Proteomics Reveals that Methylmalonyl-CoA Mutase Modulates Cell Architecture and Increases Susceptibility to Stress

 ,
,  ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
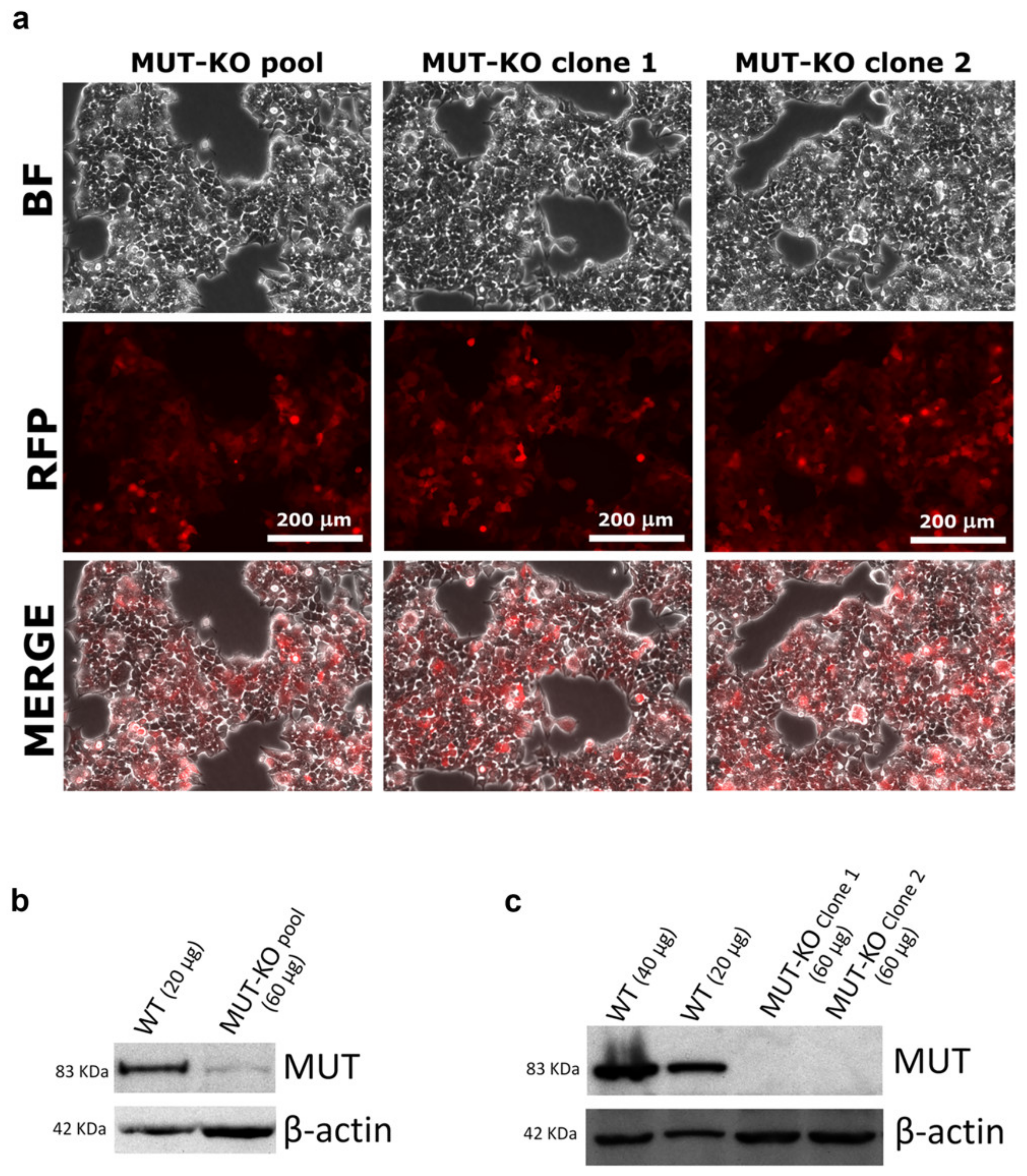
2.1. CRISPR/Cas9-Mediated MUT Gene Knockout in a HEK 293 Cell Line
2.2. Methylmalonic Acid and Propionylcarnitine Are Increased in MUT-KO Cells
2.3. MUT Knockout Does Not Affect Cell Viability and Proliferation
2.4. Label-Free Quantification for the Proteomic Analysis of MUT-KO versus WT
2.5. Bioinformatic Analysis: GO Terms Annotation
2.6. Rescuing MUT Protein Expression in a MUT-KO HEK 293 Cell Line
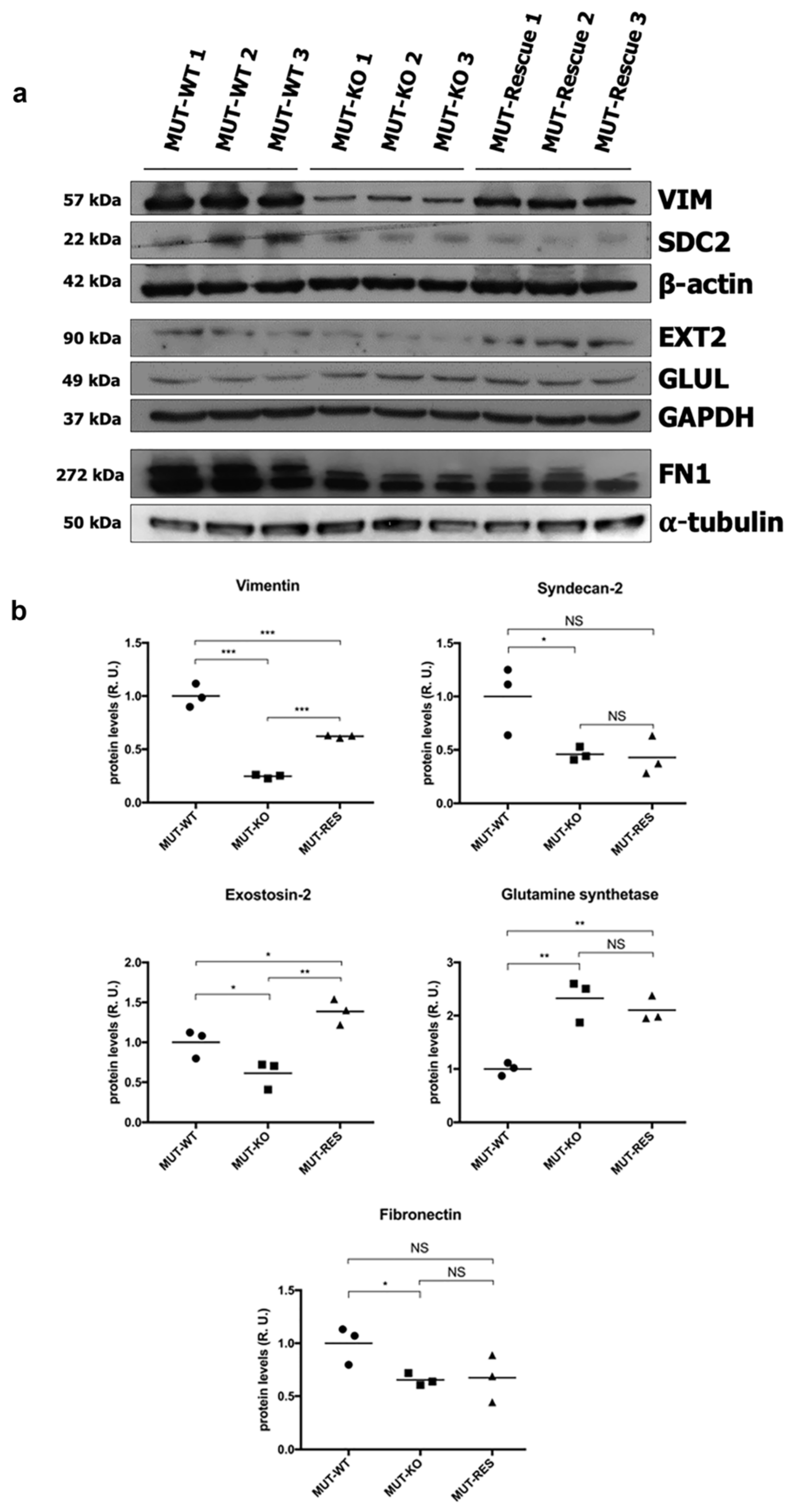
2.7. Validation of Proteomic Identifications in WT and MUT-KO and Protein Expression Levels Analysis in MUT-Rescue Cells
2.8. MUT Knockout Increases the Intracellular Levels of ROS
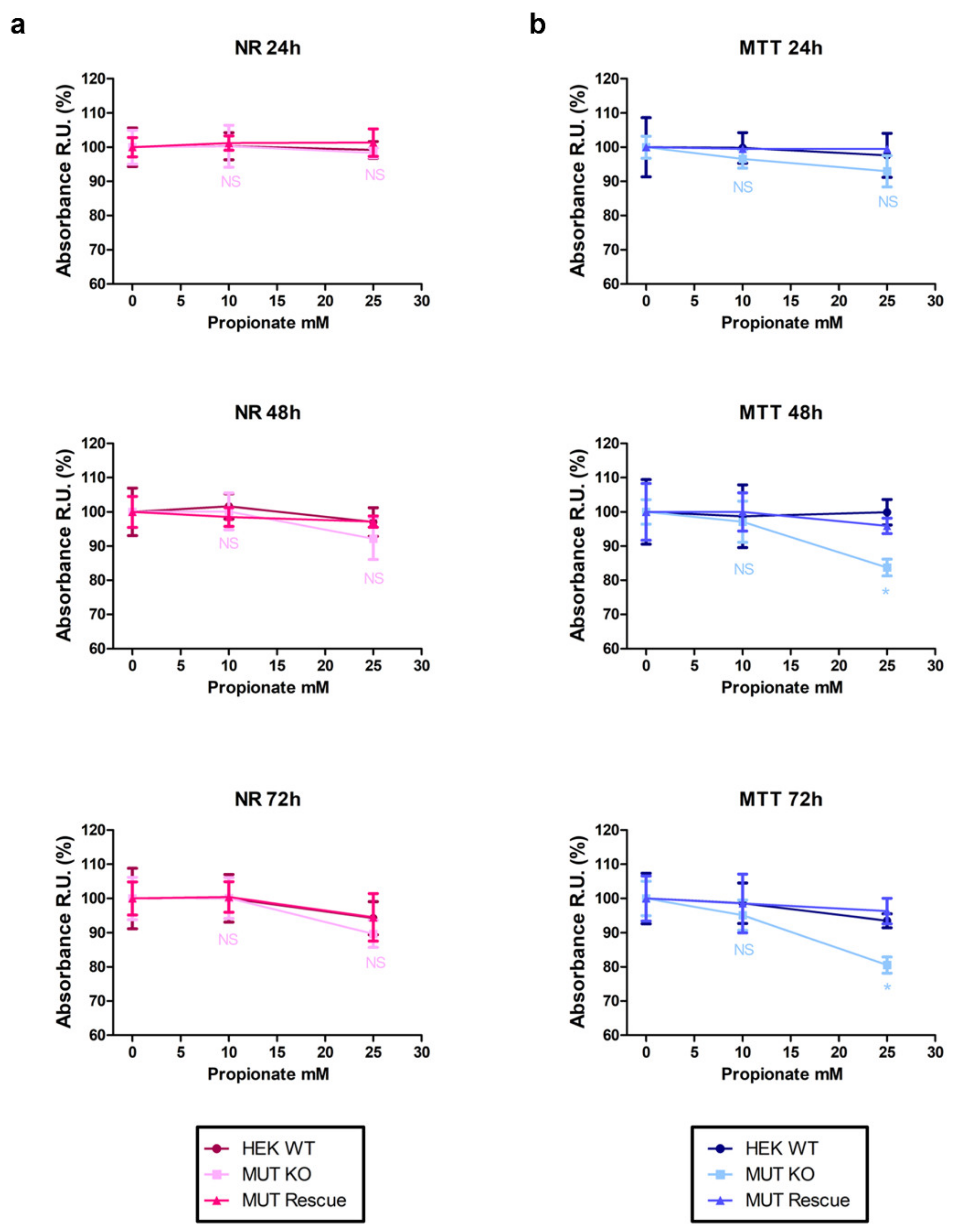
2.9. MUT Knockout Impairs Cell Viability and Mitochondrial Functionality in a Propionate-Enriched Culture Medium
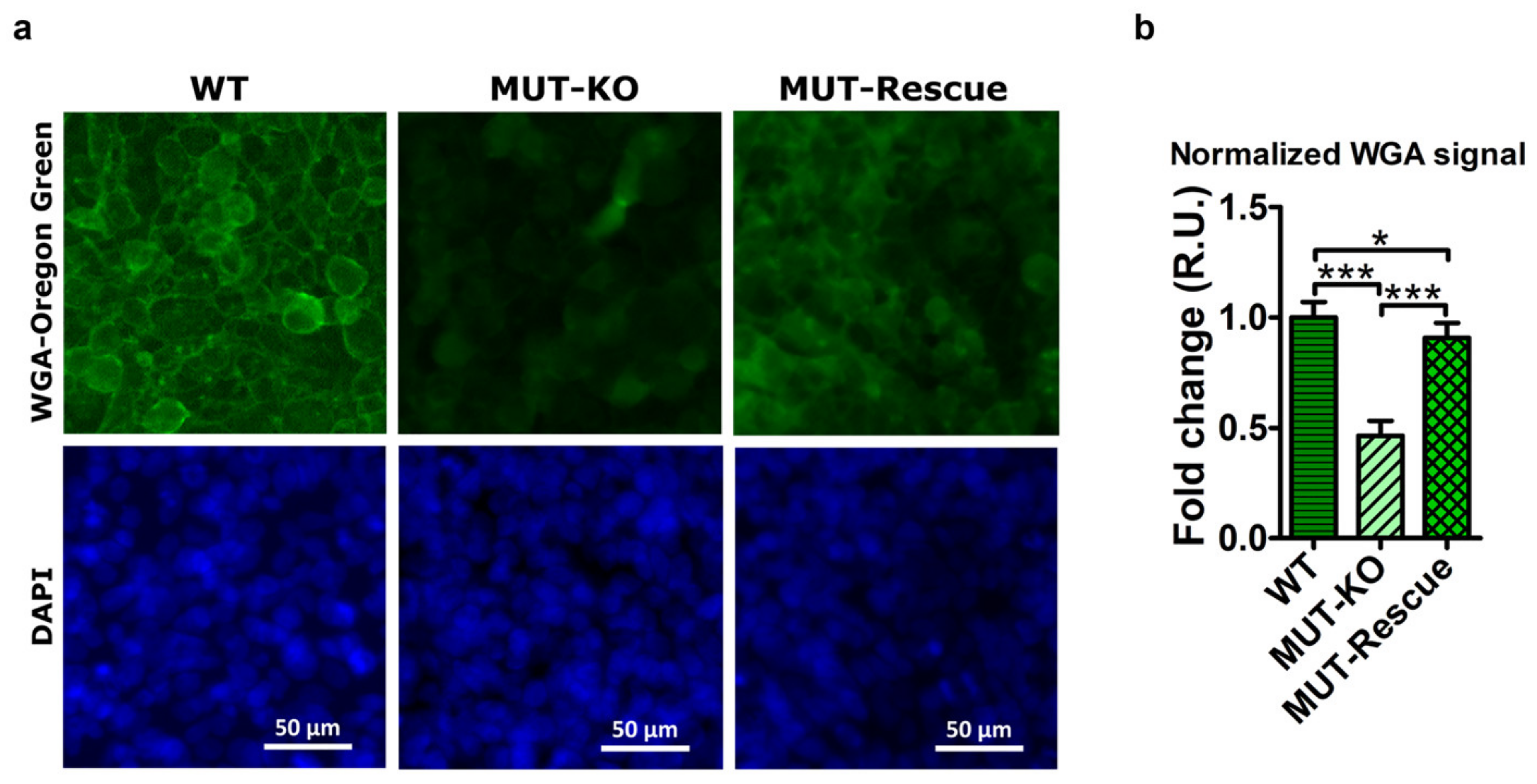
2.10. MUT Knockout Hampers Cell Structure and Morphology Impacting on Proteoglycans and Glycoproteins
3. Discussion
3.1. MUT-KO and Cell Architecture Modification: Cytoskeleton, Cell Adhesion, and Cell Junction Organization
3.2. Metal Homeostasis Unbalances in MUT-KO
3.3. Mechanisms of Stress Linked to MUT: ROS, Hyperammonemia, and Propionate
3.4. Mitochondrial Alterations in MUT-KO
3.5. Intracellular Trafficking in MUT-KO
4. Material and Methods
4.1. Cell Cultures and Treatments
4.2. Genome Editing and Transfections
4.3. Methylmalonic Acid and Propionylcarnitine Measurement
4.4. Microscopy Analysis
4.5. ROS Assay
4.6. MTT and Neutral-Red Assays
4.7. Western Blot
4.8. Quantitative Real-Time PCR
4.9. Proteomics Sample Preparation
4.10. nanoLC-MS/MS Measurements
4.11. Data Processing Following LC-MS/MS Acquisition
4.12. Bioinformatic Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chandler, R.J.; Venditti, C.P. Genetic and genomic systems to study methylmalonic acidemia. Mol. Genet. Metab. 2005, 86, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael Whitehead, V. Acquired and inherited disorders of cobalamin and folate in children. Br. J. Haematol. 2006, 134, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Okun, J.G. Methylmalonic acid—An endogenous toxin? Cell. Mol. Life Sci. 2005, 62, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Villani, G.R.; Gallo, G.; Scolamiero, E.; Salvatore, F.; Ruoppolo, M. “Classical organic acidurias”: Diagnosis and pathogenesis. Clin. Exp. Med. 2017, 17, 305–323. [Google Scholar] [CrossRef] [Green Version]
- Anzmann, A.F.; Pinto, S.; Busa, V.; Carlson, J.; McRitchie, S.; Sumner, S.; Pandey, A.; Vernon, H.J. Multi-omics studies in cellular models of methylmalonic acidemia and propionic acidemia reveal dysregulation of serine metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165538. [Google Scholar] [CrossRef]
- Scolamiero, E.; Cozzolino, C.; Albano, L.; Ansalone, A.; Caterino, M.; Corbo, G.; di Girolamo, M.G.; Di Stefano, C.; Durante, A.; Franzese, G.; et al. Targeted metabolomics in the expanded newborn screening for inborn errors of metabolism. Mol. Biosyst. 2015, 11, 1525–1535. [Google Scholar] [CrossRef]
- Haijes, H.A.; Jans, J.J.M.; Tas, S.Y.; Verhoeven-Duif, N.M.; Hasselt, P.M. Pathophysiology of propionic and methylmalonic acidemias. Part 1: Complications. J. Inherit. Metab. Dis. 2019, 42, 730–744. [Google Scholar] [CrossRef]
- Alkhunaizi, A.M.; Al-Sannaa, N. Renal Involvement in Methylmalonic Aciduria. Kidney Int. Rep. 2017, 2, 956–960. [Google Scholar] [CrossRef] [Green Version]
- Costanzo, M.; Zacchia, M.; Bruno, G.; Crisci, D.; Caterino, M.; Ruoppolo, M. Integration of Proteomics and Metabolomics in Exploring Genetic and Rare Metabolic Diseases. Kidney Dis. 2017, 3, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Knerr, I.; Weinhold, N.; Vockley, J.; Gibson, K.M. Advances and challenges in the treatment of branched-chain amino/keto acid metabolic defects. J. Inherit. Metab. Dis. 2012, 35, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Costanzo, M.; Cevenini, A.; Marchese, E.; Imperlini, E.; Raia, M.; Del Vecchio, L.; Caterino, M.; Ruoppolo, M. Label-Free Quantitative Proteomics in a Methylmalonyl-CoA Mutase-Silenced Neuroblastoma Cell Line. Int. J. Mol. Sci. 2018, 19, 3580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Ushiyama, A.; Kawakami, H.; Akimoto, Y.; Matsubara, S.; Iijima, T. Fluorescent imaging of endothelial glycocalyx layer with wheat germ agglutinin using intravital microscopy. Microsc. Res. Tech. 2016, 79, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, L.; DiBello, P.M.; Yu, M.; Miller, A.; Wang, S.; Willard, B.; Rosenblatt, D.S.; Jacobsen, D.W. The MMACHC proteome: Hallmarks of functional cobalamin deficiency in humans. Mol. Genet. Metab. 2011, 103, 226–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckes, B.; Colucci-Guyon, E.; Smola, H.; Nodder, S.; Babinet, C.; Krieg, T.; Martin, P. Impaired wound healing in embryonic and adult mice lacking vimentin. J. Cell Sci. 2000, 113, 2455–2462. [Google Scholar] [PubMed]
- Tsuruta, D. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J. Cell Sci. 2003, 116, 4977–4984. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.; Zheng, Z.; Chen, Y.; Wang, Q.; Zhang, Z.; Deng, H. Downregulation of vimentin expression increased drug resistance in ovarian cancer cells. Oncotarget 2016, 7, 45876–45888. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Shah, A.A.; Kalluri, R. Bone Morphogenic Protein-7 Induces Mesenchymal to Epithelial Transition in Adult Renal Fibroblasts and Facilitates Regeneration of Injured Kidney. J. Biol. Chem. 2005, 280, 8094–8100. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Thompson, E.W.; Williams, E.D. Mesenchymal to Epithelial Transition in Development and Disease. Cells Tissues Organs 2007, 185, 7–19. [Google Scholar] [CrossRef]
- Du, R.; Liu, B.; Zhou, L.; Wang, D.; He, X.; Xu, X.; Zhang, L.; Niu, C.; Liu, S. Downregulation of annexin A3 inhibits tumor metastasis and decreases drug resistance in breast cancer. Cell Death Dis. 2018, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- Hinck, L.; Nelson, W.; Papkoff, J. Wnt-1 modulates cell-cell adhesion in mammalian cells by stabilizing beta-catenin binding to the cell adhesion protein cadherin. J. Cell Biol. 1994, 124, 729–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bänziger, C.; Soldini, D.; Schütt, C.; Zipperlen, P.; Hausmann, G.; Basler, K. Wntless, a Conserved Membrane Protein Dedicated to the Secretion of Wnt Proteins from Signaling Cells. Cell 2006, 125, 509–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakou, E.; Hoyos-Flight, M.; Salinas, P.C. Wnt Signalling Promotes Actin Dynamics during Axon Remodelling through the Actin-Binding Protein Eps8. PLoS ONE 2015, 10, e0134976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Sun, Y.; Li, C.; Feng, J.; Li, Y.; Zhai, X.; Zhang, L.; Li, C. Ferritinophagic Flux Activation in CT26 Cells Contributed to EMT Inhibition Induced by a Novel Iron Chelator, DpdtpA. Oxid. Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Quiles del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238. [Google Scholar] [CrossRef] [Green Version]
- Moroishi, T.; Yamauchi, T.; Nishiyama, M.; Nakayama, K.I. HERC2 Targets the Iron Regulator FBXL5 for Degradation and Modulates Iron Metabolism. J. Biol. Chem. 2014, 289, 16430–16441. [Google Scholar] [CrossRef] [Green Version]
- Paterson, J.K.; Shukla, S.; Black, C.M.; Tachiwada, T.; Garfield, S.; Wincovitch, S.; Ernst, D.N.; Agadir, A.; Li, X.; Ambudkar, S.V.; et al. Human ABCB6 Localizes to Both the Outer Mitochondrial Membrane and the Plasma Membrane. Biochemistry 2007, 46, 9443–9452. [Google Scholar] [CrossRef]
- Andolfo, I.; Russo, R.; Manna, F.; De Rosa, G.; Gambale, A.; Zouwail, S.; Detta, N.; Pardo, C.L.; Alper, S.L.; Brugnara, C.; et al. Functional characterization of novel ABCB6 mutations and their clinical implications in familial pseudohyperkalemia. Haematologica 2016, 101, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Fontella, F.U.; Pulrolnik, V.; Gassen, E.; Wannmacher, C.M.D.; Klein, A.B.; Wajner, M.; Dutra-Filho, C.S. Propionic and L-methylmalonic acids induce oxidative stress in brain of young rats. Neuroreport 2000, 11, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.G.; Borges, C.G.; Seminotti, B.; Amaral, A.U.; Knebel, L.A.; Eichler, P.; de Oliveira, A.B.; Leipnitz, G.; Wajner, M. Experimental Evidence that Methylmalonic Acid Provokes Oxidative Damage and Compromises Antioxidant Defenses in Nerve Terminal and Striatum of Young Rats. Cell. Mol. Neurobiol. 2011, 31, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Luciani, A.; Schumann, A.; Berquez, M.; Chen, Z.; Nieri, D.; Failli, M.; Debaix, H.; Festa, B.P.; Tokonami, N.; Raimondi, A.; et al. Impaired mitophagy links mitochondrial disease to epithelial stress in methylmalonyl-CoA mutase deficiency. Nat. Commun. 2020, 11, 970. [Google Scholar] [CrossRef] [Green Version]
- Häberle, J.; Chakrapani, A.; Ah Mew, N.; Longo, N. Hyperammonaemia in classic organic acidaemias: A review of the literature and two case histories. Orphanet J. Rare Dis. 2018, 13, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häberle, J.; Görg, B.; Rutsch, F.; Schmidt, E.; Toutain, A.; Benoist, J.-F.; Gelot, A.; Suc, A.-L.; Höhne, W.; Schliess, F.; et al. Congenital Glutamine Deficiency with Glutamine Synthetase Mutations. N. Engl. J. Med. 2005, 353, 1926–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, R.M.; Licks, F.; Schemitt, E.G.; Colares, J.R.; do Couto Soares, M.; Zabot, G.P.; Fillmann, H.S.; Marroni, N.P. Protective effect of glutamine on the main and adjacent organs damaged by ischemia-reperfusion in rats. Protoplasma 2017, 254, 2155–2168. [Google Scholar] [CrossRef]
- Chandler, R.J.; Zerfas, P.M.; Shanske, S.; Sloan, J.; Hoffmann, V.; DiMauro, S.; Venditti, C.P. Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J. 2009, 23, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Caterino, M.; Chandler, R.J.; Sloan, J.L.; Dorko, K.; Cusmano-Ozog, K.; Ingenito, L.; Strom, S.C.; Imperlini, E.; Scolamiero, E.; Venditti, C.P.; et al. The proteome of methylmalonic acidemia (MMA): The elucidation of altered pathways in patient livers. Mol. Biosyst. 2016, 12, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Rosario, P.; Medina, J.M. Stimulation of ketogenesis by propionate in isolated rat hepatocytes: An explanation for ketosis associated with propionic acidaemia and methylmalonic acidaemia? J. Inherit. Metab. Dis. 1982, 5, 59–62. [Google Scholar] [CrossRef]
- Kim, S.; Jeon, J.M.; Kwon, O.K.; Choe, M.S.; Yeo, H.C.; Peng, X.; Cheng, Z.; Lee, M.Y.; Lee, S. Comparative Proteomic Analysis Reveals the Upregulation of Ketogenesis in Cardiomyocytes Differentiated from Induced Pluripotent Stem Cells. Proteomics 2019, 19, 1800284. [Google Scholar] [CrossRef]
- Khatri, D.; Zizioli, D.; Tiso, N.; Facchinello, N.; Vezzoli, S.; Gianoncelli, A.; Memo, M.; Monti, E.; Borsani, G.; Finazzi, D. Down-regulation of coasy, the gene associated with NBIA-VI, reduces Bmp signaling, perturbs dorso-ventral patterning and alters neuronal development in zebrafish. Sci. Rep. 2016, 6, 37660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djouadi, F.; Bastin, J. Mitochondrial Genetic Disorders: Cell Signaling and Pharmacological Therapies. Cells 2019, 8, 289. [Google Scholar] [CrossRef] [Green Version]
- Brown, Z.J.; Fu, Q.; Ma, C.; Kruhlak, M.; Zhang, H.; Luo, J.; Heinrich, B.; Yu, S.J.; Zhang, Q.; Wilson, A.; et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4+ T cell apoptosis promoting HCC development. Cell Death Dis. 2018, 9, 620. [Google Scholar] [CrossRef]
- Kølvraa, S. Inhibition of the Glycine Cleavage System by Branched-Chain Amino Acid Metabolites. Pediatr. Res. 1979, 13, 889–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simicek, M.; Lievens, S.; Laga, M.; Guzenko, D.; Aushev, V.N.; Kalev, P.; Baietti, M.F.; Strelkov, S.V.; Gevaert, K.; Tavernier, J.; et al. The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat. Cell Biol. 2013, 15, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Kimchi, A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 2012, 19, 788–797. [Google Scholar] [CrossRef]
- De Pasquale, V.; Caterino, M.; Costanzo, M.; Fedele, R.; Ruoppolo, M.; Pavone, L.M. Targeted metabolomic analysis of a mucopolysaccharidosis IIIB mouse model reveals an imbalance of branched-chain amino acid and fatty acid metabolism. Int. J. Mol. Sci. 2020, 21, 4211. [Google Scholar] [CrossRef]
- Ruoppolo, M.; Caterino, M.; Albano, L.; Pecce, R.; Di Girolamo, M.G.; Crisci, D.; Costanzo, M.; Milella, L.; Franconi, F.; Campesi, I. Targeted metabolomic profiling in rat tissues reveals sex differences. Sci. Rep. 2018, 8, 4663. [Google Scholar] [CrossRef]
- Giacco, A.; Delli Paoli, G.; Senese, R.; Cioffi, F.; Silvestri, E.; Moreno, M.; Ruoppolo, M.; Caterino, M.; Costanzo, M.; Lombardi, A.; et al. The saturation degree of fatty acids and their derived acylcarnitines determines the direct effect of metabolically active thyroid hormones on insulin sensitivity in skeletal muscle cells. FASEB J. 2019, 33, 1811–1823. [Google Scholar] [CrossRef] [Green Version]
- Cevenini, A.; Celia, C.; Orrù, S.; Sarnataro, D.; Raia, M.; Mollo, V.; Locatelli, M.; Imperlini, E.; Peluso, N.; Peltrini, R.; et al. Liposome-Embedding Silicon Microparticle for Oxaliplatin Delivery in Tumor Chemotherapy. Pharmaceutics 2020, 12, 559. [Google Scholar] [CrossRef]
- Caterino, M.; Ruoppolo, M.; Villani, G.R.D.; Marchese, E.; Costanzo, M.; Sotgiu, G.; Dore, S.; Franconi, F.; Campesi, I. Influence of Sex on Urinary Organic Acids: A Cross-Sectional Study in Children. Int. J. Mol. Sci. 2020, 21, 582. [Google Scholar] [CrossRef] [Green Version]
- De Pasquale, V.; Pezone, A.; Sarogni, P.; Tramontano, A.; Schiattarella, G.G.; Avvedimento, V.E.; Paladino, S.; Pavone, L.M. EGFR activation triggers cellular hypertrophy and lysosomal disease in NAGLU-depleted cardiomyoblasts, mimicking the hallmarks of mucopolysaccharidosis IIIB. Cell Death Dis. 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Fioretti, T.; Cevenini, A.; Zanobio, M.; Raia, M.; Sarnataro, D.; Salvatore, F.; Esposito, G. Crosstalk between 14-3-3θ and AF4 enhances MLL-AF4 activity and promotes leukemia cell proliferation. Cell. Oncol. 2019, 42, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Luber, C.A.; Cox, J.; Lauterbach, H.; Fancke, B.; Selbach, M.; Tschopp, J.; Akira, S.; Wiegand, M.; Hochrein, H.; O’Keeffe, M.; et al. Quantitative Proteomics Reveals Subset-Specific Viral Recognition in Dendritic Cells. Immunity 2010, 32, 279–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Carmody, L.C.; Blau, H.; Danis, D.; Zhang, X.A.; Gourdine, J.-P.; Vasilevsky, N.; Krawitz, P.; Thompson, M.D.; Robinson, P.N. Significantly different clinical phenotypes associated with mutations in synthesis and transamidase+remodeling glycosylphosphatidylinositol (GPI)-anchor biosynthesis genes. Orphanet J. Rare Dis. 2020, 15, 40. [Google Scholar] [CrossRef] [Green Version]
- Caterino, M.; Ruoppolo, M.; Mandola, A.; Costanzo, M.; Orrù, S.; Imperlini, E. Protein–protein interaction networks as a new perspective to evaluate distinct functional roles of voltage-dependent anion channel isoforms. Mol. Biosyst. 2017, 13, 2466–2476. [Google Scholar] [CrossRef]
- De Pasquale, V.; Costanzo, M.; Siciliano, R.; Mazzeo, M.; Pistorio, V.; Bianchi, L.; Marchese, E.; Ruoppolo, M.; Pavone, L.; Caterino, M. Proteomic Analysis of Mucopolysaccharidosis IIIB Mouse Brain. Biomolecules 2020, 10, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Term | Downregulated | Upregulated |
|---|---|---|
| BP | Zinc II ion transmembrane transport (GO:0071577) | Intermediate filament bundle assembly (GO:0045110) |
| BP | Positive regulation of cell communication (GO:0010647) | Coenzyme biosynthetic process (GO:0009108) |
| MF | Divalent inorganic cation transmembrane transporter activity (GO:0072509) | Oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor (GO:0016616) |
| CC | Autophagosome (GO:0005776) | Mitochondrion (GO:0005739) |
| CC | Autolysosome (GO:0044754) | Cytoskeleton (GO:0005856) |
| UniProt Accession | Protein Name | Gene Name | Regulation | Log2 Difference |
|---|---|---|---|---|
| P22033 | Methylmalonyl-CoA mutase, mitochondrial | MUT | DOWN | −8.4 |
| P23434 | Glycine cleavage system H protein, mitochondrial | GCSH | DOWN | −3.8 |
| Q99595 | Mitochondrial import inner membrane translocase subunit Tim17-A | TIMM17A | DOWN | −3.3 |
| Q8N4T8 | Carbonyl reductase family member 4 | CBR4 | DOWN | −3.1 |
| Q9NUQ2 | 1-acyl-sn-glycerol-3-phosphate acyltransferase epsilon | AGPAT5 | DOWN | −3.0 |
| Q9Y375 | Complex I intermediate-associated protein 30, mitochondrial | NDUFAF1 | DOWN | −2.7 |
| Q13772 | Nuclear receptor coactivator 4 | NCOA4 | DOWN | −1.1 |
| P02794 | Ferritin heavy chain | FTH1 | DOWN | −0.9 |
| P32119 | Peroxiredoxin-2 | PRDX2 | UP | 0.8 |
| P27105 | Erythrocyte band 7 integral membrane protein | STOM | UP | 0.8 |
| Q13057 | Bifunctional coenzyme A synthase | COASY | UP | 0.9 |
| Q9H2U2 | Inorganic pyrophosphatase 2, mitochondrial | PPA2 | UP | 0.9 |
| Q6YN16 | Hydroxysteroid dehydrogenase-like protein 2 | HSDL2 | UP | 1.0 |
| Q16698 | 2,4-dienoyl-CoA reductase, mitochondrial | DECR1 | UP | 1.0 |
| P35270 | Sepiapterin reductase | SPR | UP | 1.0 |
| P23786 | Carnitine O-palmitoyltransferase 2, mitochondrial | CPT2 | UP | 1.0 |
| Q8WVC6 | Dephospho-CoA kinase domain-containing protein | DCAKD | UP | 1.0 |
| P35914 | Hydroxymethylglutaryl-CoA lyase, mitochondrial | HMGCL | UP | 1.0 |
| P12532 | Creatine kinase U-type, mitochondrial | CKMT1A | UP | 1.1 |
| Q9BRX8 | Redox-regulatory protein FAM213A | FAM213A | UP | 1.2 |
| Q9NP58 | ATP-binding cassette sub-family B member 6, mitochondrial | ABCB6 | UP | 1.4 |
| Q9NRG7 | Epimerase family protein SDR39U1 | SDR39U1 | UP | 1.4 |
| Q9Y2Z9 | Ubiquinone biosynthesis monooxygenase COQ6, mitochondrial | COQ6 | UP | 1.7 |
| Q14790 | Caspase-8 | CASP8 | UP | 1.8 |
| Q86UT6 | NLR family member X1 | NLRX1 | UP | 2.1 |
| Q9BVL4 | Selenoprotein O | SELO | UP | 2.4 |
| Q6PML9 | Zinc transporter 9 | SLC30A9 | UP | 2.4 |
| O95139 | NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 6 | NDUFB6 | UP | 2.7 |
| A8MXV4 | Nucleoside diphosphate-linked moiety X motif 19, mitochondrial | NUDT19 | UP | 3.6 |
| Q8IYQ7 | Threonine synthase-like 1 | THNSL1 | UP | 3.7 |
| Q9BSF4 | Uncharacterized protein C19orf52 | C19orf52 | UP | 4.0 |
| Q9P0M9 | 39S ribosomal protein L27, mitochondrial | MRPL27 | UP | 4.5 |
| P00395 | Cytochrome c oxidase subunit 1 | COX1 | UP | 5.7 |
| UniProt Accession | Protein Name | Gene Name | Regulation | Log2 Difference | Cell Localization |
|---|---|---|---|---|---|
| Q93063 | Exostosin-2 | EXT2 | DOWN | −6.0 | ER, ERS, GA |
| O00622 | Protein CYR61 | CYR61 | DOWN | −5.9 | ER, ERS |
| Q9P2M7 | Cingulin | CGN | DOWN | −3.1 | CK, PM |
| Q2PZI1 | Probable C-mannosyltransferase DPY19L1 Alpha-1,2-mannosyltransferase ALG9 Fibronectin Mannosyl-oligosaccharide 1,2-alpha-mannosidase IA Annexin A3 Follistatin-related protein 1 | DPY19L1 | DOWN | −2.9 | N |
| Q9H6U8 | ALG9 | DOWN | −2.8 | ER | |
| P02751 | FN1 | DOWN | −2.7 | ER, ERS, PM | |
| P33908 | MAN1A1 | DOWN | −2.7 | C, ER, ERS, GA | |
| P12429 | ANXA3 | DOWN | −2.6 | C, ERS, PM | |
| Q12841 | FSTL1 | DOWN | −2.5 | ER, ERS | |
| Q12929 | Epidermal growth factor receptor kinase substrate 8 | EPS8 | DOWN | −2.1 | C, ERS, PM |
| P08670 | Vimentin | VIM | DOWN | −2.0 | C, CK, ERS, N, P, PM |
| P13591 | Neural cell adhesion molecule 1 | NCAM1 | DOWN | −1.6 | C, ERS, GA, PM |
| P34741 | Syndecan-2 | SDC2 | DOWN | −0.9 | ER, ERS, GA, L, PM |
| Q16706 | Alpha-mannosidase 2 | MAN2A1 | DOWN | −0.9 | ERS, GA |
| P23352 | Anosmin-1 | KAL1 | DOWN | −0.8 | ERS, PM |
| P10253 | Lysosomal alpha-glucosidase | GAA | UP | +0.8 | ERS, L, PM |
| P12830 | Cadherin-1 | CDH1 | UP | +3.9 | CK, E, ERS, GA, PM |
| Q6Y288 | Beta-1,3-glucosyltransferase | B3GALTL | UP | +5.8 | ER |
| UniProt Accession | Protein Name | Gene Name | Regulation | Log2 Difference | Cell Localization |
|---|---|---|---|---|---|
| Q9ULF5 | Zinc transporter ZIP10 | SLC39A10 | DOWN | −4.5 | PM |
| Q8IUH5 | Palmitoyltransferase ZDHHC17 | ZDHHC17 | DOWN | −4.2 | GA, PM |
| Q13433 | Zinc transporter ZIP6 | SLC39A6 | DOWN | −2.6 | ER, PM |
| Q9NY26 | Zinc transporter ZIP1 | SLC39A1 | DOWN | −1.1 | ER, PM |
| Q13772 | Nuclear receptor coactivator 4 | NCOA4 | DOWN | −1.1 | L, N |
| P02794 | Ferritin heavy chain | FTH1 | DOWN | −0.9 | C, ERS, L, N |
| P48200 | Iron-responsive element-binding protein 2 | IREB2 | UP | +0.9 | C, M |
| Q9NP58 | ATP-binding cassette sub-family B member 6, mitochondrial | ABCB6 | UP | +1.4 | C, E, ER, ERS, GA, M |
| Q6PML9 | Zinc transporter 9 | SLC30A9 | UP | +2.4 | CK, ER, N |
| UniProt Accession | Protein Name | Gene Name | Regulation | Log2 Difference | Cell Localization |
|---|---|---|---|---|---|
| P23434 | Glycine cleavage system H protein, mitochondrial | GCSH | DOWN | −3.8 | M |
| P35914 | Hydroxymethylglutaryl-CoA lyase, mitochondrial | HMGCL | UP | +0.9 | C, M, P |
| Q13057 | Bifunctional coenzyme A synthase | COASY | UP | +0.9 | ERS, M |
| Q16698 | 2,4-dienoyl-CoA reductase, mitochondrial | DECR1 | UP | +1.0 | C, M, N |
| P23786 | Carnitine O-palmitoyltransferase 2, mitochondrial | CPT2 | UP | +1.0 | M, N |
| Q6P1M0 A8MXV4 | Long-chain fatty acid transport protein 4 Nucleoside diphosphate-linked moiety X motif 19, mitochondrial | SLC27A4 NUDT19 | UP UP | +1.1 +3.6 | ER, PM C, P |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costanzo, M.; Caterino, M.; Cevenini, A.; Jung, V.; Chhuon, C.; Lipecka, J.; Fedele, R.; Guerrera, I.C.; Ruoppolo, M. Proteomics Reveals that Methylmalonyl-CoA Mutase Modulates Cell Architecture and Increases Susceptibility to Stress. Int. J. Mol. Sci. 2020, 21, 4998. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144998
Costanzo M, Caterino M, Cevenini A, Jung V, Chhuon C, Lipecka J, Fedele R, Guerrera IC, Ruoppolo M. Proteomics Reveals that Methylmalonyl-CoA Mutase Modulates Cell Architecture and Increases Susceptibility to Stress. International Journal of Molecular Sciences. 2020; 21(14):4998. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144998
Chicago/Turabian StyleCostanzo, Michele, Marianna Caterino, Armando Cevenini, Vincent Jung, Cerina Chhuon, Joanna Lipecka, Roberta Fedele, Ida Chiara Guerrera, and Margherita Ruoppolo. 2020. "Proteomics Reveals that Methylmalonyl-CoA Mutase Modulates Cell Architecture and Increases Susceptibility to Stress" International Journal of Molecular Sciences 21, no. 14: 4998. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144998