Intracellular Toxic Advanced Glycation End-Products Promote the Production of Reactive Oxygen Species in HepG2 Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
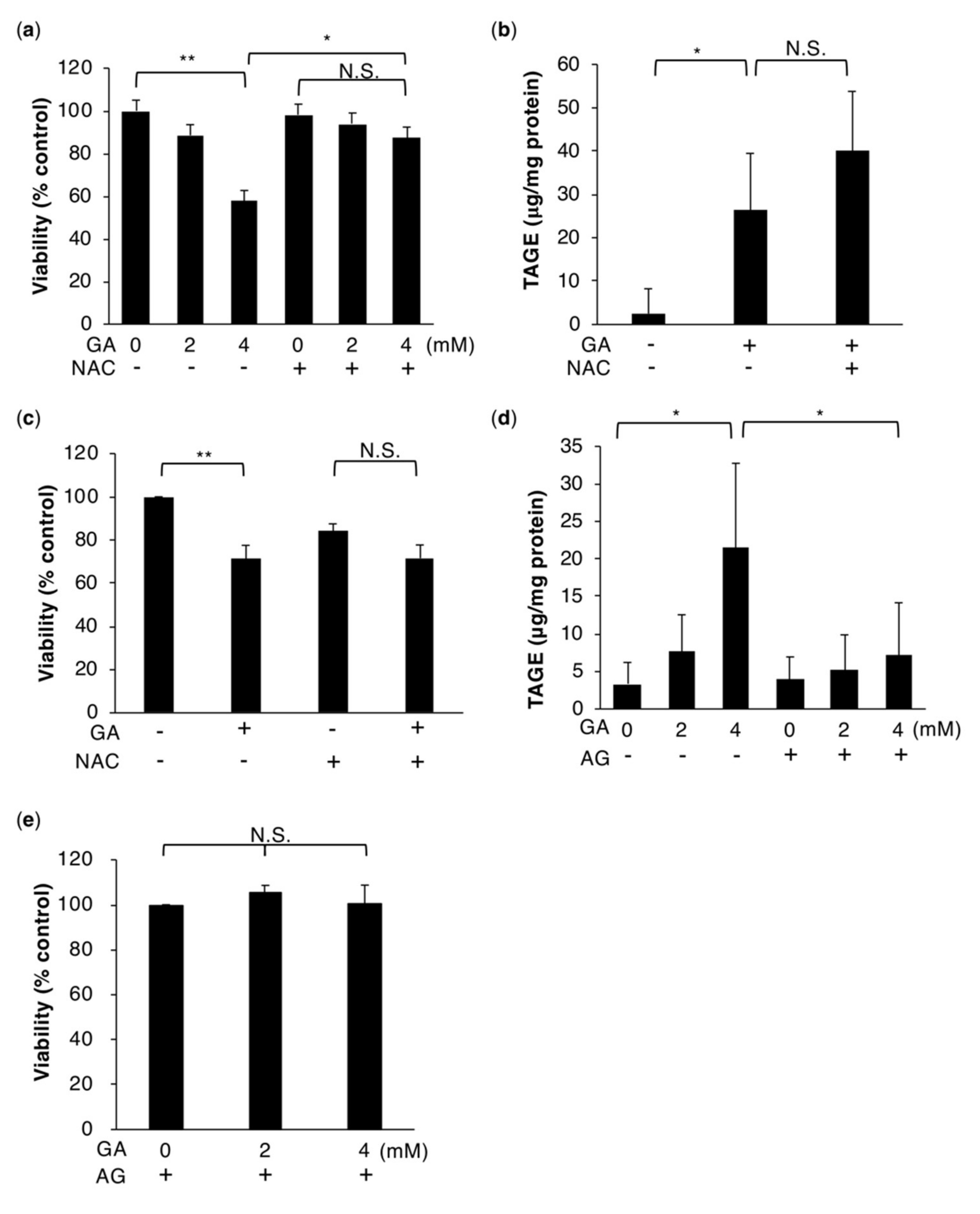
2.1. HepG2 Cell Death Associated with the Intracellular Accumulation of TAGE Is Rescued by NAC
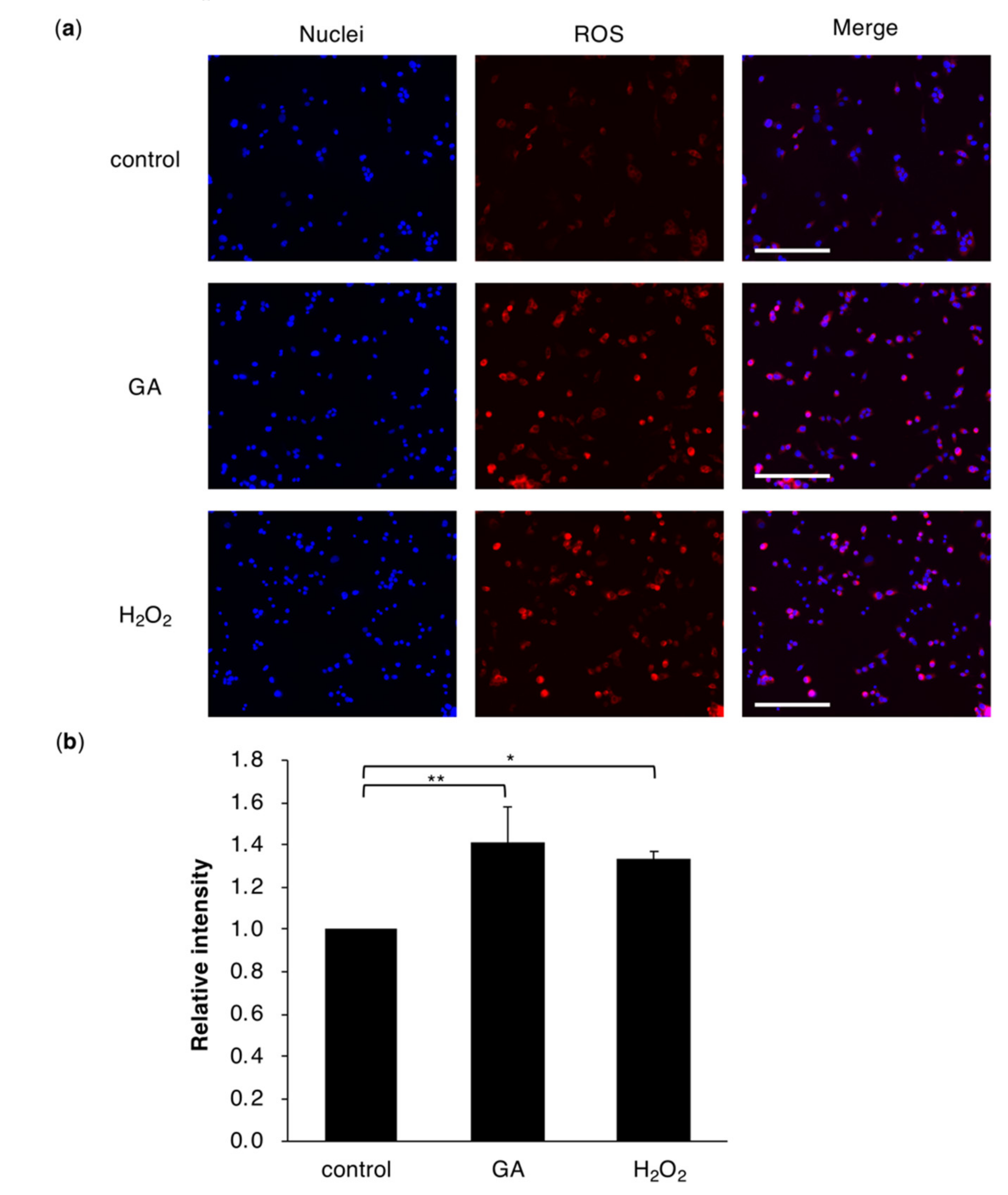
2.2. Intracellular Accumulation of TAGE Induces ROS Production in Hepatocyte Cells
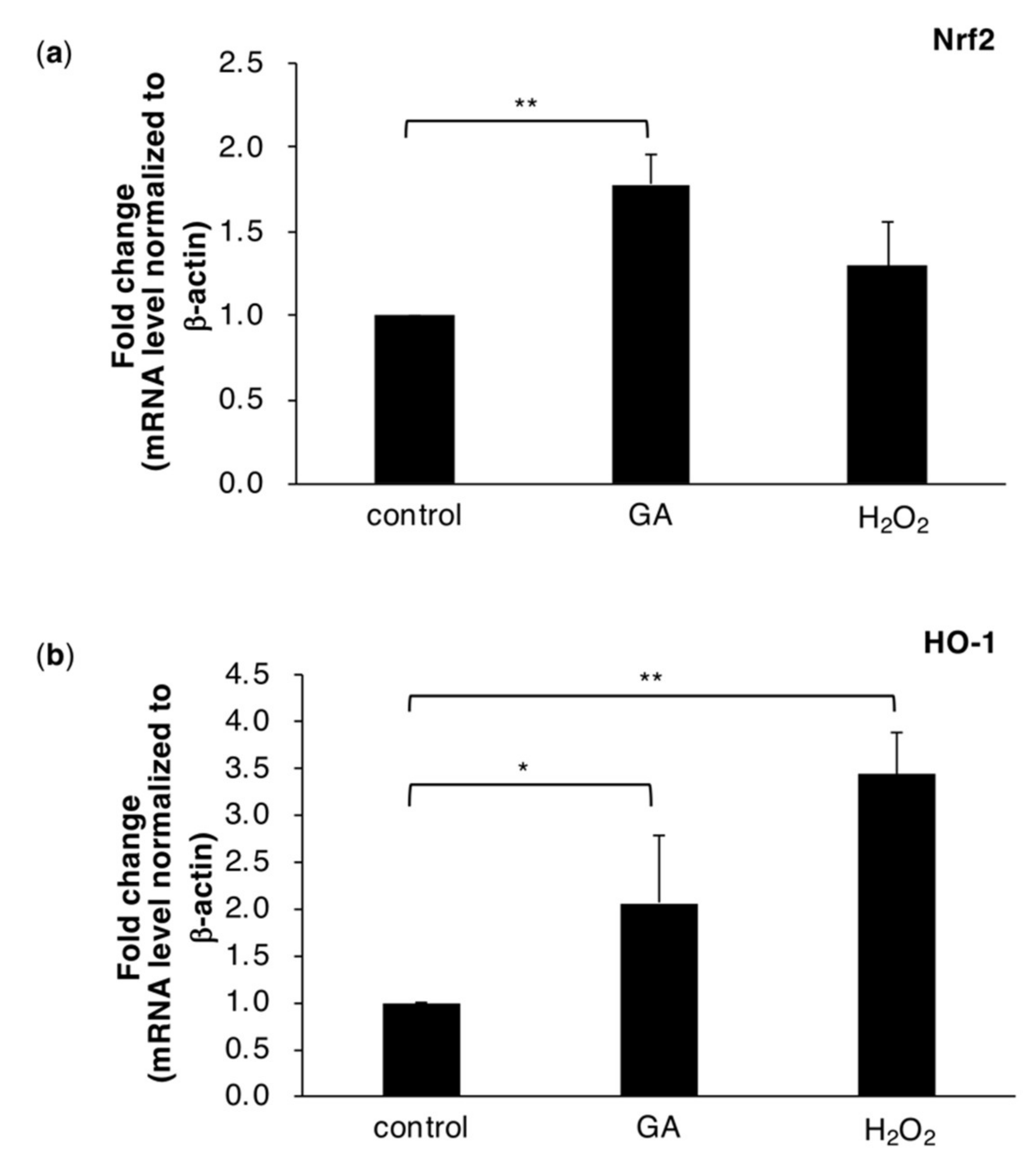
2.3. The GA Treatment Triggers Intracellular ROS Stress Responses
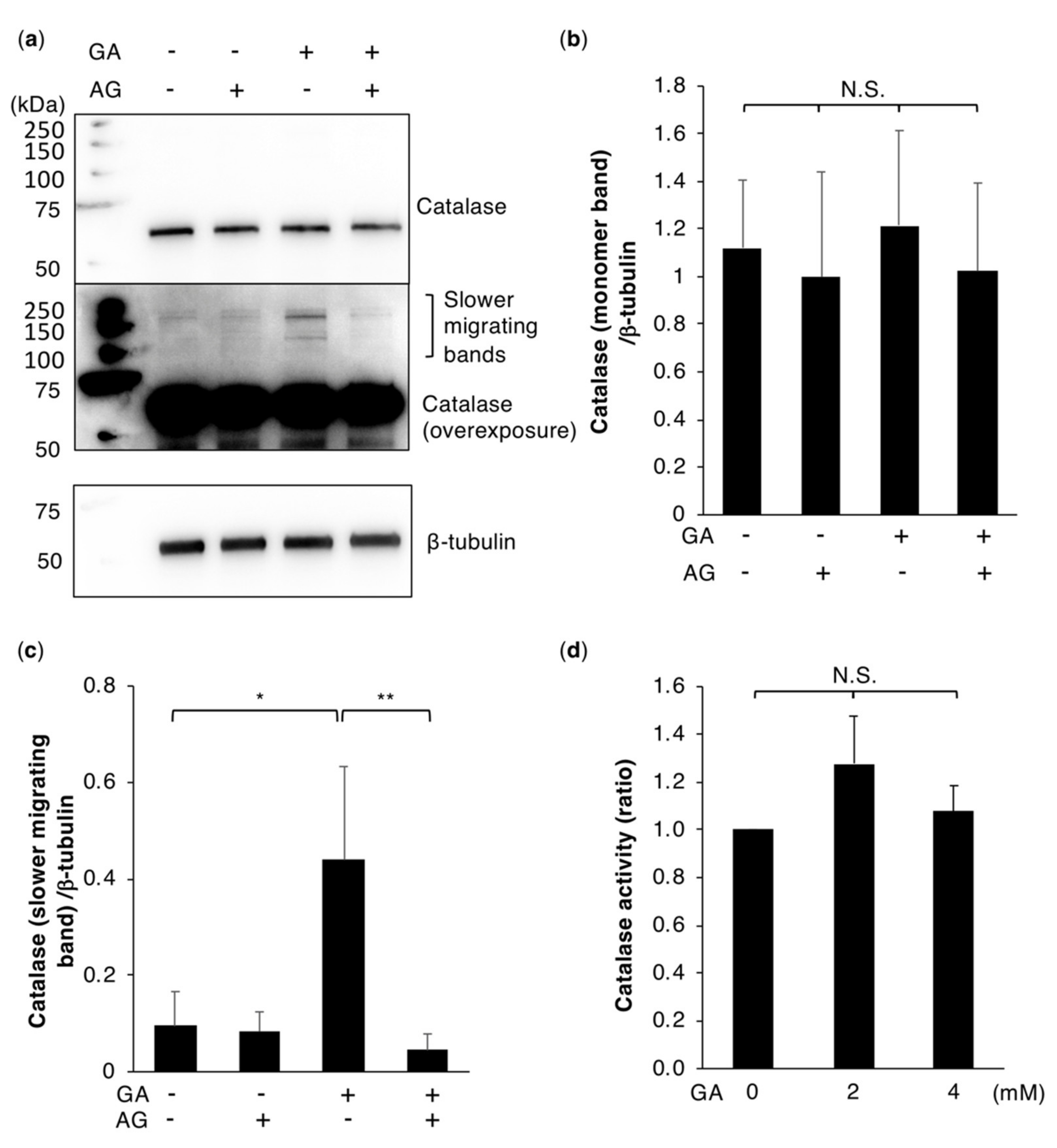
2.4. Catalase Dysfunction is Not Responsible for GA-Induced ROS Production
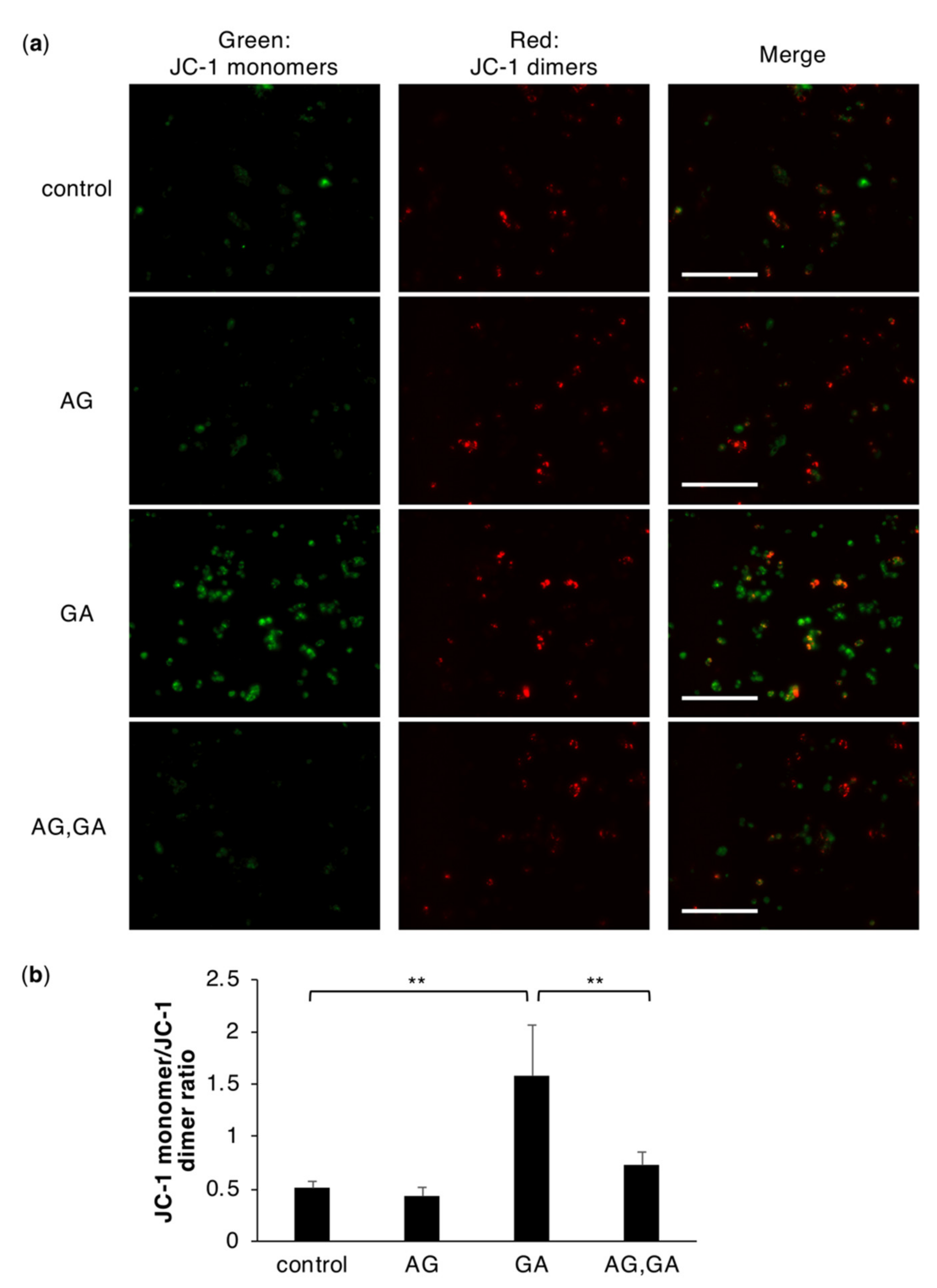
2.5. The GA Treatment Induces an Abnormal Mitochondrial Membrane Potential
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Cell Viability
4.4. Slot Blotting
4.5. ROS Detection and Measurement.
4.6. RNA Extraction and Quantitative Real-Time Reverse Transcription–PCR (qRT-PCR)
4.7. Western Blotting
4.8. Measurement of Catalase Activity
4.9. Analysis of the Mitochondrial Membrane Potential
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AG | Aminoguanidine |
| AGEs | Advanced glycation end-products |
| CRP | C-reactive protein |
| GA | Glyceraldehyde |
| HClO | Hypochlorous acid |
| HFCS | High fructose corn syrup |
| HO˙ | Hydroxyl radical |
| H2O2 | Hydrogen peroxide |
| MetS | Metabolic syndrome |
| NAC | N-acetyl-L-cysteine |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| RAGE | Receptor for AGEs |
| ROS | Reactive oxygen species |
| TAGE | Toxic AGEs |
| WB | Western blotting |
References
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism 2016, 65, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Hyogo, H.; Yamagishi, S.I.; Iwamoto, K.; Arihiro, K.; Takeuchi, M.; Sato, T.; Ochi, H.; Nonaka, M.; Nabeshima, Y.; Inoue, M.; et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2007, 22, 1112–1119. [Google Scholar] [CrossRef]
- Jinno, M.; Takeuchi, M.; Watanabe, A.; Teruya, K.; Hirohama, J.; Eguchi, N.; Miyazaki, A. Advanced glycation end-products accumulation compromises embryonic development and achievement of pregnancy by assisted reproductive technology. Hum. Reprod. 2011, 26, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.Y.; Takeuchi, M.; Hyogo, H.; Mckeown-eyssen, G.; Yamagishi, S.; Chayama, K.; Brien, P.J.O.; Ferrari, P.; Overvad, K.; Olsen, A.; et al. The association between glyceraldehyde-derived advanced glycation end-products and colorectal cancer risk. Cancer Epidem. Biomaker. Prev. 2015, 24, 1855–1863. [Google Scholar] [CrossRef] [Green Version]
- Choei, H.; Sasaki, N.; Takeuchi, M.; Yoshida, T.; Ukai, W.; Yamagishi, S.I.; Kikuchi, S.; Saito, T. Glyceraldehyde-derived advanced glycation end products in Alzheimer’s disease. Acta Neuropathol. 2004, 108, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Koriyama, Y.; Furukawa, A.; Muramatsu, M.; Takino, J.; Takeuchi, M. Glyceraldehyde caused Alzheimer’s disease-like alterations in diagnostic marker levels in SH-SY5Y human neuroblastoma cells. Sci. Rep. 2015, 5, 13313. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Ohnuma, T.; Takeuchi, M.; Katsuta, N.; Maeshima, H.; Takebayashi, Y.; Higa, M.; Nakamura, T.; Nishimon, S.; Sannohe, T.; et al. Altered serum glyceraldehyde-derived advanced glycation end product (AGE) and soluble AGE receptor levels indicate carbonyl stress in patients with schizophrenia. Neurosci. Lett. 2015, 593, 51–55. [Google Scholar] [CrossRef]
- Tahara, N.; Yamagishi, S.I.; Takeuchi, M.; Honda, A.; Tahara, A.; Nitta, Y.; Kodama, N.; Mizoguchi, M.; Kaida, H.; Ishibashi, M.; et al. Positive association between serum level of glyceraldehyde-derived advanced glycation end products and vascular inflammation evaluated by [18F]fluorodeoxyglucose positron emission tomography. Diabetes Care 2012, 35, 2618–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, S.; Yamagishi, S.; Matsui, T.; Noda, Y.; Ueda, S.; Jinnouchi, Y.; Sasaki, K.; Takeuchi, M.; Imaizumi, T. Serum levels of advanced glycation end products (AGEs) are inversely associated with the number and migratory activity of circulating endothelial progenitor cells in apparently healthy subjects. Cardiovasc. Ther. 2012, 30, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, M.; Nakashima, A.; Fujimura, N.; Maruhashi, T.; Iwamoto, Y.; Iwamoto, A.; Matsumoto, T.; Oda, N.; Hidaka, T.; Kihara, Y.; et al. Ratio of serum levels of AGEs to soluble form of RAGE is a predictor of endothelial function. Diabetes Care 2015, 38, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, M.; Oyama, J.; Takeuchi, M.; Shibata, Y.; Yamamoto, Y.; Kawasaki, T.; Komoda, H.; Kodama, K.; Sakuma, M.; Toyoda, S.; et al. Acute effects of statin on reduction of angiopoietin-like 2 and glyceraldehyde-derived advanced glycation end-products levels in patients with acute myocardial infarction: A message from SAMIT (Statin for Acute Myocardial Infarction Trial). Heart Vessel. 2016, 31, 1583–1589. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Ueda, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products in cardiomyocytes may cause cardiovascular disease. Sci. Rep. 2019, 9, 2121. [Google Scholar] [CrossRef] [Green Version]
- Takino, J.; Kobayashi, Y.; Takeuchi, M. The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J. Gastroenterol. 2010, 45, 646–655. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.; Takeuchi, M. Impact of intracellular glyceraldehyde-derived advanced glycation end-products on human hepatocyte cell death. Sci. Rep. 2017, 7, 14282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, S.; Foiles, P.; Founds, H. Therapeutic potential of breakers of advanced glycation end product-protein crosslinks. Arch. Biochem. Biophys. 2003, 419, 89–96. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.; Takeuchi, M. The relevance of toxic AGEs (TAGE) cytotoxicity to NASH pathogenesis: A mini-review. Nutrients 2019, 11, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Ueda, T.; Takino, J.; Tsutsumi, M.; Hyogo, H.; Yamagishi, S. Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker in development and progression of NASH. Med. Hypotheses 2015, 84, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Sakasai-Sakai, A.; Takata, T.; Tsutsumi, M. Toxic AGE (TAGE) theory for the pathophysiology of the onset/progression of NAFLD and ALD. Nutrients 2017, 9, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumida, Y.; Nakashima, T.; Yoh, T.; Furutani, M.; Hirohama, A.; Kakisaka, Y.; Nakajima, Y.; Ishikawa, H.; Mitsuyoshi, H.; Okanoue, T.; et al. Serum thioredoxin levels as a predictor of steatohepatitis in patients with nonalcoholic fatty liver disease. J. Hepatol. 2003, 38, 32–38. [Google Scholar] [CrossRef]
- Sumida, Y.; Niki, E.; Naito, Y.; Yoshikawa, T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic. Res. 2013, 47, 869–880. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid. Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef]
- Vasileva, L.V.; Savova, M.S.; Amirova, K.M.; Dinkova-Kostova, A.T.; Georgiev, M.I. Obesity and NRF2-mediated cytoprotection: Where is the missing link? Pharmacol. Res. 2020, 156, 104760. [Google Scholar] [CrossRef]
- Wang, C.; Cui, Y.; Li, C.; Zhang, Y.; Xu, S.; Li, X.; Li, H.; Zhang, X. Nrf2 deletion causes “benign” simple steatosis to develop into nonalcoholic steatohepatitis in mice fed a high-fat diet. Lipids Health Dis. 2013, 12, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Malaguarnera, L.; Madeddu, R.; Palio, E.; Arena, N.; Malaguarnera, M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J. Hepatol. 2005, 42, 585–591. [Google Scholar] [CrossRef]
- Yoshida, T.; Yamagishi, S.; Nakamura, K.; Matsui, T.; Imaizumi, T.; Takeuchi, M.; Ueno, T.; Sata, M. Pigment epithelium-derived factor (PEDF) inhibits advanced glycation end product (AGE)-induced C-reactive protein expression in hepatoma cells by suppressing Rac-1 activation. FEBS Lett. 2006, 580, 2788–2796. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, K.; Kanno, K.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Tazuma, S.; Chayama, K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J. Gastroenterol. 2008, 43, 298–304. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quiñones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.Y.; Sun, L.; Chen, X.P.; Zhang, D.S. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Brennan, J.P.; Southworth, R.; Medina, R.A.; Davidson, S.M.; Duchen, M.R.; Shattock, M.J. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc. Res. 2006, 72, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, D.H.; Forbes, J.M.; Angus, P.W.; Herath, C.B. Development and progression of non-alcoholic fatty liver disease: The role of advanced glycation end products. Int. J. Mol. Sci. 2019, 20, 5037. [Google Scholar] [CrossRef] [Green Version]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J. Gastroenterol. 2012, 18, 1781–1788. [Google Scholar] [CrossRef]
- Kawasaki, T.; Igarashi, K.; Koeda, T.; Sugimoto, K.; Nakagawa, K.; Hayashi, S.; Yamaji, R.; Inui, H.; Fukusato, T.; Yamanouchi, T. Rats fed fructose-enriched diets have characteristics of nonalcoholic hepatic steatosis. J. Nutr. 2009, 139, 2067–2071. [Google Scholar] [CrossRef] [Green Version]
- Takino, J.; Nagamine, K.; Takeuchi, M.; Hori, T. In vitro identification of nonalcoholic fatty liver disease-related protein hnRNPM. World J. Gastroenterol. 2015, 21, 1784–1793. [Google Scholar] [CrossRef]
- Thong-Ngam, D.; Samuhasaneeto, S.; Kulaputana, O.; Klaikeaw, N. N-acetylcysteine attenuates oxidative stress and liver pathology in rats with non-alcoholic steatohepatitis. World J. Gastroenterol. 2007, 13, 5127–5132. [Google Scholar] [CrossRef] [Green Version]
- De Andrade, K.Q.; Moura, F.A.; Dos Santos, J.M.; De Araújo, O.R.P.; de Farias Santos, J.C.; Goulart, M.O.F. Oxidative stress and inflammation in hepatic diseases: Therapeutic possibilities of N-acetylcysteine. Int. J. Mol. Sci. 2015, 16, 30269–30308. [Google Scholar] [CrossRef]
- Bataille, A.M.; Manautou, J.E. Nrf2 a potential target for new therapeutics in liver disease. Clin. Pharmacol. Ther. 2012, 92, 340–348. [Google Scholar] [CrossRef]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.J.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, J.; Pang, X.; Wang, S.; Wu, D.; Zhang, X.; Feng, L. Angiotensin II induces C-reactive protein expression via AT1-ROS-MAPK-NF-κB signal pathway in hepatocytes. Cell. Physiol. Biochem. 2013, 32, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the mitochondrial membrane potential using the cationic JC-1 dye as a sensitive fluorescent probe. Bio-Protocol 2019, 9, 3128. [Google Scholar] [CrossRef]
- Takahashi, H.; Tran, P.O.; LeRoy, E.; Harmon, J.S.; Tanaka, Y.; Robertson, R.P. D-Glyceraldehyde causes production of intracellular peroxide in pancreatic islets, oxidative stress, and defective beta cell function via non-mitochondrial pathways. J. Biol. Chem. 2004, 279, 37316–37323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, M.; Makita, Z.; Bucala, R.; Suzuki, T.; Koike, T.; Kameda, Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol. Med. 2000, 6, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Iwase, T.; Tajima, A.; Sugimoto, S.; Okuda, K.I.; Hironaka, I.; Kamata, Y.; Takada, K.; Mizunoe, Y. A simple assay for measuring catalase activity: A visual approach. Sci. Rep. 2013, 3, 3081. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. Intracellular Toxic Advanced Glycation End-Products Promote the Production of Reactive Oxygen Species in HepG2 Cells. Int. J. Mol. Sci. 2020, 21, 4861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144861
Sakasai-Sakai A, Takata T, Takeuchi M. Intracellular Toxic Advanced Glycation End-Products Promote the Production of Reactive Oxygen Species in HepG2 Cells. International Journal of Molecular Sciences. 2020; 21(14):4861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144861
Chicago/Turabian StyleSakasai-Sakai, Akiko, Takanobu Takata, and Masayoshi Takeuchi. 2020. "Intracellular Toxic Advanced Glycation End-Products Promote the Production of Reactive Oxygen Species in HepG2 Cells" International Journal of Molecular Sciences 21, no. 14: 4861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144861