11, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Zhu, Q.; Zhu, L.; Liu, Z.; Karki, R.; Malik, A.; Sharma, D.; Li, L.; Malireddi, R.K.; Gurung, P.; et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 2015, 162, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Goss, C.; Anz, D.; Simanski, M.; Glaser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar] [CrossRef] [Green Version]
- Javierre, B.M.; Fernandez, A.F.; Richter, J.; Al-Shahrour, F.; Martin-Subero, J.I.; Rodriguez-Ubreva, J.; Berdasco, M.; Fraga, M.F.; O’Hanlon, T.P.; Rider, L.G.; et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010, 20, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.H.; Jo, S.I.; Kim, S.J.; Lee, J.M.; Jeong, J.H.; Kang, J.S.; Cho, N.J.; Kim, S.S.; Lee, E.Y.; Moon, J.S. Circulating Cell-Free mtDNA Contributes to AIM2 Inflammasome-Mediated Chronic Inflammation in Patients with Type 2 Diabetes. Cells 2019, 8, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, R.; Rauch, I. The NAIP/NLRC4 inflammasome in infection and pathology. Mol. Aspects Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Haloupek, N.; Grob, P.; Tenthorey, J.; Vance, R.E.; Nogales, E. Cryo-EM studies of NAIP-NLRC4 inflammasomes. Methods Enzymol. 2019, 625, 177–204. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhou, Q.; Zhang, C.; Fan, S.; Cheng, W.; Zhao, Y.; Shao, F.; Wang, H.W.; Sui, S.F.; Chai, J. Structural and biochemical basis for induced self-propagation of NLRC4. Science 2015, 350, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, S.; Ruan, J.; Wu, J.; Tong, A.B.; Yin, Q.; Li, Y.; David, L.; Lu, A.; Wang, W.L.; et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 2015, 350, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Moghaddas, F.; Zeng, P.; Zhang, Y.; Schutzle, H.; Brenner, S.; Hofmann, S.R.; Berner, R.; Zhao, Y.; Lu, B.; Chen, X.; et al. Autoinflammatory mutation in NLRC4 reveals a leucine-rich repeat (LRR)-LRR oligomerization interface. J. Allergy Clin. Immunol. 2018, 142, 1956–1967e1956. [Google Scholar] [CrossRef] [Green Version]
- Hausmann, A.; Bock, D.; Geiser, P.; Berthold, D.L.; Fattinger, S.A.; Furter, M.; Bouman, J.A.; Barthel-Scherrer, M.; Lang, C.M.; Bakkeren, E.; et al. Intestinal epithelial NAIP/NLRC4 restricts systemic dissemination of the adapted pathogen Salmonella Typhimurium due to site-specific bacterial PAMP expression. Mucosal Immunol. 2020, 13, 530–544. [Google Scholar] [CrossRef] [Green Version]
- West, T.E.; Myers, N.D.; Chantratita, N.; Chierakul, W.; Limmathurotsakul, D.; Wuthiekanun, V.; Miao, E.A.; Hajjar, A.M.; Peacock, S.J.; Liggitt, H.D.; et al. NLRC4 and TLR5 each contribute to host defense in respiratory melioidosis. PLoS Negl. Trop. Dis. 2014, 8, e3178. [Google Scholar] [CrossRef]
- Tenthorey, J.L.; Chavez, R.A.; Thompson, T.W.; Deets, K.A.; Vance, R.E.; Rauch, I. NLRC4 inflammasome activation is NLRP3- and phosphorylation-independent during infection and does not protect from melanoma. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Yang, Q.; Xiao, L.; Shen, H.M.; Liou, Y.C. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun. 2019, 10, 2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhujabal, Z.; Birgisdottir, A.B.; Sjottem, E.; Brenne, H.B.; Overvatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Szargel, R.; Shani, V.; Abd Elghani, F.; Mekies, L.N.; Liani, E.; Rott, R.; Engelender, S. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum. Mol. Genet. 2016, 25, 3476–3490. [Google Scholar] [CrossRef] [Green Version]
- Ambivero, C.T.; Cilenti, L.; Main, S.; Zervos, A.S. Mulan E3 ubiquitin ligase interacts with multiple E2 conjugating enzymes and participates in mitophagy by recruiting GABARAP. Cell. Signal. 2014, 26, 2921–2929. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Novak, I. Mitophagy: A complex mechanism of mitochondrial removal. Antioxid. Redox Signal. 2012, 17, 794–802. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Cali, T. PINK1/Parkin Mediated Mitophagy, Ca(2+) Signalling, and ER-Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef] [Green Version]
- Bingol, B.; Sheng, M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic. Biol. Med. 2016, 100, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Rodger, C.E.; McWilliams, T.G.; Ganley, I.G. Mammalian mitophagy—From in vitro molecules to in vivo models. FEBS J. 2018, 285, 1185–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Rogov, V.V. A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 2019, 76, 268–285. [Google Scholar] [CrossRef]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238 e210. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Desaubry, L.; Song, Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2020, 16, 419–434. [Google Scholar] [CrossRef]
- Shi, R.Y.; Zhu, S.H.; Li, V.; Gibson, S.B.; Xu, X.S.; Kong, J.M. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushima, M.; Fujiwara, T.; Takahashi, E.; Minaguchi, T.; Eguchi, Y.; Tsujimoto, Y.; Suzumori, K.; Nakamura, Y. Isolation, mapping, and functional analysis of a novel human cDNA (BNIP3L) encoding a protein homologous to human NIP3. Genes Chromosomes Cancer 1998, 21, 230–235. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 2015, 43, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Esteban-Martinez, L.; Boya, P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy 2018, 14, 915–917. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Wu, H.; Xue, D.; Chen, G.; Han, Z.; Huang, L.; Zhu, C.; Wang, X.; Jin, H.; Wang, J.; Zhu, Y.; et al. The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy 2014, 10, 1712–1725. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Diaz, A.G.; Cardona-Munoz, E.G.; Pacheco-Moises, F.P. The Role of Cardiolipin and Mitochondrial Damage in Kidney Transplant. Oxid. Med. Cell. Longev. 2019, 2019, 3836186. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Anton, Z.; Landajuela, A.; Hervas, J.H.; Montes, L.R.; Hernandez-Tiedra, S.; Velasco, G.; Goni, F.M.; Alonso, A. Human Atg8-cardiolipin interactions in mitophagy: Specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy 2016, 12, 2386–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.T.; Bayir, H.; Kagan, V.E. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: Implications for Parkinson disease. Autophagy 2014, 10, 376–378. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell. Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Li, Y.; Gasparski, A.N.; Abeliovich, H.; Greenberg, M.L. Cardiolipin Regulates Mitophagy through the Protein Kinase C Pathway. J. Biol. Chem. 2017, 292, 2916–2923. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.; Liu, X.; Zhang, J.; Wang, H.G.; Ye, J.M.; Shi, Y. Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 2015, 11, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation - a two-way street. Mol. Med. 2017, 23, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Piscianz, E.; Loganes, C.; Vecchi Brumatti, L.; Knowles, A.; Bilel, S.; Tommasini, A.; Bortul, R.; Zweyer, M. Innovative Target Therapies Are Able to Block the Inflammation Associated with Dysfunction of the Cholesterol Biosynthesis Pathway. Int. J. Mol. Sci. 2015, 17, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.S.; Suh, H.W.; Kim, S.J.; Jo, E.K. Mitochondrial Control of Innate Immunity and Inflammation. Immune Netw. 2017, 17, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Helal, J.N.; Van Hoecke, J.; Garapon-Bar, C.; Goubel, F. Surface myoelectric signals during ergocycle exrcises at various mechanical powers and pedalling rates. J. Electromyogr. Kinesiol. 1992, 2, 242–251. [Google Scholar] [CrossRef]
- Tan, T.; Zimmermann, M.; Reichert, A.S. Controlling quality and amount of mitochondria by mitophagy: Insights into the role of ubiquitination and deubiquitination. Biol. Chem. 2016, 397, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020, 16, 3–17. [Google Scholar] [CrossRef]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 2017, 16, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Harrington, J.S.; Choi, A.M.K.; Nakahira, K. Mitochondrial DNA in Sepsis. Curr. Opin. Crit. Care 2017, 23, 284–290. [Google Scholar] [CrossRef]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell. Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Schlattner, U.; Tokarska-Schlattner, M.; Ramirez, S.; Tyurina, Y.Y.; Amoscato, A.A.; Mohammadyani, D.; Huang, Z.; Jiang, J.; Yanamala, N.; Seffouh, A.; et al. Dual function of mitochondrial Nm23-H4 protein in phosphotransfer and intermembrane lipid transfer: A cardiolipin-dependent switch. J. Biol. Chem. 2013, 288, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacombe, M.L.; Tokarska-Schlattner, M.; Epand, R.F.; Boissan, M.; Epand, R.M.; Schlattner, U. Interaction of NDPK-D with cardiolipin-containing membranes: Structural basis and implications for mitochondrial physiology. Biochimie 2009, 91, 779–783. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [Green Version]
- Yeon, S.H.; Yang, G.; Lee, H.E.; Lee, J.Y. Oxidized phosphatidylcholine induces the activation of NLRP3 inflammasome in macrophages. J. Leukoc. Biol. 2017, 101, 205–215. [Google Scholar] [CrossRef]
- Zhang, Q.; Kuang, H.; Chen, C.; Yan, J.; Do-Umehara, H.C.; Liu, X.Y.; Dada, L.; Ridge, K.M.; Chandel, N.S.; Liu, J. The kinase Jnk2 promotes stress-induced mitophagy by targeting the small mitochondrial form of the tumor suppressor ARF for degradation. Nat. Immunol. 2015, 16, 458–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Xu, X.; Ma, J.; Wu, J.; Wang, Y.; Zhou, R.; Han, J. Gene deletion of Gabarap enhances Nlrp3 inflammasome-dependent inflammatory responses. J. Immunol. 2013, 190, 3517–3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Lopez, E.; Zhong, Z.; Stubelius, A.; Sweeney, S.R.; Booshehri, L.M.; Antonucci, L.; Liu-Bryan, R.; Lodi, A.; Terkeltaub, R.; Lacal, J.C.; et al. Choline Uptake and Metabolism Modulate Macrophage IL-1beta and IL-18 Production. Cell Metab. 2019, 29, 1350–1362 e1357. [Google Scholar] [CrossRef]
- Li, S.; Wu, H.; Han, D.; Ma, S.; Fan, W.; Wang, Y.; Zhang, R.; Fan, M.; Huang, Y.; Fu, X.; et al. A Novel Mechanism of Mesenchymal Stromal Cell-Mediated Protection against Sepsis: Restricting Inflammasome Activation in Macrophages by Increasing Mitophagy and Decreasing Mitochondrial ROS. Oxid. Med. Cell. Longev. 2018, 2018, 3537609. [Google Scholar] [CrossRef]
- Cho, D.H.; Kim, J.K.; Jo, E.K. Mitophagy and Innate Immunity in Infection. Mol. Cells 2020, 43, 10–22. [Google Scholar] [CrossRef]
- Jabir, M.S.; Hopkins, L.; Ritchie, N.D.; Ullah, I.; Bayes, H.K.; Li, D.; Tourlomousis, P.; Lupton, A.; Puleston, D.; Simon, A.K.; et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 2015, 11, 166–182. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Fang, L.; Tan, S.; Yu, M.; Li, X.; He, S.; Wei, Y.; Li, G.; Jiang, J.; Wu, M. Type I CRISPR-Cas targets endogenous genes and regulates virulence to evade mammalian host immunity. Cell Res. 2016, 26, 1273–1287. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, B.; Zhou, C.; Lin, P.; Qin, S.; Gao, P.; Wang, Z.; Xia, Z.; Wu, M. Bacterial Type I CRISPR-Cas systems influence inflammasome activation in mammalian host by promoting autophagy. Immunology 2019, 158, 240–251. [Google Scholar] [CrossRef]
- Kader, M.; Alaoui-El-Azher, M.; Vorhauer, J.; Kode, B.B.; Wells, J.Z.; Stolz, D.; Michalopoulos, G.; Wells, A.; Scott, M.; Ismail, N. MyD88-dependent inflammasome activation and autophagy inhibition contributes to Ehrlichia-induced liver injury and toxic shock. PLoS Pathog. 2017, 13, e1006644. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Yu, J.; He, T.; Liu, X.; Su, J.; Wang, M.; Wang, J.; Zhu, Y. Lactobacillus johnsonii L531 ameliorates enteritis via elimination of damaged mitochondria and suppression of SQSTM1-dependent mitophagy in a Salmonella infantis model of piglet diarrhea. FASEB J. 2020, 34, 2821–2839. [Google Scholar] [CrossRef] [Green Version]
- Lupfer, C.; Thomas, P.G.; Anand, P.K.; Vogel, P.; Milasta, S.; Martinez, J.; Huang, G.; Green, M.; Kundu, M.; Chi, H.; et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 2013, 14, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Ojeda, D.S.; Grasso, D.; Urquiza, J.; Till, A.; Vaccaro, M.I.; Quarleri, J. Cell Death Is Counteracted by Mitophagy in HIV-Productively Infected Astrocytes but Is Promoted by Inflammasome Activation Among Non-productively Infected Cells. Front. Immunol. 2018, 9, 2633. [Google Scholar] [CrossRef] [Green Version]
- Rawat, P.; Teodorof-Diedrich, C.; Spector, S.A. Human immunodeficiency virus Type-1 single-stranded RNA activates the NLRP3 inflammasome and impairs autophagic clearance of damaged mitochondria in human microglia. Glia 2019, 67, 802–824. [Google Scholar] [CrossRef] [PubMed]
- Thangaraj, A.; Periyasamy, P.; Liao, K.; Bendi, V.S.; Callen, S.; Pendyala, G.; Buch, S. HIV-1 TAT-mediated microglial activation: Role of mitochondrial dysfunction and defective mitophagy. Autophagy 2018, 14, 1596–1619. [Google Scholar] [CrossRef] [Green Version]
- Chivero, E.T.; Guo, M.L.; Periyasamy, P.; Liao, K.; Callen, S.E.; Buch, S. HIV-1 Tat Primes and Activates Microglial NLRP3 Inflammasome-Mediated Neuroinflammation. J. Neurosci. 2017, 37, 3599–3609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamik, M.K.; Hui, E.; Branton, W.G.; McKenzie, B.A.; Chisholm, J.; Cohen, E.A.; Power, C. HIV-1 Viral Protein R Activates NLRP3 Inflammasome in Microglia: Implications for HIV-1 Associated Neuroinflammation. J. Neuroimmune Pharmacol. 2017, 12, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, C.; Long, F.; Yang, D.; Liu, X.; Hu, Y.; Wu, C.; Wang, B.; Wang, M.; Chen, Y.; et al. Parkin Impairs Antiviral Immunity by Suppressing the Mitochondrial Reactive Oxygen Species-Nlrp3 Axis and Antiviral Inflammation. iScience 2019, 16, 468–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Xia, C.; Li, S.; Du, L.; Zhang, L.; Zhou, R. Defective mitophagy driven by dysregulation of rheb and KIF5B contributes to mitochondrial reactive oxygen species (ROS)-induced nod-like receptor 3 (NLRP3) dependent proinflammatory response and aggravates lipotoxicity. Redox Biol. 2014, 3, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Durga Devi, T.; Babu, M.; Makinen, P.; Kaikkonen, M.U.; Heinaniemi, M.; Laakso, H.; Yla-Herttuala, E.; Rieppo, L.; Liimatainen, T.; Naumenko, N.; et al. Aggravated Postinfarct Heart Failure in Type 2 Diabetes Is Associated with Impaired Mitophagy and Exaggerated Inflammasome Activation. Am. J. Pathol. 2017, 187, 2659–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, B.; Jantuan, E.; Shen, F.; Chiu, B.; Sergi, C. Autophagy-Inflammasome Interplay in Heart Failure: A Systematic Review on Basics, Pathways, and Therapeutic Perspectives. Ann. Clin. Lab. Sci. 2017, 47, 243–252. [Google Scholar]
- Ghosh, R.; Pattison, J.S. Macroautophagy and Chaperone-Mediated Autophagy in Heart Failure: The Known and the Unknown. Oxidative Med. Cell. Longev. 2018, 2018, 8602041. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Miao, W.; Ma, J.; Xv, Z.; Bo, H.; Li, J.; Zhang, Y.; Ji, L.L. Acute Exercise-Induced Mitochondrial Stress Triggers an Inflammatory Response in the Myocardium via NLRP3 Inflammasome Activation with Mitophagy. Oxidative Med. Cell. Longev. 2016, 2016, 1987149. [Google Scholar] [CrossRef] [Green Version]
- Onat, U.I.; Yildirim, A.D.; Tufanli, O.; Cimen, I.; Kocaturk, B.; Veli, Z.; Hamid, S.M.; Shimada, K.; Chen, S.; Sin, J.; et al. Intercepting the Lipid-Induced Integrated Stress Response Reduces Atherosclerosis. J. Am. Coll. Cardiol. 2019, 73, 1149–1169. [Google Scholar] [CrossRef]
- Zhang, N.P.; Liu, X.J.; Xie, L.; Shen, X.Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Invest. 2019, 99, 749–763. [Google Scholar] [CrossRef]
- Gao, Y.; Li, J.; Li, J.; Hu, C.; Zhang, L.; Yan, J.; Li, L.; Zhang, L. Tetrahydroxy stilbene glycoside alleviated inflammatory damage by mitophagy via AMPK related PINK1/Parkin signaling pathway. Biochem. Pharmacol. 2020, 177, 113997. [Google Scholar] [CrossRef]
- Hu, Z.L.; Sun, T.; Lu, M.; Ding, J.H.; Du, R.H.; Hu, G. Kir6.1/K-ATP channel on astrocytes protects against dopaminergic neurodegeneration in the MPTP mouse model of Parkinson’s disease via promoting mitophagy. Brain. Behav. Immun. 2019, 81, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef] [PubMed]
- Su, S.H.; Wu, Y.F.; Lin, Q.; Wang, D.P.; Hai, J. URB597 protects against NLRP3 inflammasome activation by inhibiting autophagy dysfunction in a rat model of chronic cerebral hypoperfusion. J. Neuroinflammation 2019, 16, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, J.; Xu, W.; Ding, F.; Ding, W. Prohibitin 2-mediated mitophagy attenuates renal tubular epithelial cells injury by regulating mitochondrial dysfunction and NLRP3 inflammasome activation. Am. J. Physiol. Renal Physiol. 2019, 316, F396–F407. [Google Scholar] [CrossRef]
- Kang, R.; Zeng, L.; Xie, Y.; Yan, Z.; Zhou, B.; Cao, L.; Klionsky, D.J.; Tracey, K.J.; Li, J.; Wang, H.; et al. A novel PINK1- and PARK2-dependent protective neuroimmune pathway in lethal sepsis. Autophagy 2016, 12, 2374–2385. [Google Scholar] [CrossRef] [Green Version]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441–455 e448. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, Y.; Siraj, S.; Jin, H.; Fan, Y.; Yang, X.; Huang, X.; Wang, X.; Wang, J.; Liu, L.; et al. FUN14 Domain-Containing 1-Mediated Mitophagy Suppresses Hepatocarcinogenesis by Inhibition of Inflammasome Activation in Mice. Hepatology 2019, 69, 604–621. [Google Scholar] [CrossRef]
- Bhansali, S.; Bhansali, A.; Dutta, P.; Walia, R.; Dhawan, V. Metformin upregulates mitophagy in patients with T2DM: A randomized placebo-controlled study. J. Cell. Mol. Med. 2020, 24, 2832–2846. [Google Scholar] [CrossRef]
- Iacano, A.J.; Lewis, H.; Hazen, J.E.; Andro, H.; Smith, J.D.; Gulshan, K. Miltefosine increases macrophage cholesterol release and inhibits NLRP3-inflammasome assembly and IL-1beta release. Sci. Rep. 2019, 9, 11128. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxidative Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Shrestha, S.; Li, J.; Yu, X.; Chen, J.; Yan, F.; Ying, G.; Gu, C.; Wang, L.; Chen, G. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci. Rep. 2017, 7, 2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019, 864, 172715. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1beta and IL-18 and Arrests CKD. J. Am. Soc. Nephrol. 2017, 28, 1437–1449. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Bae, S.H.; Ryu, J.C.; Kwon, Y.; Oh, J.H.; Kwon, J.; Moon, J.S.; Kim, K.; Miyawaki, A.; Lee, M.G.; et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar] [CrossRef] [Green Version]
- Mai, C.T.; Wu, M.M.; Wang, C.L.; Su, Z.R.; Cheng, Y.Y.; Zhang, X.J. Palmatine attenuated dextran sulfate sodium (DSS)-induced colitis via promoting mitophagy-mediated NLRP3 inflammasome inactivation. Mol Immunol. 2019, 105, 76–85. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Yang, Y.; Liu, X.; Zhu, Y.; Zou, J.; Peng, S.; Le, T.H.; Chen, Y.; Zhao, S.; et al. Ginsenoside Rd ameliorates colitis by inducing p62-driven mitophagy-mediated NLRP3 inflammasome inactivation in mice. Biochem. Pharmacol. 2018, 155, 366–379. [Google Scholar] [CrossRef]
- Guo, W.; Sun, Y.; Liu, W.; Wu, X.; Guo, L.; Cai, P.; Wu, X.; Wu, X.; Shen, Y.; Shu, Y.; et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 2014, 10, 972–985. [Google Scholar] [CrossRef]
- Wu, J.; Li, X.; Zhu, G.; Zhang, Y.; He, M.; Zhang, J. The role of Resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Exp. Cell Res. 2016, 341, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.S.; Chen, L.; Lu, Y.Y.; Lei, S.Q.; Peng, M.; Xia, Z.Y. Honokiol-Mediated Mitophagy Ameliorates Postoperative Cognitive Impairment Induced by Surgery/Sevoflurane via Inhibiting the Activation of NLRP3 Inflammasome in the Hippocampus. Oxidative Med. Cell. Longev. 2019, 2019, 8639618. [Google Scholar] [CrossRef] [PubMed]
- Su, S.H.; Wang, Y.Q.; Wu, Y.F.; Wang, D.P.; Lin, Q.; Hai, J. Cannabinoid receptor agonist WIN55,212-2 and fatty acid amide hydrolase inhibitor URB597 may protect against cognitive impairment in rats of chronic cerebral hypoperfusion via PI3K/AKT signaling. Behav. Brain Res. 2016, 313, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Meng, J.; Xu, Q.; Long, T.; Bi, F.; Chang, C.; Liu, W. Rapamycin improves the neuroprotection effect of inhibition of NLRP3 inflammasome activation after TBI. Brain Res. 2019, 1710, 163–172. [Google Scholar] [CrossRef]
- He, Q.; Li, Z.; Meng, C.; Wu, J.; Zhao, Y.; Zhao, J. Parkin-Dependent Mitophagy is Required for the Inhibition of ATF4 on NLRP3 Inflammasome Activation in Cerebral Ischemia-Reperfusion Injury in Rats. Cells 2019, 8, 897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yumnamcha, T.; Devi, T.S.; Singh, L.P. Auranofin Mediates Mitochondrial Dysregulation and Inflammatory Cell Death in Human Retinal Pigment Epithelial Cells: Implications of Retinal Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1065. [Google Scholar] [CrossRef]
- Pillai, V.B.; Samant, S.; Sundaresan, N.R.; Raghuraman, H.; Kim, G.; Bonner, M.Y.; Arbiser, J.L.; Walker, D.I.; Jones, D.P.; Gius, D.; et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 2015, 6, 6656. [Google Scholar] [CrossRef]
- Woldt, E.; Sebti, Y.; Solt, L.A.; Duhem, C.; Lancel, S.; Eeckhoute, J.; Hesselink, M.K.; Paquet, C.; Delhaye, S.; Shin, Y.; et al. Rev-erb-alpha modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat. Med. 2013, 19, 1039–1046. [Google Scholar] [CrossRef]
- Wang, S.; Lin, Y.; Yuan, X.; Li, F.; Guo, L.; Wu, B. REV-ERBalpha integrates colon clock with experimental colitis through regulation of NF-kappaB/NLRP3 axis. Nat. Commun. 2018, 9, 4246. [Google Scholar] [CrossRef] [Green Version]
- Pourcet, B.; Zecchin, M.; Ferri, L.; Beauchamp, J.; Sitaula, S.; Billon, C.; Delhaye, S.; Vanhoutte, J.; Mayeuf-Louchart, A.; Thorel, Q.; et al. Nuclear Receptor Subfamily 1 Group D Member 1 Regulates Circadian Activity of NLRP3 Inflammasome to Reduce the Severity of Fulminant Hepatitis in Mice. Gastroenterology 2018, 154, 1449–1464. [Google Scholar] [CrossRef]
- Zhou, Z.; Lin, Y.; Gao, L.; Yang, Z.; Wang, S.; Wu, B. Circadian pharmacological effects of berberine on chronic colitis in mice: Role of the clock component Rev-erbalpha. Biochem. Pharmacol. 2020, 172, 113773. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.J.; Alibhai, F.J.; Khatua, T.N.; Rasouli, M.; Bridle, B.W.; Burris, T.P.; Martino, T.A. SR9009 administered for one day after myocardial ischemia-reperfusion prevents heart failure in mice by targeting the cardiac inflammasome. Commun. Biol. 2019, 2, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Fang, X.; Xu, Y.; Xiao, H.; Huang, T.; Zhang, Y.; Ge, Y.; Li, Y.; Zong, L.; Gao, J. Rev-erbalpha can regulate the NF-kappaB/NALP3 pathway to modulate lipopolysaccharide-induced acute lung injury and inflammation. Int. Immunopharmacol. 2019, 73, 312–320. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Inflammasome activation. (i) A priming signal (signal 1) is necessary for NLRP3 inflammasome activation in response to secondary signal (assembly signal 2). The priming signal through various innate and cytokine receptors such as TLR, IL-1R, and TNFR which activates the transcriptional factor NF-κB to increase the expression of NLRP3 and pro-forms of IL-1β and IL-18. The signal 2 activation by P2X7 receptors, pore forming toxins induce NLRP3 inflammasome assembly and caspase-1 activation leading to processing of pro-forms of IL-1β and IL-18, and the secretion of mature cytokines. Lysosomal destabilization, and mitochondrial damage also induces the NLRP3 activation. A serine-threonine kinase NEK7 facilitates NLRP3 oligomerization. (ii) AIM2 inflammasome activation is triggered by sensing the presence of double-stranded DNA. The positively charged surface of the HIN-200 domain binds to the DNA, and the pyrin domain engages to ASC for inflammasome complex assembly. AIM2 can sense the self-DNA and are associated with various pathological conditions. (iii) For the activation of NLRC4 inflammasome, NAIP acts as cytosolic receptor for a variety of bacterial protein (T3SS, Flagellin, etc.) and co-assemble into NLRC4 inflammasome complex. All the inflammasome activation lead to pyroptosis through gasdermin D that acts as a key substrate of inflammatory caspases. TLR, Toll-like receptor; IL-1R, interleukin 1 receptor; TNFR, tumor necrosis factor receptor; P2X7, P2X purinoreceptor 7; NEK7, NIMA related kinase 7; T3SS, type III secretion systems. Upward arrow (↑) in red represents increase/induction; all other black arrows represent signaling events.
Figure 1.
Inflammasome activation. (i) A priming signal (signal 1) is necessary for NLRP3 inflammasome activation in response to secondary signal (assembly signal 2). The priming signal through various innate and cytokine receptors such as TLR, IL-1R, and TNFR which activates the transcriptional factor NF-κB to increase the expression of NLRP3 and pro-forms of IL-1β and IL-18. The signal 2 activation by P2X7 receptors, pore forming toxins induce NLRP3 inflammasome assembly and caspase-1 activation leading to processing of pro-forms of IL-1β and IL-18, and the secretion of mature cytokines. Lysosomal destabilization, and mitochondrial damage also induces the NLRP3 activation. A serine-threonine kinase NEK7 facilitates NLRP3 oligomerization. (ii) AIM2 inflammasome activation is triggered by sensing the presence of double-stranded DNA. The positively charged surface of the HIN-200 domain binds to the DNA, and the pyrin domain engages to ASC for inflammasome complex assembly. AIM2 can sense the self-DNA and are associated with various pathological conditions. (iii) For the activation of NLRC4 inflammasome, NAIP acts as cytosolic receptor for a variety of bacterial protein (T3SS, Flagellin, etc.) and co-assemble into NLRC4 inflammasome complex. All the inflammasome activation lead to pyroptosis through gasdermin D that acts as a key substrate of inflammatory caspases. TLR, Toll-like receptor; IL-1R, interleukin 1 receptor; TNFR, tumor necrosis factor receptor; P2X7, P2X purinoreceptor 7; NEK7, NIMA related kinase 7; T3SS, type III secretion systems. Upward arrow (↑) in red represents increase/induction; all other black arrows represent signaling events.

Figure 2.
An overview of mitophagy. PINK1/Parkin-mediated mitophagy involves the recognization of dysfunctional mitochondria by PINK1 and activation of Parkin. This leads to ubiquitination of outer mitochondrial membrane-associated proteins, which are then recognized by autophagic receptors such as p62, OPTN, NDP52, and NBR1. These receptors can bridge the cargos with LC3/GABARAP to activate selective autophagy. Inner mitochondrial membrane protein Prohibitin 2 promotes PINK1/Parkin-mediated mitophagy through PGAM5. Mitophagy receptors such as NIX, BNIP3, FUNDC1 present in OMM can interact with LC3 to activate the selective autophagy. FUNDC1 mediates the mitophagy via its dephosphorylation (represented as ‘-‘) at Ser-13 by PGAM5 and phosphorylation (represented as ‘+’) at Ser-13 and Tyr-18 by CK2 and Src kinase respectively. Cardiolipin, by its transmembrane distribution, can activate mitophagy through binding to LC3 or GABARAP. OPTN, optineurin; OMM, outer mitochondrial membrane-associated proteins; GABARAP, γ-aminobutyric acid A receptor-associated protein; PGAM5, PGAM family member 5; CK2, Casein kinase 2; Δψ, mitochondrial membrane potential; ROS, reactive oxygen species; NIX, NIP3-like protein X; FUNDC1, FUN14 domain containing 1.
Figure 2.
An overview of mitophagy. PINK1/Parkin-mediated mitophagy involves the recognization of dysfunctional mitochondria by PINK1 and activation of Parkin. This leads to ubiquitination of outer mitochondrial membrane-associated proteins, which are then recognized by autophagic receptors such as p62, OPTN, NDP52, and NBR1. These receptors can bridge the cargos with LC3/GABARAP to activate selective autophagy. Inner mitochondrial membrane protein Prohibitin 2 promotes PINK1/Parkin-mediated mitophagy through PGAM5. Mitophagy receptors such as NIX, BNIP3, FUNDC1 present in OMM can interact with LC3 to activate the selective autophagy. FUNDC1 mediates the mitophagy via its dephosphorylation (represented as ‘-‘) at Ser-13 by PGAM5 and phosphorylation (represented as ‘+’) at Ser-13 and Tyr-18 by CK2 and Src kinase respectively. Cardiolipin, by its transmembrane distribution, can activate mitophagy through binding to LC3 or GABARAP. OPTN, optineurin; OMM, outer mitochondrial membrane-associated proteins; GABARAP, γ-aminobutyric acid A receptor-associated protein; PGAM5, PGAM family member 5; CK2, Casein kinase 2; Δψ, mitochondrial membrane potential; ROS, reactive oxygen species; NIX, NIP3-like protein X; FUNDC1, FUN14 domain containing 1.

Figure 3.
Inflammasome and mitophagy in health and disease. Balance between inflammasome and mitophagy activation is required to prevent the harmful inflammatory responses of microbial or other danger signals, and to maintain protective immunity and good health. Any disproportion between activation of inflammasome and mitophagy causes sustained mitochondrial stress leading to pyroptosis and pathological inflammation.
Figure 3.
Inflammasome and mitophagy in health and disease. Balance between inflammasome and mitophagy activation is required to prevent the harmful inflammatory responses of microbial or other danger signals, and to maintain protective immunity and good health. Any disproportion between activation of inflammasome and mitophagy causes sustained mitochondrial stress leading to pyroptosis and pathological inflammation.


{kind=link}
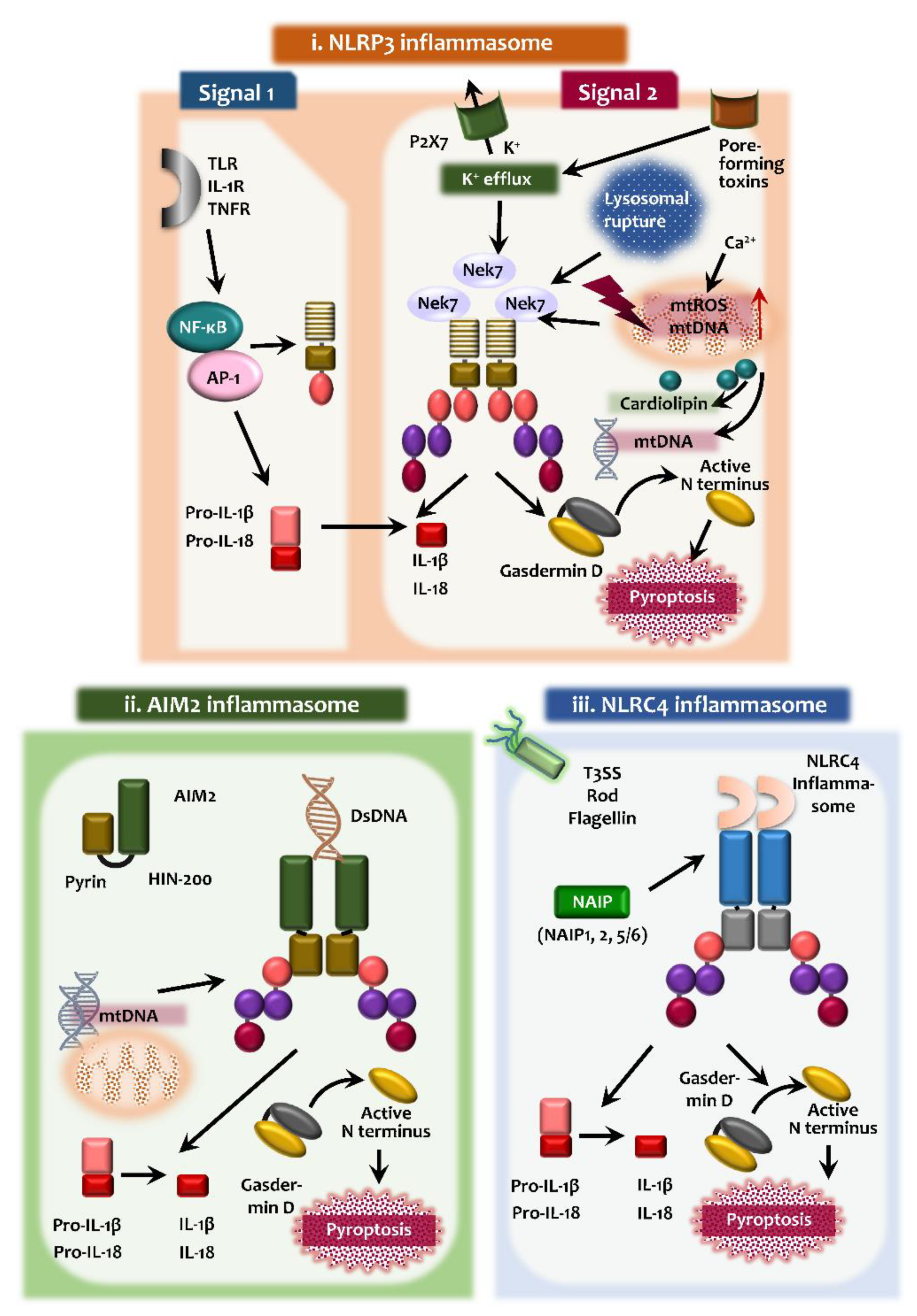{kind=link}
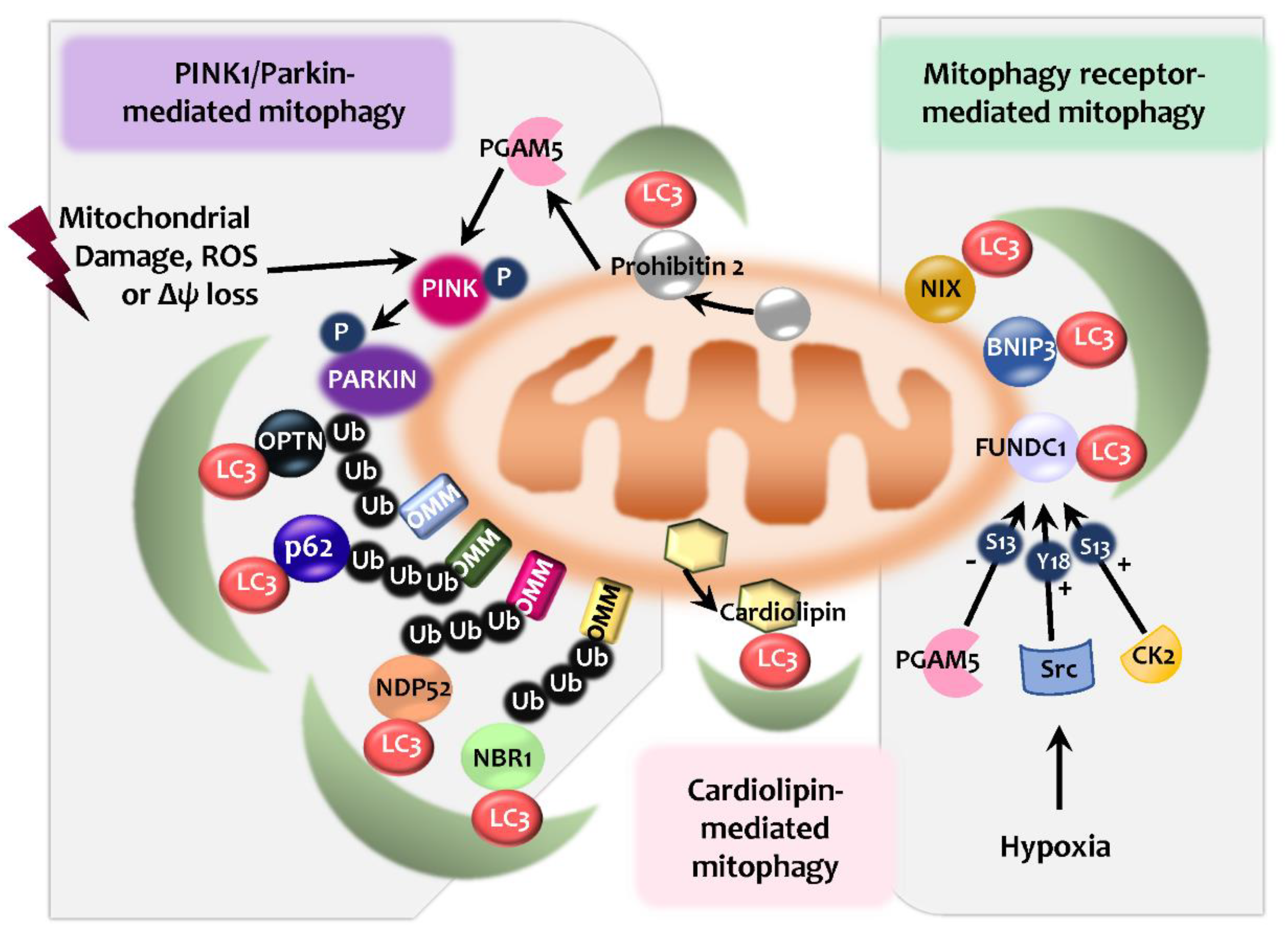{kind=link}
{kind=link}
Table 1.
Therapeutic candidates based on the regulation of mitophagy-inflammasome connection in human diseases.
Table 1.
Therapeutic candidates based on the regulation of mitophagy-inflammasome connection in human diseases.
| Agents/Drugs | Disease/Model | Functions | Underlying Mechanisms | Experimental Objects | Reference |
|---|---|---|---|---|---|
| Metformin | Type 2 diabetes | ↓ Mitochondrial oxidative stress and distortions in mitochondrial morphology | ↑ Protein expression of p-AMPKα (T172), and NLRP3 ↑ Protein expression of mitophagy-related markers (PINK1, PARKIN, MFN2, NIX, LC3-II, LAMP2) | Patient samples, PBMCs | [171] |
| Miltefosine | Atherosclerosis | ↑ ABCA1-mediated cholesterol release ↓ LPS-mediated NLRP3 inflammasome assembly and IL-1β release ↑ Mitophagy induction | ↑ Disruption of lipid-rafts ↑ Inhibition of phosphatidylserine flip ↓ LPS-mediated mitochondrial ROS generation ↓ Loss of mitochondrial membrane potential in murine macrophages treated with LPS | HEK293-ABCA1-GFP cells, BHK cells, RAW264.7, BMDM | [172] |
| Melatonin | Atherosclerosis | ↓ Atherosclerotic plaque progression and serum IL-1β level in vivo murine models ↓ NLRP3 inflammasome activation in atherosclerotic lesions and in Ox-LDL-treated murine macrophages | ↑ Sirt3-dependent mitophagy induction and reactive oxygen species (ROS) scavenging ↑ Parkin-mediated mitophagy process through Sirt3-FOXO3a pathway | RAW264.7, ApoE−/− mice | [173] |
| Subarachnoid hemorrhage | ↓ Brain edema and neurological dysfunction in rat receiving SAH ↓ SAH-induced neuronal cell death in the ipsilateral basal cortex ↓ Pathological changes in mitochondria | ↑ Protein expression of autophagy markers (LC3-II/LC3-I and Atg5) and mitophagy markers (Parkin and PINK-1) ↓ ROS and pro-inflammatory cytokines generation ↓ NLRP3 inflammasome activation | Endovascular perforation-induced SAH rat model, Brain tissue | [174] | |
| Liraglutide | Non-alcoholic steatohepatitis | ↓ Lipid accumulation in hepatocytes ↓ NLRP3 inflammasome-induced hepatocyte pyroptosis ↑ Mitophagy induction | ↓ Mitochondrial dysfunction and reactive oxygen species generation ↑ Co-localization of PINK1/Parkin and mitochondria and OPTN and NIX expressions in HepG2 cells treated with LPS and PA | HepG2 cell line | [175] |
| URB597 | Chronic cerebral hypoperfusion | ↓ CCH-induced NLRP3 inflammasome activation, impaired autophagy, and defective mitophagy in rat hippocampus | ↑ The restoration of lysosomal function ↓ CCH-induced microglial overactivation ↓ CCH-induced ROS accumulation | BCCAo-induced CCH rat model, Hippocampus | [163] |
| Elamipretide (SS-31) | Bilateral renal ischemia | ↓ Ischemia-induced tubulointerstitial fibrosis and glomerulosclerosis ↓ Inflammatory Response and endothelial Injury after Ischemia | ↓ NLRP3 inflammasome activation and upregulation of IL-18, IL-1β, and TNF induced by ischemia ↑ Mitochondrial integrity and autophagic vacuoles containing organelles and cytoplasmic contents in podocytes | Ischemia-reperfusion experimental rat model, Kidney tissue | [176] |
| SESN2 | Sepsis | ↓ Clearance of damaged mitochondria in sesn2−/− macrophages ↑ IL1β and IL18 production in serum and lung tissue of sesn2−/− mice after CLP ↑ Mortality rate in sesn2−/− mice | ↑ Mitochondrial priming by mediating aggregation of SQSTM1 and its binding to lysine 63-linked ubiquitins on the mitochondrial surface ↑ Activation of autophagic machinery for the degradation of primed mitochondria via an increase of ULK1 protein ↓ Prolonged NLRP3 inflammasome activation by inducing mitophagy | sesn2−/− mice, sesn2−/− GFPM -AP1-LC3 mice, nos2−/− mice, nlrp3−/− mice, BMDM | [177] |
| Palmatine | Ulcerative colitis | ↓ Pathological and histological scores in DSS mice · ↓ Colonic inflammation in DSS mice | ↓ NLRP3 inflammasomes activation in DSS mice and THP-1 ↑ PINK1 and Parkin-driven mitophagy in DSS mice and THP-1 | DSS-induced UC model using C57BL/6 mice, THP-1 cell | [178] |
| Ginsenoside Rd | Ulcerative colitis | ↓ Body weight loss, stool consistency alterations and blood loss in DSS mice ↓ Recruitment of inflammatory cell into colonic tissue ↓ NLRP3 inflammasome activation in colon tissue and THP-1 cells | ↓ LPS plus ATP-induced mitochondrial perturbation and mtDNA release in THP-1 ↑ p62 expression and its translocation to mitochondria ↑ p62-mediated autophagy activation ↑ AMPK/ULK1signaling pathway in vitro and in vivo | DSS-induced UC model using BALB/C mice, THP-1 cell, Colonic cell suspension | [179] |
| Pramipexole | Sepsis | ↑ Survival rate in septic pink1-/- and park2-/- mice ↓ Inflammasome-dependent cytokines (IL-1β and HMGB1) in septic pink1-/- and park2-/- mice | ↑ Circulating dopamine levels in septic pink1-/- and park2-/- mice ↓ Circulating lactate levels in septic pink1-/- and park2-/- mice ↓ Sepsis-induced Hif1α, Ldha, and Pdk1 as well as GSH depletion in the brain from pink1-/- and park2-/- mice ↓ Hif1α-mediated aerobic glycolysis involved in inflammasome activation in pink1-/- and park2-/- mice | CLP sepsis in pink1-/-, park2-/-, nlrp3-/-, hif1α-/- mice | [167] |
| Andrographolide | Colitis-associated cancer | ↓ Tumorigenesis and inflammation in a colitis-associated colorectal cancer model ↓ Progression of DSS-induced experimental colitis in mice | ↓ NLRP3 inflammasome activation in LPS-primed macrophages treated with ATP or in mice with DSS-induced colitis ↓ ATP-induced collapse of the mitochondria membrane potential and mitochondria fragmentation in LPS-primed macrophages ↑ Mitophagy induction in macrophages | THP-1 cell, Peritoneal macrophages, BMDM, In vivo CAC mice models | [180] |
| Resveratrol | Peritoneal mesothelial cells inflammatory injury | ↑ Protection from ROS-NLRP3-mediated inflammatory injury | ↑ Mitophagy/autophagy via AMPK activation | Human peritoneal mesothelial cell line (HMrSV5) | [181] |
| Honokiol | Behavioral and cognitive impairment | ↑ Cognitive recovery | ↑ Mitophagy ↓ mtROS NLRP3 and microglia activation in the hippocampus ↓ Neuronal apoptosis | Surgery/anesthesia (sevoflurane) induced cognitive impairment in mice | [182] |
PBMC: Peripheral blood mononuclear cell; AMPK: AMP-activated protein kinase; NLRP3: NLR Family Pyrin Domain Containing 3; PINK1: PTEN Induced Kinase 1; MFN2: Mitofusin 2; LPS: Lipopolysaccharide; ROS: Reactive oxygen species; BMDM: Bone marrow-derived macrophages; HepG2: Human hepatocellular carcinoma cell line; PA: Palmitic acid; OPTN: Optineurin; NIX: NIP3-like protein X; ox-LDL: oxidized low-density lipoprotein; CCH: Chronic cerebral hypoperfusion; BCCAo: Bilateral common carotid artery occlusion; AAV: Adeno-associated virus; MCAO: middle cerebral artery occlusion-reperfusion; SAH: Subarachnoid hemorrhage; Atg: Autophagy-related gene; SESN2: Sestrin 2; CLP: Cecal ligation and puncture; SQSTM1: sequestosome 1; ULK1: Unc-51 like kinase 1; UC: Ulcerative colitis; HMGB1: High mobility group box 1; DSS: Dextran sulfate sodium; Andro: Andrographolide; CAC: Colitis-associated cancer; mtROS: mitochondrial reactive oxygen species; ↑, increase; ↓, decrease.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yuk, J.-M.; Silwal, P.; Jo, E.-K. Inflammasome and Mitophagy Connection in Health and Disease. Int. J. Mol. Sci. 2020, 21, 4714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134714
AMA Style
Yuk J-M, Silwal P, Jo E-K. Inflammasome and Mitophagy Connection in Health and Disease. International Journal of Molecular Sciences. 2020; 21(13):4714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134714
Chicago/Turabian StyleYuk, Jae-Min, Prashanta Silwal, and Eun-Kyeong Jo. 2020. "Inflammasome and Mitophagy Connection in Health and Disease" International Journal of Molecular Sciences 21, no. 13: 4714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21134714
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.