Approaching Sex Differences in Cardiovascular Non-Coding RNA Research

 ,
,  , , , , ,
, , , , ,  ,
,  and
on behalf of the EU-CardioRNA COST Action
and
on behalf of the EU-CardioRNA COST Action
Abstract
:1. Introduction
2. Sex-Differences in Cardiovascular Susceptibility
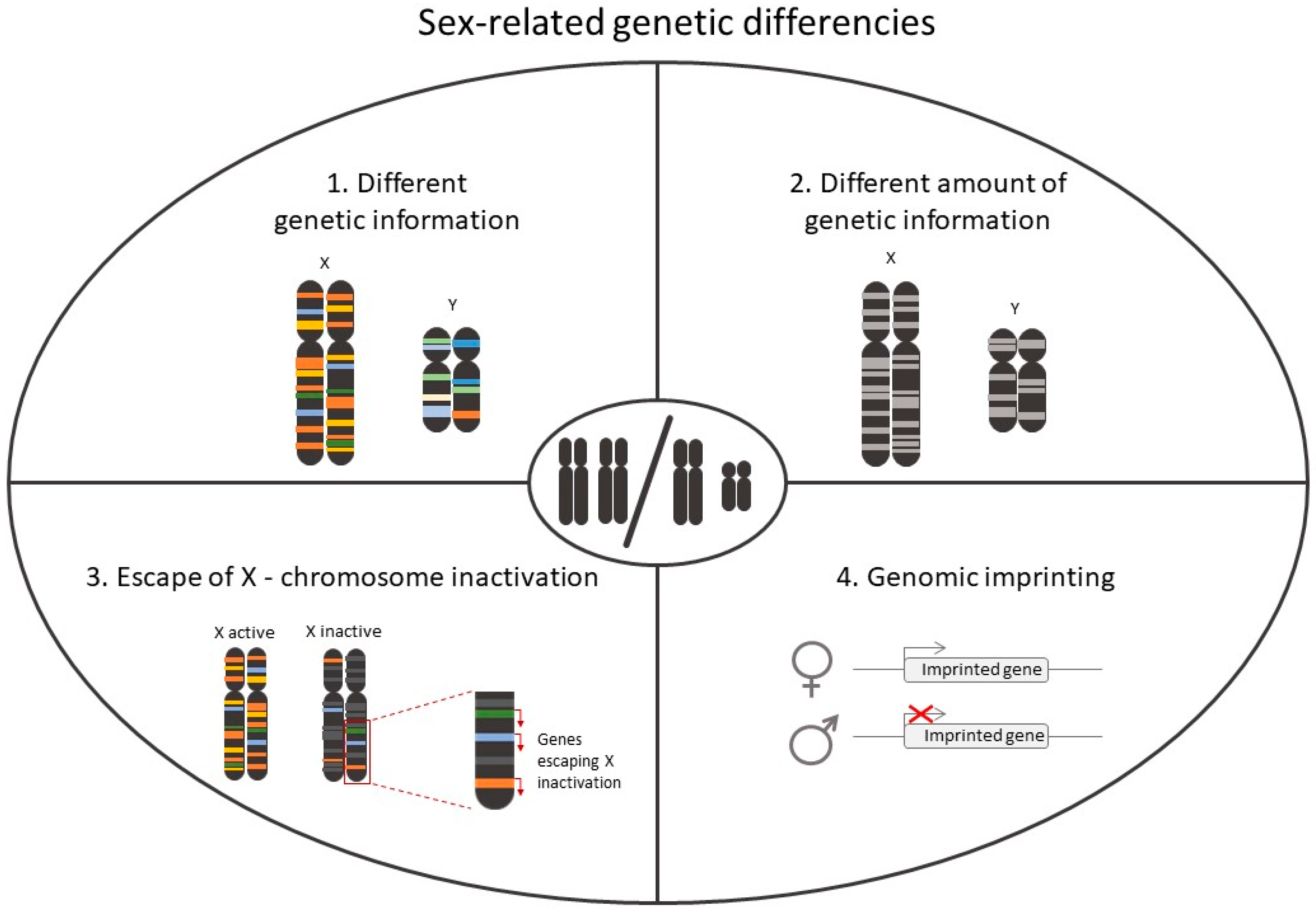
2.1. Role of Chromosomes in Sex-Differences in CVD
2.2. Role of Hormonal Status in Sex-Differences in CVD
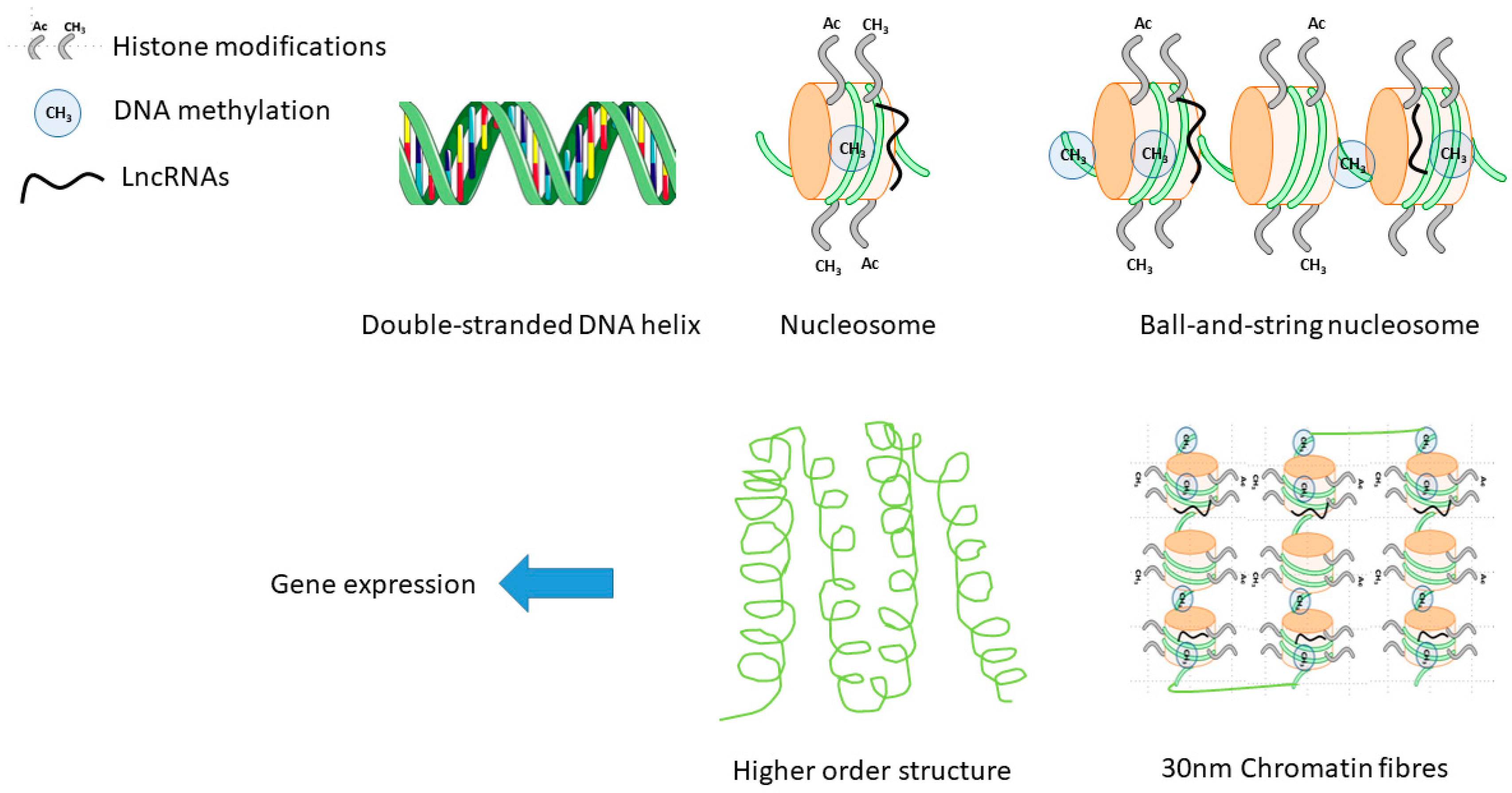
2.3. Epigenetics in Sex-Differences in CVD
2.3.1. DNA Methylation
2.3.2. Histone Modifications
2.3.3. RNA Modifications and Sex Differences
2.3.4. X Chromosome Reactivation
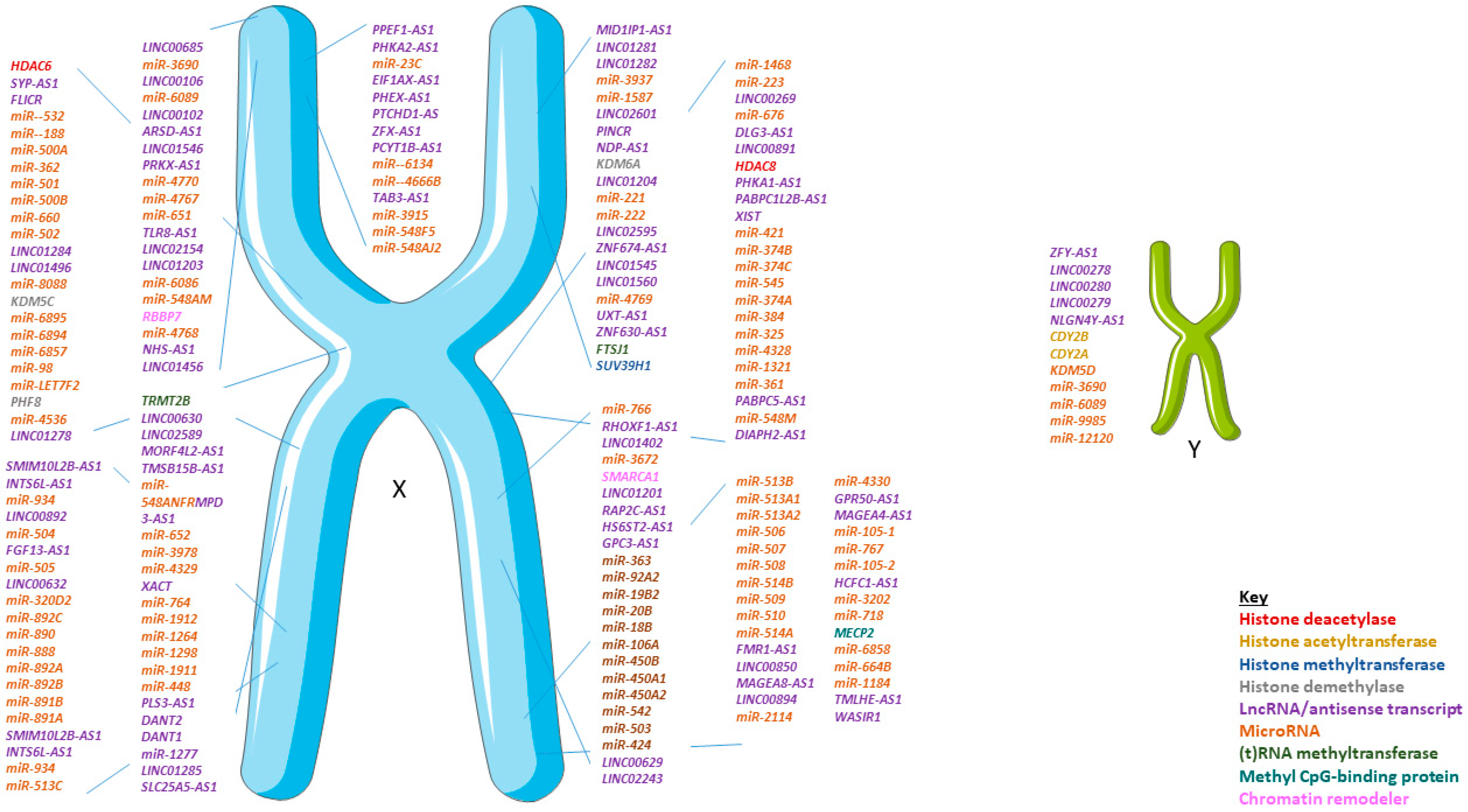
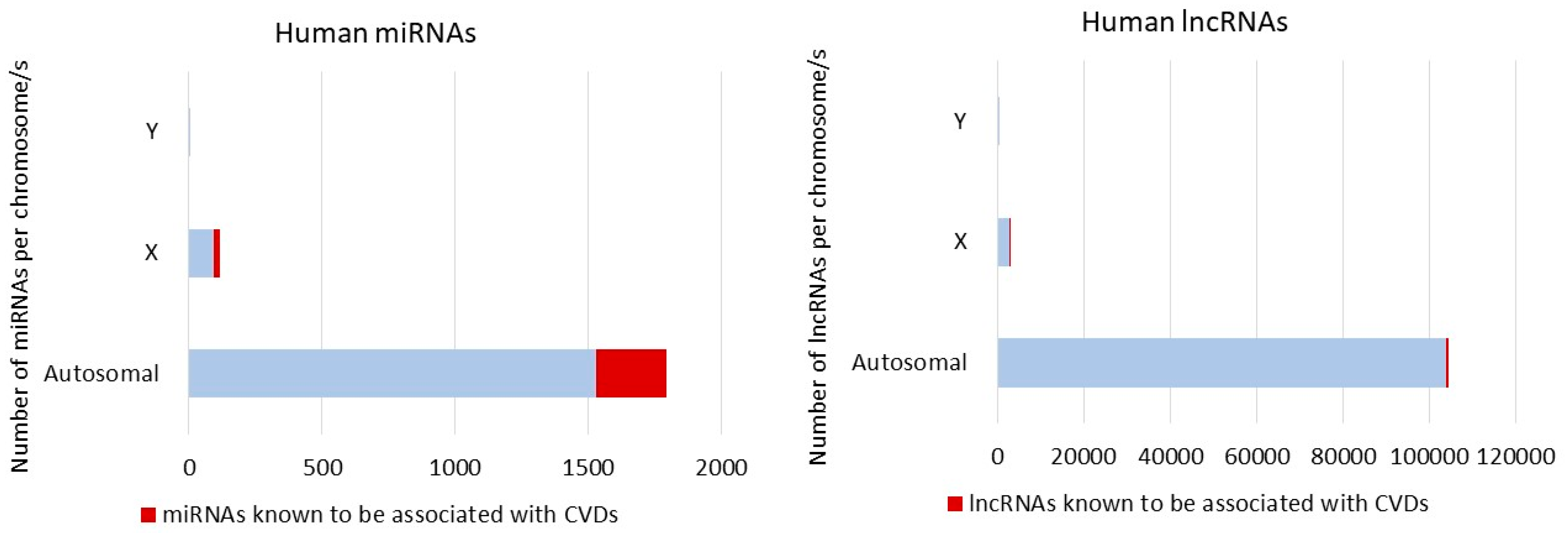
3. Non-Coding RNA and Sex Differences
4. Experimental Models to Study Sex-Related ncRNA Differences in CVD
4.1. Including Both Sexes in CVD Models Will Refine RNA Research
4.2. Animal Models in Translational Cardiovascular Research
4.3. Need for Adequate Experimental Models
5. Mouse and Cell Experimental Models to Address Sex Differences in Epigenetics
5.1. Mouse Models
The “Four Core Genotypes” Model
5.2. Vascular Cell Cultures
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | Androgen receptor |
| ARVM | Adult rat ventricular myocytes |
| CVD | Cardiovascular disease |
| eNOS | Endothelial nitric oxide |
| ER | Estrogen receptor |
| FCG | Four core genotype |
| FSH | Follicle stimulating hormone |
| HDAC | Histone deacetylase |
| HRT | Hormone replacement therapy |
| Kb | kilobase |
| LncRNA | Long non-coding RNA |
| MI | Myocardial infarction |
| miRNA/miR | MicroRNA |
| ncRNA | Non-coding RNA |
| ORX | Orchiectomy |
| OVX | Ovariectomy |
| RNA-seq | RNA-sequencing |
| SERM | Selective estrogen receptor modulators |
| SRY | Sex determining region Y |
| XCI | X chromosome inactivation |
References
- Garcia, M.; Mulvagh, S.L.; Merz, C.N.B.; Buring, J.E.; Manson, J.E. Cardiovascular Disease in Women: Clinical Perspectives. Circ. Res. 2016, 118, 1273–1293. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, M.C.; Zekavat, S.M.; Aragam, K.; Finneran, P.; Klarin, D.; Bhatt, D.L.; Januzzi, J.L.; Scott, N.S.; Natarajan, P. Association of Premature Natural and Surgical Menopause With Incident Cardiovascular Disease. JAMA 2019, 322, 2411. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-H.; Vemuganti, R. Effect of Sex and Age Interactions on Functional Outcome after Stroke. CNS Neurosci. Ther. 2014, 21, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.P.; Cassis, L.A.; Eghbali, M.; Reue, K.; Sandberg, K. Sex Hormones and Sex Chromosomes Cause Sex Differences in the Development of Cardiovascular Diseases. Arter. Thromb. Vasc. Biol. 2017, 37, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannenbaum, C.; Ellis, R.P.; Eyssel, F.; Zou, J.; Schiebinger, L. Sex and gender analysis improves science and engineering. Nature 2019, 575, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, J.A.; Collins, F.S. Policy: NIH to balance sex in cell and animal studies. Nature 2014, 509, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic Modifications. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Robinson, E.L.; Gomes, C.P.D.C.; Potočnjak, I.; Hellemans, J.; Betsou, F.; De Gonzalo-Calvo, D.; Stoll, M.; Yilmaz, M.B.; Ágg, B.; Beis, D.; et al. A Year in the Life of the EU-CardioRNA COST Action: CA17129 Catalysing Transcriptomics Research in Cardiovascular Disease. Non-Coding RNA 2020, 6, 17. [Google Scholar] [CrossRef]
- Khramtsova, E.A.; Davis, L.K.; Stranger, B.E. Author Correction: The role of sex in the genomics of human complex traits. Nat. Rev. Genet. 2019, 20, 494. [Google Scholar] [CrossRef]
- Accounting for Sex in the Genome. Available online: https://0-www-nature-com.brum.beds.ac.uk/articles/nm.4445 (accessed on 10 June 2020).
- Orlowska-Baranowska, E.; Gora, J.; Baranowski, R.; Stoklosa, P.; Betka, L.G.V.; Pędzich-Placha, E.; Milkowska, M.; Koblowska, M.; Hryniewiecki, T.; Gaciong, Z.; et al. Association of the Common Genetic Polymorphisms and Haplotypes of the Chymase Gene with Left Ventricular Mass in Male Patients with Symptomatic Aortic Stenosis. PLoS ONE 2014, 9, e96306. [Google Scholar] [CrossRef]
- Urata, H.; Kinoshita, A.; Misono, K.S.; Bumpus, F.M.; Husain, A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J. Biol. Chem. 1990, 265, 22348–22357. [Google Scholar] [PubMed]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reckelhoff, J.F. Gender differences in hypertension. Curr. Opin. Nephrol. Hypertens. 2018, 27, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H.; Maki, K.C.; Karp, S.K.; Ingram, K.A. Management of hypercholesterolaemia in postmenopausal women. Drugs Aging 2002, 19, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Ely, D.L.; Turner, M.E. Hypertension in the spontaneously hypertensive rat is linked to the Y chromosome. Hypertension 1990, 16, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Ely, D.; Turner, M.; Milsted, A. Review of the Y chromosome and hypertension. Braz. J. Med Biol. Res. 2000, 33, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Charchar, F.J.; Bloomer, L.D.; A Barnes, T.; Cowley, M.J.; Nelson, C.P.; Wang, Y.; Denniff, M.; Debiec, R.; Christofidou, P.; Nankervis, S.; et al. Inheritance of coronary artery disease in men: An analysis of the role of the Y chromosome. Lancet 2012, 379, 915–922. [Google Scholar] [CrossRef] [Green Version]
- Suto, J.-I.; Satou, K. Effect of the Y chromosome on plasma high-density lipoprotein-cholesterol levels in Y-chromosome-consomic mouse strains. BMC Res. Notes 2014, 7, 393. [Google Scholar] [CrossRef] [Green Version]
- Link, J.C.; Chen, X.; Prien, C.; Borja, M.S.; Hammerson, B.; Oda, M.N.; Arnold, A.P.; Reue, K. Increased high-density lipoprotein cholesterol levels in mice with XX versus XY sex chromosomes. Arter. Thromb. Vasc. Biol. 2015, 35, 1778–1786. [Google Scholar] [CrossRef] [Green Version]
- Severino, P.; D’Amato, A.; Netti, L.; Pucci, M.; Mariani, M.V.; Cimino, S.; I Birtolo, L.; Infusino, F.; De Orchi, P.; Palmirotta, R.; et al. Susceptibility to ischaemic heart disease: Focusing on genetic variants for ATP-sensitive potassium channel beyond traditional risk factors. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef]
- Severino, P.; D’Amato, A.; Pucci, M.; Infusino, F.; Birtolo, L.I.; Mariani, M.; LaValle, C.; Maestrini, V.; Mancone, M.; Fedele, F. Ischemic Heart Disease and Heart Failure: Role of Coronary Ion Channels. Int. J. Mol. Sci. 2020, 21, 3167. [Google Scholar] [CrossRef] [PubMed]
- Stone, G.; Choi, A.; Meritxell, O.; Gorham, J.; Heydarpour, M.; Seidman, C.E.; Seidman, J.G.; Aranki, S.F.; Body, S.C.; Carey, V.J.; et al. Sex differences in gene expression in response to ischemia in the human left ventricular myocardium. Hum. Mol. Genet. 2019, 28, 1682–1693. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V.; Lehmkuhl, E.; Lehmkuhl, H.B.; Hetzer, R. Gender aspects in heart failure. Pathophysiology and medical therapy. Arch. Mal. Coeur Vaiss. 2004, 97, 899–908. [Google Scholar]
- Heidecker, B.; Kittleson, M.M.; Kasper, E.K.; Wittstein, I.S.; Champion, H.C.; Russell, S.D.; Baughman, K.L.; Hare, J.M. Transcriptomic Analysis Identifies the Effect of Beta-Blocking Agents on a Molecular Pathway of Contraction in the Heart and Predicts Response to Therapy. JACC Basic Transl. Sci. 2016, 1, 107–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.A.; Korach, K.S. Estrogen receptors and human disease: An update. Arch. Toxicol. 2012, 86, 1491–1504. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, M.E.; Yamamoto, Y.; Brady, M.P.; Lu, Z.P.; Maziasz, P.J.; Liu, C.T.; Pint, B.A.; More, K.L.; Meyer, H.M.; Payzant, E.A. Molecular and Cellular Basis of Cardiovascular Gender Differences. Science 2005, 308, 1583–1587. [Google Scholar] [CrossRef] [Green Version]
- Vrtacnik, P.; Ostanek, B.; Mencej-Bedrac, S.; Marc, J. The many faces of estrogen signaling. Biochem. Medica 2014, 24, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Mikkola, T.; Tuomikoski, P.; Lyytinen, H.; Korhonen, P.; Hoti, F.; Vattulainen, P.; Gissler, M.; Ylikorkala, O. Estradiol-based postmenopausal hormone therapy and risk of cardiovascular and all-cause mortality. Menopause 2015, 22, 1–983. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Prentice, R.L.; Manson, J.E.; Wu, L.; Barad, D.; Barnabei, V.M.; Ko, M.; LaCroix, A.Z.; Margolis, K.L.; Stefanick, M.L. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007, 297, 1465–1477. [Google Scholar] [CrossRef]
- Lobo, R.A. Surgical menopause and cardiovascular risks. Menopause 2007, 14, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Novella, S.; Dantas, A.P.; Segarra, G.; Medina, P.; Hermenegildo, C. Vascular Aging in Women: Is Estrogen the Fountain of Youth? Front. Physiol. 2012, 3, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarkson, T.B.; Meléndez, G.C.; Appt, S.E. Timing hypothesis for postmenopausal hormone therapy: Its origin, current status, and future. Menopause 2013, 20, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Azodi, M.; Kamps, R.; Heymans, S.; Robinson, E.L. The Missing “lnc” between Genetics and Cardiac Disease. Non-Coding RNA 2020, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Jjingo, D.; Conley, A.B.; Yi, S.V.; Lunyak, V.; Jordan, I.K. On the presence and role of human gene-body DNA methylation. Oncotarget 2012, 3, 462–474. [Google Scholar] [CrossRef] [Green Version]
- Haas, J.; Frese, K.S.; Park, Y.J.; Keller, A.; Vogel, B.; Lindroth, A.M.; Weichenhan, D.; Franke, J.; Fischer, S.; Bauer, A.; et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med. 2013, 5, 413–429. [Google Scholar] [CrossRef]
- Movassagh, M.; Choy, M.-K.; Goddard, M.; Bennett, M.R.; Down, T.A.; Foo, R.S.-Y. Differential DNA Methylation Correlates with Differential Expression of Angiogenic Factors in Human Heart Failure. PLoS ONE 2010, 5, e8564. [Google Scholar] [CrossRef]
- Vujic, A.; Robinson, E.L.; Ito, M.; Haider, S.; Ackers-Johnson, M.; See, K.; Methner, C.; Figg, N.; Brien, P.; Roderick, H.L.; et al. Experimental heart failure modelled by the cardiomyocyte-specific loss of an epigenome modifier, DNMT3B. J. Mol. Cell. Cardiol. 2015, 82, 174–183. [Google Scholar] [CrossRef]
- Nührenberg, T.G.; Hammann, N.; Schnick, T.; Preißl, S.; Witten, A.; Stoll, M.; Gilsbach, R.; Neumann, F.-J.; Hein, L. Cardiac Myocyte De Novo DNA Methyltransferases 3a/3b Are Dispensable for Cardiac Function and Remodeling after Chronic Pressure Overload in Mice. PLoS ONE 2015, 10, e0131019. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Tan, X.; Tampe, B.; Nyamsuren, G.; Liu, X.; Maier, L.S.; Sossalla, S.; Kalluri, R.; Zeisberg, M.; Hasenfuss, G.; et al. Epigenetic balance of aberrant Rasal1 promoter methylation and hydroxymethylation regulates cardiac fibrosis. Cardiovasc. Res. 2015, 105, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, C.; Kunderfranco, P.; Rubino, M.; Larcher, V.; Carullo, P.; Anselmo, A.; Kurz, K.; Carell, T.; Angius, A.; Latronico, M.V.G.; et al. DNA hydroxymethylation controls cardiomyocyte gene expression in development and hypertrophy. Nat. Commun. 2016, 7, 12418. [Google Scholar] [CrossRef] [PubMed]
- El Khattabi, L.A.; Backer, S.; Pinard, A.; Dieudonné, M.-N.; Tsatsaris, V.; Vaiman, D.; Dandolo, L.; Bloch-Gallego, E.; Jammes, H.; Barbaux, S. A genome-wide search for new imprinted genes in the human placenta identifies DSCAM as the first imprinted gene on chromosome 21. Eur. J. Hum. Genet. 2018, 27, 49–60. [Google Scholar] [CrossRef]
- Wolffe, A.P. Transcriptional control: Imprinting insulation. Curr. Biol. 2000, 10, R463–R465. [Google Scholar] [CrossRef] [Green Version]
- White, V.; Jawerbaum, A.; Mazzucco, M.B.; Gauster, M.; Desoye, G.; Hiden, U. IGF2 stimulates fetal growth in a sex- and organ-dependent manner. Pediatr. Res. 2017, 83, 183–189. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zheng, D.; Wang, L.; Huang, Y.; Liu, H.; Xu, L.; Liao, Q.; Liu, P.; Shi, X.; Wang, Z.; et al. Elevated PLA2G7 Gene Promoter Methylation as a Gender-Specific Marker of Aging Increases the Risk of Coronary Heart Disease in Females. PLoS ONE 2013, 8, e59752. [Google Scholar] [CrossRef]
- Guo, T.-M.; Huang, L.-L.; Liu, K.; Ke, L.; Luo, Z.-J.; Li, Y.-Q.; Chen, X.-L.; Cheng, B. Pentraxin 3 (PTX3) promoter methylation associated with PTX3 plasma levels and neutrophil to lymphocyte ratio in coronary artery disease. J. Geriatr. Cardiol. 2016, 13, 712–717. [Google Scholar]
- Cash, H.L.; McGarvey, S.T.; Houseman, E.A.; Marsit, C.J.; Hawley, N.L.; Lambert-Messerlian, G.; Viali, S.; Tuitele, J.; Kelsey, K.T. Cardiovascular disease risk factors and DNA methylation at the LINE-1 repeat region in peripheral blood from Samoan islanders. Epigenetics 2011, 6, 1257–1264. [Google Scholar] [CrossRef] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Guillemette, B.; Drogaris, P.; Lin, H.-H.S.; Armstrong, H.; Hamada, K.; Imhof, A.; Bonneil, E.; Thibault, P.; Verreault, A.; Festenstein, R. H3 Lysine 4 Is Acetylated at Active Gene Promoters and Is Regulated by H3 Lysine 4 Methylation. PLoS Genet. 2011, 7, e1001354. [Google Scholar] [CrossRef] [Green Version]
- Backs, J.; Song, K.; Bezprozvannaya, S.; Chang, S.; Olson, E.N. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Investig. 2006, 116, 1853–1864. [Google Scholar] [CrossRef]
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002, 110, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Antos, C.L.; McKinsey, T.A.; Dreitz, M.; Hollingsworth, L.M.; Zhang, C.-L.; Schreiber, K.; Rindt, H.; Olson, E.N.; Zhang, C.-L.; Gorczynski, R.J. Dose-dependent Blockade to Cardiomyocyte Hypertrophy by Histone Deacetylase Inhibitors. J. Biol. Chem. 2003, 278, 28930–28937. [Google Scholar] [CrossRef] [Green Version]
- Jebessa, Z.H.; Shanmukha, K.D.; Dewenter, M.; Lehmann, L.H.; Xu, C.; Schreiter, F.; Siede, D.; Gong, X.-M.; Worst, B.C.; Federico, G.; et al. The lipid-droplet-associated protein ABHD5 protects the heart through proteolysis of HDAC4. Nat. Metab. 2019, 1, 1157–1167. [Google Scholar] [CrossRef]
- Qian, H.; Chen, Y.; Nian, Z.; Su, L.; Yu, H.; Chen, F.-J.; Zhang, X.; Xu, W.; Zhou, L.; Liu, J.; et al. HDAC6-mediated acetylation of lipid droplet–binding protein CIDEC regulates fat-induced lipid storage. J. Clin. Investig. 2017, 127, 1353–1369. [Google Scholar] [CrossRef] [Green Version]
- Thienpont, B.; Aronsen, J.M.; Robinson, E.L.; Okkenhaug, H.; Loche, E.; Ferrini, A.; Brien, P.; Alkass, K.; Tomasso, A.; Agrawal, A.; et al. The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J. Clin. Investig. 2017, 127, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.-W.; Grant, P.A.; Rissman, E. Sex differences in histone modifications in the neonatal mouse brain. Epigenetics 2009, 4, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Keiser, A.; Wood, M.A. Examining the contribution of histone modification to sex differences in learning and memory. Learn. Mem. 2019, 26, 318–331. [Google Scholar] [CrossRef]
- Hussain, S.; Tuorto, F.; Menon, S.; Blanco, S.; Cox, C.; Flores, J.V.; Watt, S.; Kudo, N.; Lyko, F.; Frye, M. The mouse cytosine-5 RNA methyltransferase NSun2 is a component of the chromatoid body and required for testis differentiation. Mol. Cell. Biol. 2013, 33, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R. MTA Is an Arabidopsis Messenger RNA Adenosine Methylase and Interacts with a Homolog of a Sex-Specific Splicing Factor[W][OA]. Plant Cell 2008, 20, 1278–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shvetsova, E.; BIOS Consortium; Sofronova, A.; Monajemi, R.; Gagalova, K.; Draisma, H.; White, S.J.; Santen, G.W.E.; Lopes, S.M.C.D.S.; Heijmans, B.T.; et al. Skewed X-inactivation is common in the general female population. Eur. J. Hum. Genet. 2018, 27, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Syrett, C.; Kramer, M.C.; Basu, A.; Atchison, M.L.; Anguera, M.C. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc. Natl. Acad. Sci. USA 2016, 113, E2029–E2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voskuhl, R.R. Sex differences in autoimmune diseases. Biol. Sex Differ. 2011, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, P.; Wang, Z.; Feng, Q.; Pintilie, G.D.; Foulds, C.; Lanz, R.B.; Ludtke, S.J.; Schmid, M.F.; Chiu, W.; O’Malley, B.W. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol. Cell 2015, 57, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiRenzo, J.; Shang, Y.; Phelan, M.; Sif, S.; Myers, M.; Kingston, R.; Brown, M. BRG-1 Is Recruited to Estrogen-Responsive Promoters and Cooperates with Factors Involved in Histone Acetylation. Mol. Cell. Biol. 2000, 20, 7541–7549. [Google Scholar] [CrossRef] [Green Version]
- Stamova, B.; Tian, Y.; Jickling, G.; Bushnell, C.; Zhan, X.; Liu, D.; Ander, B.P.; Verro, P.; Patel, V.; Pevec, W.C.; et al. The X-chromosome has a different pattern of gene expression in women compared with men with ischemic stroke. Stroke 2011, 43, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Eghbali, M. Influence of sex differences on microRNA gene regulation in disease. Biol. Sex Differ. 2014, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Volders, P.-J.; Verheggen, K.; Menschaert, G.; Vandepoele, K.; Martens, L.; Vandesompele, J.; Mestdagh, P. An update on LNCipedia: A database for annotated human lncRNA sequences. Nucleic Acids Res. 2015, 43, 4363–4364. [Google Scholar] [CrossRef] [Green Version]
- Beale, A.L.; Meyer, P.; Marwick, T.H.; Lam, C.S.; Kaye, D.M. Sex Differences in Cardiovascular Pathophysiology: Why Women Are Overrepresented in Heart Failure With Preserved Ejection Fraction. Circulation 2018, 138, 198–205. [Google Scholar] [CrossRef]
- Eisenberg, E.; Di Palo, K.E.; Piña, I.L. Sex differences in heart failure. Clin. Cardiol. 2018, 41, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Aimo, A.; Vergaro, G.; Barison, A.; Maffei, S.; Borrelli, C.; Morrone, D.; Cameli, M.; Palazzuoli, A.; Ambrosio, G.; Coiro, S.; et al. Sex-related differences in chronic heart failure. Int. J. Cardiol. 2018, 255, 145–151. [Google Scholar] [CrossRef]
- Hermans-Beijnsberger, S.; Van Bilsen, M.; Schroen, B. Long non-coding RNAs in the failing heart and vasculature. Non-Coding RNA Res. 2018, 3, 118–130. [Google Scholar] [CrossRef]
- Gomes, C.P.D.C.; Schroen, B.; Kuster, G.M.; Robinson, E.L.; Ford, K.; Squire, I.B.; Heymans, S.; Martelli, F.; Emanueli, C.; Devaux, Y.; et al. Regulatory RNAs in Heart Failure. Circulation 2020, 141, 313–328. [Google Scholar] [CrossRef]
- Ramirez, F.D.; Motazedian, P.; Jung, R.G.; Di Santo, P.; Macdonald, Z.; Simard, T.; Clancy, A.; Russo, J.J.; Welch, V.; Wells, G.A.; et al. Sex Bias Is Increasingly Prevalent in Preclinical Cardiovascular Research: Implications for Translational Medicine and Health Equity for Women: A Systematic Assessment of Leading Cardiovascular Journals Over a 10-Year Period. Circulation 2017, 135, 625–626. [Google Scholar] [CrossRef] [Green Version]
- Hartman, R.J.G.; Huisman, S.E.; Ruijter, H.M.D. Sex differences in cardiovascular epigenetics-a systematic review. Biol. Sex Differ. 2018, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Li, W.; Yang, J.; Shang, C.; Lin, C.-H.; Cheng, W.; Hang, C.T.; Cheng, H.-L.; Chen, C.-H.; Wong, J.; et al. Epigenetic response to environmental stress: Assembly of BRG1–G9a/GLP–DNMT3 repressive chromatin complex on Myh6 promoter in pathologically stressed hearts. Biochim. Biophys. Acta (BBA)—Bioenerg. 2016, 1863, 1772–1781. [Google Scholar] [CrossRef]
- Harikrishnan, K.N.; Okabe, J.; Mathiyalagan, P.; Khan, A.W.; Jadaan, S.A.; Sarila, G.; Ziemann, M.; Khurana, I.; Maxwell, S.S.; Du, X.-J.; et al. Sex-Based Mhrt Methylation Chromatinizes MeCP2 in the Heart. iScience 2019, 17, 288–301. [Google Scholar] [CrossRef] [Green Version]
- Teeuw, W.J.; Laine, M.L.; Bizzarro, S.; Loos, B.G. A Lead ANRIL Polymorphism Is Associated with Elevated CRP Levels in Periodontitis: A Pilot Case-Control Study. PLoS ONE 2015, 10, e0137335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vausort, M.; Salgado-Somoza, A.; Zhang, L.; Leszek, P.; Scholz, M.; Teren, A.; Burkhardt, R.; Thiery, J.; Wagner, D.R.; Devaux, Y. Myocardial Infarction-Associated Circular RNA Predicting Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2016, 68, 1247–1248. [Google Scholar] [CrossRef]
- Higashimoto, K.; Soejima, H.; Saito, T.; Okumura, K.; Mukai, T. Imprinting disruption of the CDKN1C/KCNQ1OT1 domain: The molecular mechanisms causing Beckwith-Wiedemann syndrome and cancer. Cytogenet. Genome Res. 2006, 113, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Coto, E.; Calvo, D.; Reguero, J.R.; Morís, C.; Rubín, J.M.; Díaz-Corte, C.; Gil-Peña, H.; Alosno, B.; Iglesias, S.; Gómez, J. Differential methylation of lncRNA KCNQ1OT1 promoter polymorphism was associated with symptomatic cardiac long QT. Epigenomics 2017, 9, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Lalem, T.; Zhang, L.; Scholz, M.; Burkhardt, R.; Saccheti, V.; Teren, A.; Thiery, J.; Devaux, Y. Cyclin dependent kinase inhibitor 1 C is a female-specific marker of left ventricular function after acute myocardial infarction. Int. J. Cardiol. 2019, 274, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Bartolomei, M.S. X-Inactivation, Imprinting, and Long Noncoding RNAs in Health and Disease. Cell 2013, 152, 1308–1323. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafrenière, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2018, 47, D155–D162. [Google Scholar] [CrossRef]
- Su, Q.; Lv, X.; Sun, Y.; Ye, Z.; Kong, B.; Qin, Z. Role of TLR4/MyD88/NF-κB signaling pathway in coronary microembolization-induced myocardial injury prevented and treated with nicorandil. Biomed. Pharmacoth. 2018, 106, 776–784. [Google Scholar] [CrossRef]
- Yan, S.; Wang, P.; Wang, J.; Yang, J.; Lu, H.; Jin, C.; Cheng, M.; Xu, D. Long Non-coding RNA HIX003209 Promotes Inflammation by Sponging miR-6089 via TLR4/NF-κB Signaling Pathway in Rheumatoid Arthritis. Front Immunol. 2019, 10, 2218. [Google Scholar] [CrossRef]
- Song, R.; Ro, S.; Michaels, J.D.; Park, C.; McCarrey, J.R.; Yan, W. Many X-linked microRNAs escape meiotic sex chromosome inactivation. Nat. Genet. 2009, 41, 488–493. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Lalem, T.; Devaux, Y. Circulating microRNAs to predict heart failure after acute myocardial infarction in women. Clin. Biochem. 2019, 70, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Florijn, B.W.; Bijkerk, R.; Van Der Veer, E.P.; Van Zonneveld, A.J. Gender and cardiovascular disease: Are sex-biased microRNA networks a driving force behind heart failure with preserved ejection fraction in women? Cardiovasc Res. 2018, 114, 210–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-T.; Tsai, P.-C.; Liao, Y.-C.; Hsu, C.-Y.; Juo, S.-H.H. Circulating microRNAs have a sex-specific association with metabolic syndrome. J. Biomed. Sci. 2013, 20, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Buchan, R.; Cook, S.A. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc. Res. 2010, 86, 410–420. [Google Scholar] [CrossRef]
- Hinkel, R.; Penzkofer, D.; Zühlke, S.; Fischer, A.; Husada, W.; Xu, Q.-F.; Baloch, E.; Van Rooij, E.; Zeiher, A.; Kupatt, C.; et al. Inhibition of MicroRNA-92a Protects Against Ischemia/Reperfusion Injury in a Large-Animal Model. Circulation 2013, 128, 1066–1075. [Google Scholar] [CrossRef] [Green Version]
- Bonauer, A.; Carmona, G.; Iwasaki, M.; Mione, M.C.; Koyanagi, M.; Fischer, A.; Burchfield, J.; Fox, H.; Doebele, C.; Ohtani, K.; et al. MicroRNA-92a Controls Angiogenesis and Functional Recovery of Ischemic Tissues in Mice. Science 2009, 324, 1710–1713. [Google Scholar] [CrossRef] [Green Version]
- Verjans, R.; Peters, T.; Beaumont, F.J.; Van Leeuwen, R.; Van Herwaarden, T.; Verhesen, W.; Munts, C.; Bijnen, M.; Henkens, M.; Diez, J.; et al. MicroRNA-221/222 Family Counteracts Myocardial Fibrosis in Pressure Overload-Induced Heart Failure. Hypertension 2018, 71, 280–288. [Google Scholar] [CrossRef]
- Howard, E.W.; Yang, X. microRNA Regulation in Estrogen Receptor-Positive Breast Cancer and Endocrine Therapy. Biol. Proced. Online 2018, 20, 17. [Google Scholar] [CrossRef]
- Volders, P.-J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2018, 47, D135–D139. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-J.; Wang, Y.-M.; Hu, Y.; Lin, Q.; Chen, R.; Liu, H.; Cao, W.-Z.; Zhu, H.-F.; Tong, C.; Li, L.; et al. HDncRNA: A comprehensive database of non-coding RNAs associated with heart diseases. Database 2018, 2018, bay067. [Google Scholar] [CrossRef] [Green Version]
- Selvamani, A.; Williams, M.H.; Miranda, R.C.; Sohrabji, F. Circulating miRNA profiles provide a biomarker for severity of stroke outcomes associated with age and sex in a rat model. Clin. Sci. 2014, 127, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biolleau, A.; Somoza, A.S.; Dankiewicz, J.; Stammet, P.; Gilje, P.; Erlinge, D.; Hassager, C.; Wise, M.P.; Kuiper, M.; Friberg, H.; et al. Circulating Levels of miR-574-5p Are Associated with Neurological Outcome after Cardiac Arrest in Women: A Target Temperature Management (TTM) Trial Substudy. Dis. Markers 2019, 2019, 1802879–1802910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, M.; Kawasaki, T.; Matsuda, T.; Arai, T.; Gojo, S.; Takeuchi, J.K. Sexual dimorphisms of mRNA and miRNA in human/murine heart disease. PLoS ONE 2017, 12, e0177988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudink, E.A.; Florijn, B.W.; Weijs, B.; Duijs, J.M.; Luermans, J.G.; Peeters, F.E.; Schurgers, L.J.; Wildberger, J.; Schotten, U.; Bijkerk, R.; et al. Vascular Calcification and not Arrhythmia in Idiopathic Atrial Fibrillation Associates with Sex Differences in Diabetic Microvascular Injury miRNA Profiles. MicroRNA 2019, 8, 127–134. [Google Scholar] [CrossRef]
- Vausort, M.; Wagner, D.R.; Devaux, Y. Long Noncoding RNAs in Patients With Acute Myocardial Infarction. Circ. Res. 2014, 115, 668–677. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.; Arnold, A.P.; Berkley, K.J.; Blaustein, J.D.; Eckel, L.A.; Hampson, E.; Herman, J.P.; Marts, S.; Sadee, W.; Steiner, M.; et al. Strategies and Methods for Research on Sex Differences in Brain and Behavior. Endocrinology 2005, 146, 1650–1673. [Google Scholar] [CrossRef]
- Reusch, J.E.B.; Kumar, T.R.; Regensteiner, J.G.; Zeitler, P.S.; Arany, Z.; Merz, C.N.B.; Barrett-Connor, E.; Boyle, K.E.; Brown, L.; Clegg, D.; et al. Conference Participants Identifying the Critical Gaps in Research on Sex Differences in Metabolism Across the Life Span. Endocrinology 2017, 159, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K. Sex as an important biological variable in biomedical research. BMB Rep. 2018, 51, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.E.; Giraldo, C.V.; Schaible, N.S.; Zakeri, R.; Miller, V.M. Reporting of sex as a variable in cardiovascular studies using cultured cells. Biol. Sex Differ. 2011, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- Leong, X.-F.; Ng, C.-Y.; Jaarin, K. Animal Models in Cardiovascular Research: Hypertension and Atherosclerosis. BioMed Res. Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.B.; Koob, G.F. Sex Differences in Animal Models: Focus on Addiction. Pharmacol. Rev. 2016, 68, 242–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, P.A.; Leinwand, L.A. Dietary phytoestrogens present in soy dramatically increase cardiotoxicity in male mice receiving a chemotherapeutic tyrosine kinase inhibitor. Mol. Cell. Endocrinol. 2014, 399, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.M.; Li, H.; Zhu, X.; Shah, A.S.; Lu, L.J.; Davidson, W.S. A Comparison of the Mouse and Human Lipoproteome: Suitability of the Mouse Model for Studies of Human Lipoproteins. J. Proteome Res. 2015, 14, 2686–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blenck, C.L.; Harvey, P.A.; Reckelhoff, J.F.; Leinwand, L.A. The Importance of Biological Sex and Estrogen in Rodent Models of Cardiovascular Health and Disease. Circ. Res. 2016, 118, 1294–1312. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Dworatzek, E.; Seeland, U.; Kararigas, G.; Arnal, J.-F.; Brunelleschi, S.; Carpenter, T.C.; Erdmann, J.; Franconi, F.; Giannetta, E.; et al. Sex in basic research: Concepts in the cardiovascular field. Cardiovasc. Res. 2017, 113, 711–724. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.K.; Matkovich, S.J.; Nerbonne, J.M. Regional Differences in mRNA and lncRNA Expression Profiles in Non-Failing Human Atria and Ventricles. Sci. Rep. 2018, 8, 13919. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Yang, W.; Shi, J.; Zhou, Y.; Yang, J.; Cui, Q.; Zhou, Y. Identification and Analysis of Human Sex-biased MicroRNAs. Genom. Proteom. Bioinform. 2018, 16, 200–211. [Google Scholar] [CrossRef]
- Trexler, C.; Odell, A.T.; Jeong, M.Y.; Dowell, R.D.; Leinwand, L.A. Transcriptome and Functional Profile of Cardiac Myocytes Is Influenced by Biological Sex. Circ. Cardiovasc. Genet. 2017, 10, e001770. [Google Scholar] [CrossRef]
- Matarrese, P.; Tieri, P.; Anticoli, S.; Ascione, B.; Conte, M.; Franceschi, C.; Malorni, W.; Salvioli, S.; Ruggieri, A. X-chromosome-linked miR548am-5p is a key regulator of sex disparity in the susceptibility to mitochondria-mediated apoptosis. Cell Death Dis. 2019, 10, 1–12. [Google Scholar]
- Dubois-Deruy, E.; Cuvelliez, M.; Fiedler, J.; Charrier, H.; Mulder, P.; Hebbar, E.; Pfanne, A.; Beseme, O.; Chwastyniak, M.; Amouyel, P.; et al. MicroRNAs regulating superoxide dismutase 2 are new circulating biomarkers of heart failure. Sci. Rep. 2017, 7, 14747. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Gou, Y.; Zhang, H.; Zuo, H.; Zhang, H.; Liu, Z.; Yao, D. Estradiol improves cardiovascular function through up-regulation of SOD2 on vascular wall. Redox Biol. 2014, 3, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Ruderisch, H.; Queirós, A.M.; Fliegner, D.; Eschen, C.; Kararigas, G.; Regitz-Zagrosek, V. Sex-specific regulation of cardiac microRNAs targeting mitochondrial proteins in pressure overload. Biol. Sex Differ. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stary, C.M.; Xu, L.; Li, L.; Sun, X.; Ouyang, Y.-B.; Xiong, X.; Zhao, J.; Giffard, R.G. Inhibition of miR-181a protects female mice from transient focal cerebral ischemia by targeting astrocyte estrogen receptor-α. Mol. Cell. Neurosci. 2017, 82, 118–125. [Google Scholar] [CrossRef]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijay, V.; Han, T.; Moland, C.L.; Kwekel, J.C.; Fuscoe, J.C.; Desai, V.G. Sexual Dimorphism in the Expression of Mitochondria-Related Genes in Rat Heart at Different Ages. PLoS ONE 2015, 10, e0117047. [Google Scholar] [CrossRef]
- Demonacos, C.; Karayanni, N.; Chatzoglou, E.; Tsiriyiotis, C.; Spandidos, D.; Sekeris, C.E. Mitochondrial genes as sites of primary action of steroid hormones. Steroids 1996, 61, 226–232. [Google Scholar] [CrossRef]
- Schwend, T.; Gustafsson, J.Å. False positives in MALDI-TOF detection of ERβ in mitochondria. Biochem. Biophys. Res. Commun. 2006, 343, 707–711. [Google Scholar] [CrossRef]
- Yang, S.-H.; Liu, R.; Perez, E.J.; Wen, Y.; Stevens, S.M., Jr.; Valencia, T.; Brun-Zinkernagel, A.-M.; Prokai, P.; Will, Y.; Dykens, J.; et al. Mitochondrial Localization of Estrogen Receptor Beta. Proc. Natl. Acad. Sci. USA 2004, 101, 4130–4135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psarra, A.-M.G.; Sekeris, C.E. Steroid and thyroid hormone receptors in mitochondria. IUBMB Life 2008, 60, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Solakidi, S.; Psarra, A.-M.G.; Nikolaropoulos, S.; Sekeris, C.E. Estrogen receptors alpha and beta (ERalpha and ERbeta) and androgen receptor (AR) in human sperm: Localization of ERbeta and AR in mitochondria of the midpiece. Hum Reprod. 2005, 20, 3481–3487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jusic, A.; Devaux, Y. Mitochondrial noncoding RNA—Regulatory network in cardiovascular disease. Basic Res. Cardiol. 2020, 115, 23. [Google Scholar] [CrossRef] [PubMed]
- Stammet, P.; Goretti, E.; Vausort, M.; Zhang, L.; Wagner, D.R.; Devaux, Y. Circulating microRNAs after cardiac arrest*. Crit. Care Med. 2012, 40, 3209–3214. [Google Scholar] [CrossRef]
- Jusic, A.; Devaux, Y.; EU-CardioRNA COST Action (CA17129). Noncoding RNAs in Hypertension. Hypertension 2019, 74, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Biolleau, A.; Cardenas, C.L.L.; Courtois, A.; Zhang, L.; Rodosthenous, R.S.; Das, S.; Sakalihasan, N.; Michel, J.-B.; Lindsay, M.E.; Devaux, Y.; et al. MiR-574-5p: A Circulating Marker of Thoracic Aortic Aneurysm. Int. J. Mol. Sci. 2019, 20, 3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rooij, E.; Olson, E.N. MicroRNA therapeutics for cardiovascular disease: Opportunities and obstacles. Nat. Rev. Drug Discov. 2012, 11, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Ooi, J.; Matsumoto, A.; Tham, Y.K.; Singla, S.; Kiriazis, H.; Patterson, N.L.; Sadoshima, J.; Obad, S.; Lin, R.C.Y.; et al. Sex differences in response to miRNA-34a therapy in mouse models of cardiac disease: Identification of sex-, disease- and treatment-regulated miRNAs. J. Physiol. 2016, 594, 5959–5974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patten, R.D. Models of gender differences in cardiovascular disease. Drug Discov. Today Dis. Model. 2007, 4, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Ventura-Clapier, R.; Moulin, M.; Piquereau, J.; Lemaire, C.; Mericskay, M.; Veksler, V.; Garnier, A. Mitochondria: A central target for sex differences in pathologies. Clin. Sci. 2017, 131, 803–822. [Google Scholar] [CrossRef]
- Ounzain, S.; Micheletti, R.; Beckmann, T.; Schroen, B.; Alexanian, M.; Pezzuto, I.; Crippa, S.; Nemir, M.; Sarre, A.; Johnson, R.; et al. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur. Hear. J. 2014, 36, 353–368. [Google Scholar] [CrossRef]
- Arnold, A.P.; Chen, X. What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Front. Neuroendocr. 2009, 30, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Yin, Y.-Z.; Xiao, J. An In Vivo Estrogen Deficiency Mouse Model for Screening Exogenous Estrogen Treatments of Cardiovascular Dysfunction After Menopause. J. Vis. Exp. 2019, 150, e59536. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.E.; Vandenput, L.; Tivesten, Å.; Norlén, A.-K.; Lagerquist, M.; Windahl, S.H.; Börjesson, A.E.; Farman, H.H.; Poutanen, M.; Benrick, A.; et al. Measurement of a Comprehensive Sex Steroid Profile in Rodent Serum by High-Sensitive Gas Chromatography-Tandem Mass Spectrometry. Endocrinology 2015, 156, 2492–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koebele, S.V.; Bimonte-Nelson, H.A. Modeling menopause: The utility of rodents in translational behavioral endocrinology research. Maturitas 2016, 87, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, V.R.; Mendes, E.; Casaro, M.; Antiorio, A.T.F.B.; Oliveira, F.A.; Ferreira, C.M. Description of Ovariectomy Protocol in Mice. Methods Mol Biol. 2019, 1916, 303–309. [Google Scholar] [CrossRef]
- Novella, S.; Dantas, A.P.; Segarra, G.; Novensà, L.; Bueno, C.; Heras, M.; Hermenegildo, C.; Medina, P. Gathering of aging and estrogen withdrawal in vascular dysfunction of senescent accelerated mice. Exp. Gerontol. 2010, 45, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Yousefzadeh, N.; Kashfi, K.; Jeddi, S.; Ghasemi, A. Ovariectomized rat model of osteoporosis: A practical guide. EXCLI J. 2020, 19, 89–107. [Google Scholar]
- Vidal-Gómez, X.; Novella, S.; Pérez-Monzó, I.; Garabito, M.; Dantas, A.P.; Segarra, G.; Hermenegildo, C.; Medina, P. Decreased bioavailability of nitric oxide in aorta from ovariectomized senescent mice. Role of cyclooxygenase. Exp. Gerontol. 2016, 76, 1–8. [Google Scholar] [CrossRef]
- Jänne, M.; Deol, H.K.; Power, S.G.A.; Yee, S.-P.; Hammond, G. Human Sex Hormone-Binding Globulin Gene Expression in Transgenic Mice. Mol. Endocrinol. 1998, 12, 123–136. [Google Scholar] [CrossRef] [Green Version]
- McNamara, K.M.; Harwood, D.; Simanainen, U.; Walters, K.A.; Jimenez, M.; Handelsman, D.J. Measurement of sex steroids in murine blood and reproductive tissues by liquid chromatography–tandem mass spectrometry. J. Steroid Biochem. Mol. Biol. 2010, 121, 611–618. [Google Scholar] [CrossRef]
- Nelson, J.F.; Felicio, L.S.; Randall, P.K.; Sims, C.; Finch, C.E. A longitudinal study of estrous cyclicity in aging C57BL/6J mice: I. Cycle frequency, length and vaginal cytology. Biol. Reprod. 1982, 27, 327–339. [Google Scholar] [CrossRef]
- Mohamed, M.; Abdel-Rahman, A.A. Effect of long-term ovariectomy and estrogen replacement on the expression of estrogen receptor gene in female rats. Eur. J. Endocrinol. 2000, 142, 307–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, L.P.; Dyer, C.A.; Eastgard, R.L.; Hoyer, P.B.; Banka, C.L. Atherosclerotic Lesion Development in a Novel Ovary-Intact Mouse Model of Perimenopause. Arter. Thromb. Vasc. Biol. 2005, 25, 1910–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, H.; Pollow, D.P.; Hoyer, P.B. The VCD Mouse Model of Menopause and Perimenopause for the Study of Sex Differences in Cardiovascular Disease and the Metabolic Syndrome. Physiology 2016, 31, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habibi, P.; Alihemmati, A.; Nasirzadeh, M.; Yousefi, H.; Habibi, M.; Ahmadiasl, N. Involvement of microRNA-133 and -29 in cardiac disturbances in diabetic ovariectomized rats. Iran J. Basic Med. Sci. 2016, 19, 1177–1185. [Google Scholar]
- Wang, N.; Sun, L.-Y.; Zhang, S.-C.; Wei, R.; Xie, F.; Liu, J.; Yan, Y.; Duan, M.-J.; Sun, L.-L.; Sun, Y.-H.; et al. MicroRNA-23a Participates in Estrogen Deficiency Induced Gap Junction Remodeling of Rats by Targeting GJA1. Int. J. Biol. Sci. 2015, 11, 390–403. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmson, A.S.; Rodriguez, M.L.; Eriksson, E.S.; Johansson, I.; Fogelstrand, P.; Stubelius, A.; Lindgren, S.; Fagman, J.B.; Hansson, G.K.; Carlsten, H.; et al. Testosterone Protects Against Atherosclerosis in Male Mice by Targeting Thymic Epithelial Cells—Brief Report. Arter. Thromb. Vasc. Biol. 2018, 38, 1519–1527. [Google Scholar] [CrossRef] [Green Version]
- Movérare-Skrtic, S.; Venken, K.; Andersson, N.; Lindberg, M.K.; Svensson, J.; Swanson, C.; Vanderschueren, D.; Oscarsson, J.; Gustafsson, J.-A.; Ohlsson, C. Dihydrotestosterone Treatment Results in Obesity and Altered Lipid Metabolism in Orchidectomized Mice*. Obesity 2006, 14, 662–672. [Google Scholar] [CrossRef]
- Wu, J.; Hadoke, P.W.F.; Mair, I.; Lim, W.G.; Miller, E.; Denvir, M.A.; Smith, L.B. Modulation of neointimal lesion formation by endogenous androgens is independent of vascular androgen receptor. Cardiovasc. Res. 2014, 103, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Chodari, L.; Dariushnejad, H.; Ghorbanzadeh, V. Voluntary wheel running and testosterone replacement increases heart angiogenesis through miR-132 in castrated diabetic rats. Physiol. Int. 2019, 106, 48–58. [Google Scholar] [CrossRef]
- Markiewicz, M.; Znoyko, S.; Stawski, L.; Ghatnekar, A.; Gilkeson, G.; Trojanowska, M. A role for estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) in collagen biosynthesis in mouse skin. J. Investig. Dermatol. 2013, 133, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Carreau, S.; Genissel, C.; Bilinska, B.; Levallet, J. Sources of oestrogen in the testis and reproductive tract of the male. Int. J. Androl. 1999, 22, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Deng, C.; Lu, W.; Xiao, J.; Ma, D.; Guo, M.; Recker, R.R.; Gatalica, Z.; Wang, Z.; Xiao, G.G. let-7 MicroRNAs Induce Tamoxifen Sensitivity by Downregulation of Estrogen Receptor α Signaling in Breast Cancer. Mol. Med. 2011, 17, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Gong, S.; Zhou, J.; Zhong, M.; Su, G. microRNA regulation of the expression of the estrogen receptor in endometrial cancer. Mol. Med. Rep. 2010, 3, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Devanathan, S.; Whitehead, T.; Schweitzer, G.G.; Fettig, N.; Kovacs, A.; Korach, K.S.; Finck, B.N.; Shoghi, K.I. An Animal Model with a Cardiomyocyte-Specific Deletion of Estrogen Receptor Alpha: Functional, Metabolic, and Differential Network Analysis. PLoS ONE 2014, 9, e101900. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.; Ekblad, E.; Heine, T.; Gustafsson, J. Increased magnitude of relaxation to oestrogen in aorta from oestrogen receptor beta knock-out mice. J. Endocrinol. 2000, 166, R5–R9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.; Prossnitz, E.R.; Barton, M. GPER/GPR30 and Regulation of Vascular Tone and Blood Pressure. Immunol. Endocr. Metab. Agents Med. Chem. 2011, 11, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Billon-Galés, A.; Krust, A.; Fontaine, C.; Abot, A.; Flouriot, G.; Toutain, C.; Berges, H.; Gadeau, A.-P.; Lenfant, F.; Gourdy, P.; et al. Activation function 2 (AF2) of estrogen receptor-α is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc. Natl. Acad. Sci. USA 2011, 108, 13311–13316. [Google Scholar] [CrossRef] [Green Version]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.-L.; Boudou, F.; Sautier, L.; Vessières, E.; Kim, S.H.; et al. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. USA 2013, 111, E283–E290. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Mackie, R.; Kampf, K.; Domadia, S.; Brown, J.D.; O’Neill, R.J.; Arnold, A.P. Four Core Genotypes mouse model: Localization of the Sry transgene and bioassay for testicular hormone levels. BMC Res. Notes 2015, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Manwani, B.; Liu, F.; Scranton, V.; Hammond, M.D.; Sansing, L.H.; McCullough, L.D. Differential effects of aging and sex on stroke induced inflammation across the lifespan. Exp. Neurol. 2013, 249, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Huxley, V.H.; Kemp, S.S.; Schramm, C.; Sieveking, S.; Bingaman, S.; Yu, Y.; Zaniletti, I.; Stockard, K.; Wang, J. Sex differences influencing micro- and macrovascular endothelial phenotype in vitro. J. Physiol. 2018, 596, 3929–3949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, K.; McCormack, C.E.; Bradbury, N.A. Do you know the sex of your cells? Am. J. Physiol Cell-Physiol. 2013, 306, C3–C18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, H.R.; Rawat, N.; Das, S. Alternatives to amelogenin markers for sex determination in humans and their forensic relevance. Mol. Biol. Rep. 2020, 47, 2347–2360. [Google Scholar] [CrossRef] [PubMed]
- Settin, A.; Elsobky, E.; Hammad, A.; Al-Erany, A. Rapid Sex Determination Using PCR Technique Compared to Classic Cytogenetics. Int. J. Heal. Sci. 2008, 2, 49–52. [Google Scholar]
- Hassan, F.M.; Razik, H.A.A.; Wadie, M.S.; Abdelfattah, D.S. XIST and RPS4Y1 long non-coding RNA transcriptome as sex biomarkers in different body fluids. Egypt. J. Forensic Sci. 2019, 9, 16. [Google Scholar] [CrossRef]
- Boese, A.C.; Kim, S.C.; Yin, K.-J.; Lee, J.-P.; Hamblin, M.H. Sex differences in vascular physiology and pathophysiology: Estrogen and androgen signaling in health and disease. Am. J. Physiol. Circ. Physiol. 2017, 313, H524–H545. [Google Scholar] [CrossRef]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen Signaling Multiple Pathways to Impact Gene Transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Novella, S.; Pérez-Cremades, D.; Mompeón, A.; Hermenegildo, C. Mechanisms underlying the influence of oestrogen on cardiovascular physiology in women. J. Physiol. 2019, 597, 4873–4886. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Young, B.D.; Bender, J.R. Endothelial estrogen receptor isoforms and cardiovascular disease. Mol. Cell. Endocrinol. 2014, 389, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Begam, A.J.; Selvaraj, J.; Nanjan, M. Estrogen receptor agonists/antagonists in breast cancer therapy: A critical review. Bioorganic Chem. 2017, 71, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Martinkovich, S.; Planey, S.L.; Shah, D. Selective estrogen receptor modulators: Tissue specificity and clinical utility. Clin. Interv. Aging 2014, 9, 1437–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, L.; A Kroon, P.; Rimm, E.B.; Cohn, J.S.; Harvey, I.; A Le Cornu, K.; Ryder, J.J.; Hall, W.L.; Cassidy, A. Flavonoids, flavonoid-rich foods, and cardiovascular risk: A meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2008, 88, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Arnal, J.-F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef]
- Miller, M.M.; McMullen, P.D.; Andersen, M.E.; Clewell, R.A. Multiple receptors shape the estrogen response pathway and are critical considerations for the future of in vitro-based risk assessment efforts. Crit. Rev. Toxicol. 2017, 47, 570–586. [Google Scholar] [CrossRef] [Green Version]
- Sołtysik, K.; Czekaj, P. ERα36—Another piece of the estrogen puzzle. Eur. J. Cell Biol. 2015, 94, 611–625. [Google Scholar] [CrossRef]
- Stevis, P.E.; Deecher, D.C.; Suhadolnik, L.; Mallis, L.M.; Frail, D.E. Differential effects of estradiol and estradiol-BSA conjugates. Endocrinology 1999, 140, 5455–5458. [Google Scholar] [CrossRef]
- Harrington, W.R.; Kim, S.H.; Funk, C.C.; Madak-Erdogan, Z.; Schiff, R.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Estrogen Dendrimer Conjugates that Preferentially Activate Extranuclear, NongenomicVersusGenomic Pathways of Estrogen Action. Mol. Endocrinol. 2006, 20, 491–502. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Wu, Q.; Oltmann, S.; Konaniah, E.S.; Umetani, M.; Korach, K.S.; Thomas, G.; Mineo, C.; Yuhanna, I.S.; Kim, S.H.; et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J. Clin. Investig. 2010, 120, 2319–2330. [Google Scholar] [CrossRef] [Green Version]
- Madak-Erdogan, Z.; Kim, S.H.; Gong, P.; Zhao, Y.C.; Zhang, H.; Chambliss, K.L.; Carlson, K.E.; Mayne, C.G.; Shaul, P.W.; Korach, K.S.; et al. Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci. Signal. 2016, 9, ra53. [Google Scholar] [CrossRef] [Green Version]
- Sikora, M.J.; Johnson, M.D.; Lee, A.V.; Oesterreich, S. Endocrine Response Phenotypes Are Altered by Charcoal-Stripped Serum Variability. Endocrinology 2016, 157, 3760–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welshons, W.V.; Grady, L.H.; Engler, K.S.; Judy, B.M. Control of proliferation of MCF-7 breast cancer cells in a commercial preparation of charcoal-stripped adult bovine serum. Breast Cancer Res. Treat. 1992, 23, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Welshons, W.V.; Wolf, M.F.; Murphy, C.S.; Jordan, V. Estrogenic activity of phenol red. Mol. Cell. Endocrinol. 1988, 57, 169–178. [Google Scholar] [CrossRef]
- Berthois, Y.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Phenol red in tissue culture media is a weak estrogen: Implications concerning the study of estrogen-responsive cells in culture. Proc. Natl. Acad. Sci. USA 1986, 83, 2496–2500. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MiRNA Identifier | CVD | Specificity | Regulation | Chr | Organism Studied | References |
|---|---|---|---|---|---|---|
| miR-103 | coronary artery calcification | ♂ | ? | 20–5 | Human | PMID:30465521 |
| miR-106a/b | cardiac fibrosis | ♂ | Hormonal | X–7 | Mouse | PMID:24157234 |
| miR-125a-5p | coronary artery calcification | ♂ | ? | 19 | Human | PMID:30465521 |
| miR-126 | coronary artery calcification | ♀ | ? | 9 | Human | PMID:30465521 |
| miR-136 | post-stroke | ♀ | ? | 14 | Rat | PMID:24428837 |
| miR-142-3p | postmenopausal HRT * | ♀ | Hormonal | 17 | Human, mouse | PMID:25040542 |
| miR-15a | post-stroke | ♀ | ? | 13 | Rat | PMID:24428837 |
| miR-182 | postmenopausal HRT * | ♀ | Hormonal | 7 | Human, mouse | PMID:25040542 |
| miR-199a-3p | post-stroke | ♀ | ? | 1 | Rat | PMID:24428837 |
| miR-19b | post-stroke | ♀ | ? | 13–X | Rat | PMID:24428837 |
| miR-212 | descending aorta calcification | ♀ | ? | 17 | Human | PMID:30465521 |
| miR-221 | coronary artery calcification/metabolic syndrome * | ♂/♀ | ? | X | Human | PMID:30465521/ PMID:24093444 |
| miR-222 | insulin resistance in gestational diabetes mellitus * | ♀ | Hormonal | X | Human | PMID:24601884 |
| miR-223 | coronary artery calcification/postmenopausal HRT * | ♂/♀ | ?/Hormonal | X | Human, mouse | PMID:30465521/ PMID:25040542 |
| miR-23a | post-stroke/post-menopause related arrhythmia | ♀ | Apoptotic/Hormonal | 19 | Mouse/rat | PMID:21709246/ PMID:25798059 |
| miR-24 | cardiac fibrosis | ♂ | Hormonal | 9–19 | Mouse | PMID:24157234 |
| miR-27a/b | coronary artery calcification/cardiac fibrosis | ♀/♂ | ?/Hormonal | 19–9 | Human/mouse | PMID:30465521/ PMID:24157234 |
| miR-32 | post-stroke | ♀ | ? | 9 | Rat | PMID:24428837 |
| miR-34a | dilated cardiomyopathy | ♀ | ? | 1 | Mouse | PMID:27270487 |
| miR-363-3p | neuroprotective for stroke | ♀ | Apoptotic | X | Rat | PMID:27773791 |
| miR-574-5p | cardiac arrest | ♀ | ? | 4 | Human | PMID:31275442 |
| miR-let7g | metabolic syndrome * | ♀ | ? | X | Human | PMID:24093444 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jusic, A.; Salgado-Somoza, A.; Paes, A.B.; Stefanizzi, F.M.; Martínez-Alarcón, N.; Pinet, F.; Martelli, F.; Devaux, Y.; Robinson, E.L.; Novella, S., on behalf of the EU-CardioRNA COST Action. Approaching Sex Differences in Cardiovascular Non-Coding RNA Research. Int. J. Mol. Sci. 2020, 21, 4890. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144890
Jusic A, Salgado-Somoza A, Paes AB, Stefanizzi FM, Martínez-Alarcón N, Pinet F, Martelli F, Devaux Y, Robinson EL, Novella S on behalf of the EU-CardioRNA COST Action. Approaching Sex Differences in Cardiovascular Non-Coding RNA Research. International Journal of Molecular Sciences. 2020; 21(14):4890. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144890
Chicago/Turabian StyleJusic, Amela, Antonio Salgado-Somoza, Ana B. Paes, Francesca Maria Stefanizzi, Núria Martínez-Alarcón, Florence Pinet, Fabio Martelli, Yvan Devaux, Emma Louise Robinson, and Susana Novella on behalf of the EU-CardioRNA COST Action. 2020. "Approaching Sex Differences in Cardiovascular Non-Coding RNA Research" International Journal of Molecular Sciences 21, no. 14: 4890. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144890