The Adenosine System at the Crossroads of Intestinal Inflammation and Neoplasia

 , ,
, ,
Abstract
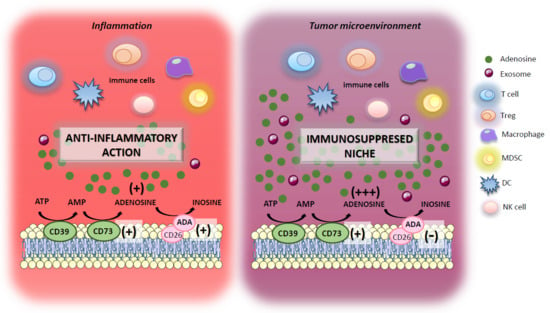
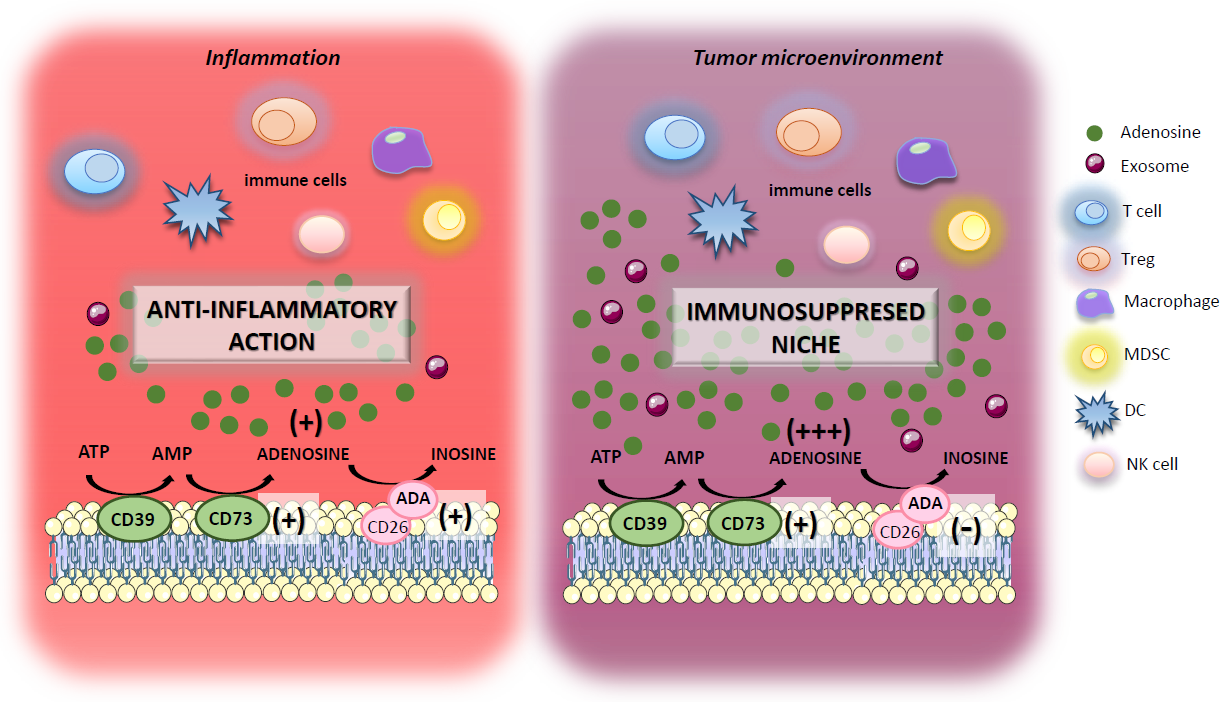
:
{kind=link}
{kind=link}
{kind=link}
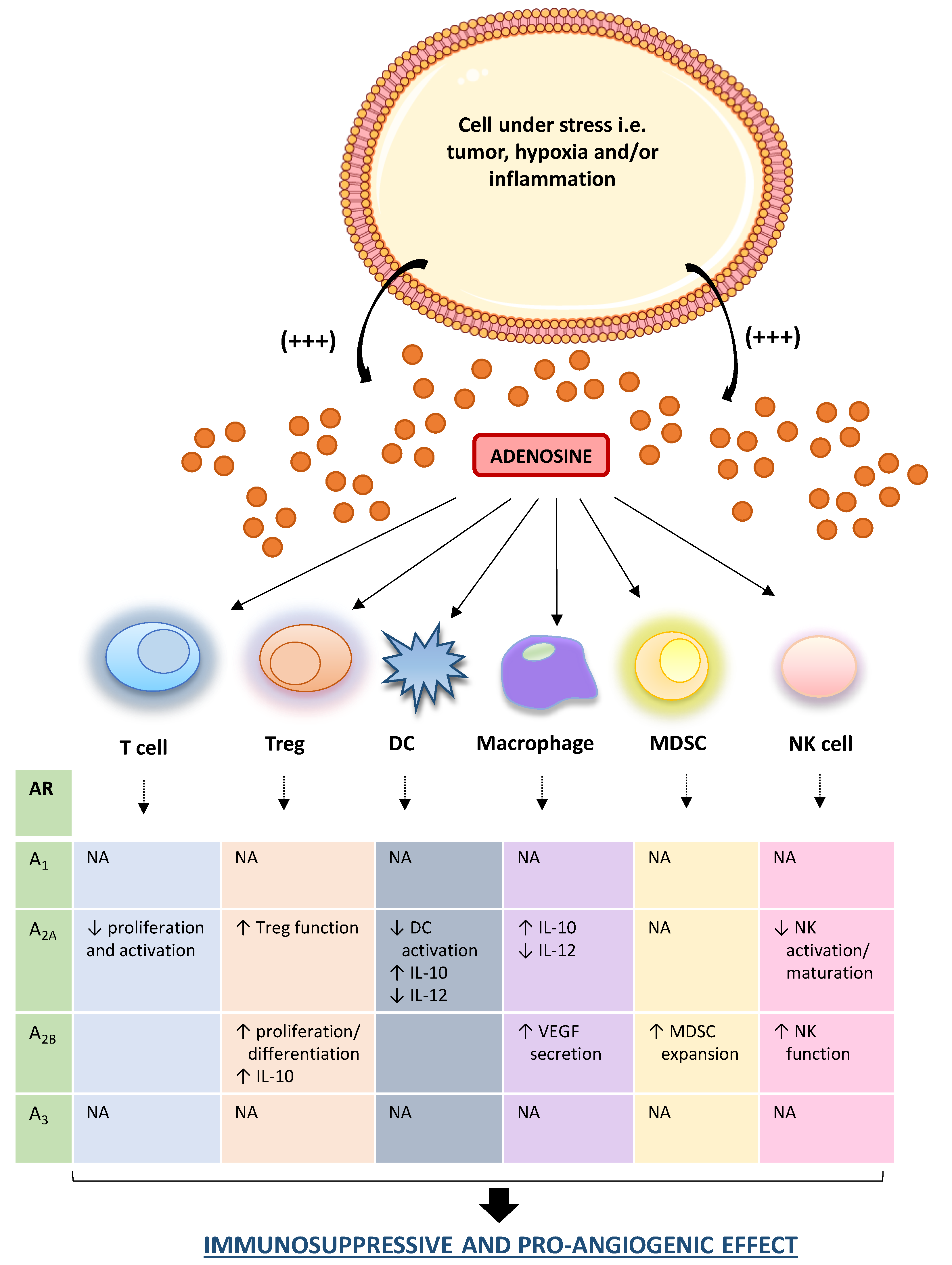{kind=link}
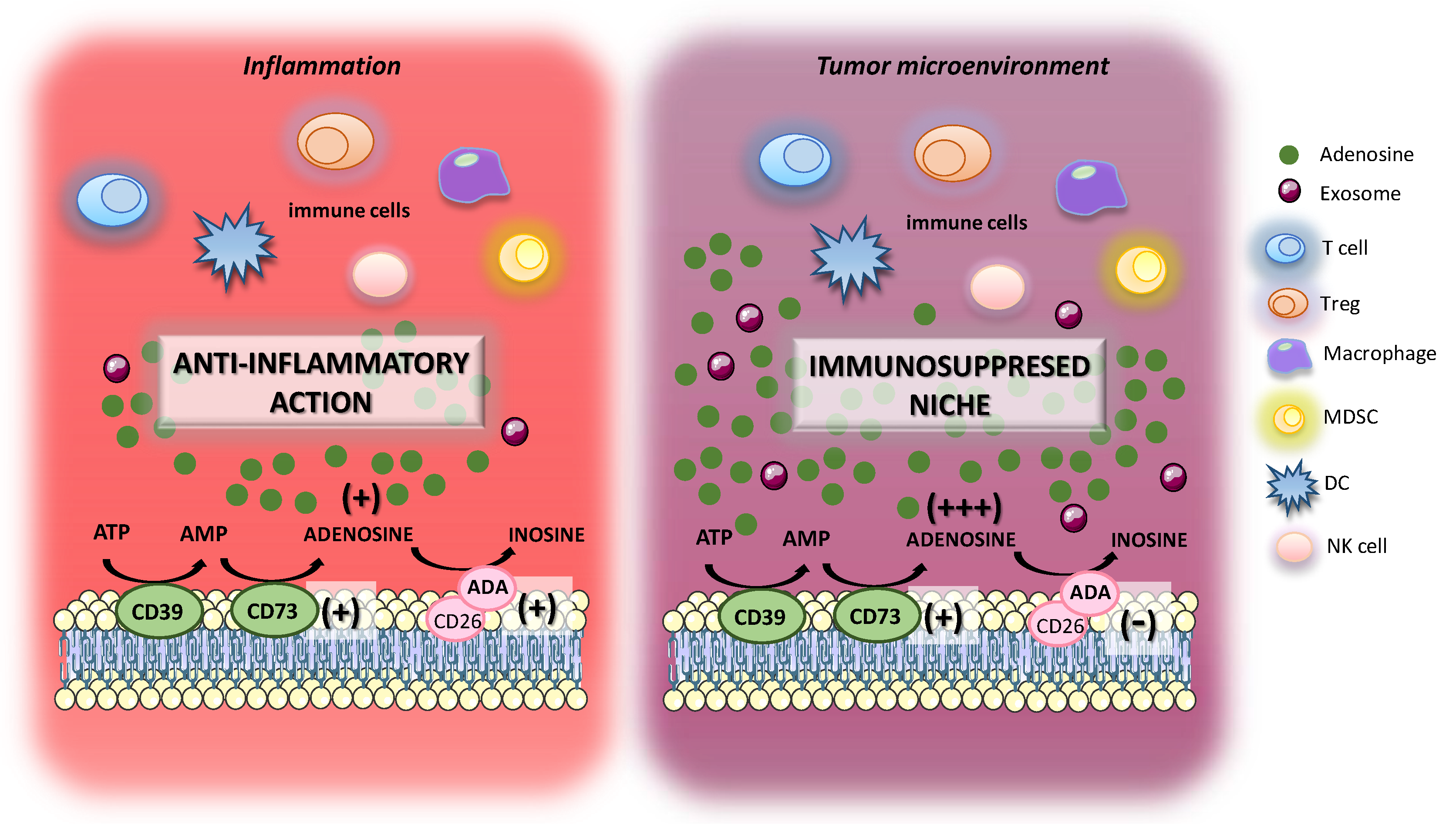{kind=link}
1. Introduction
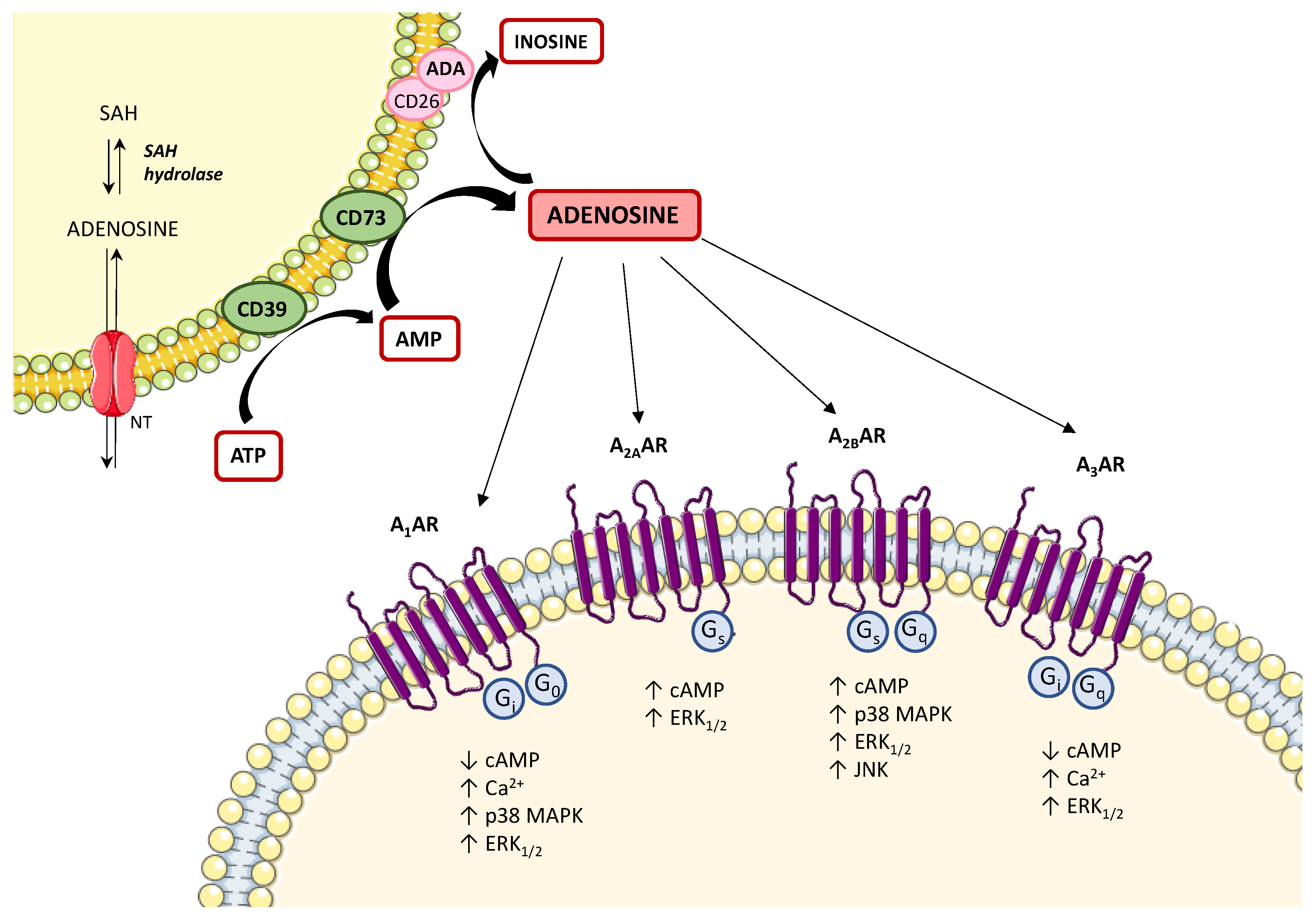
2. Adenosine System: Enzymes, Transporters, and Receptors
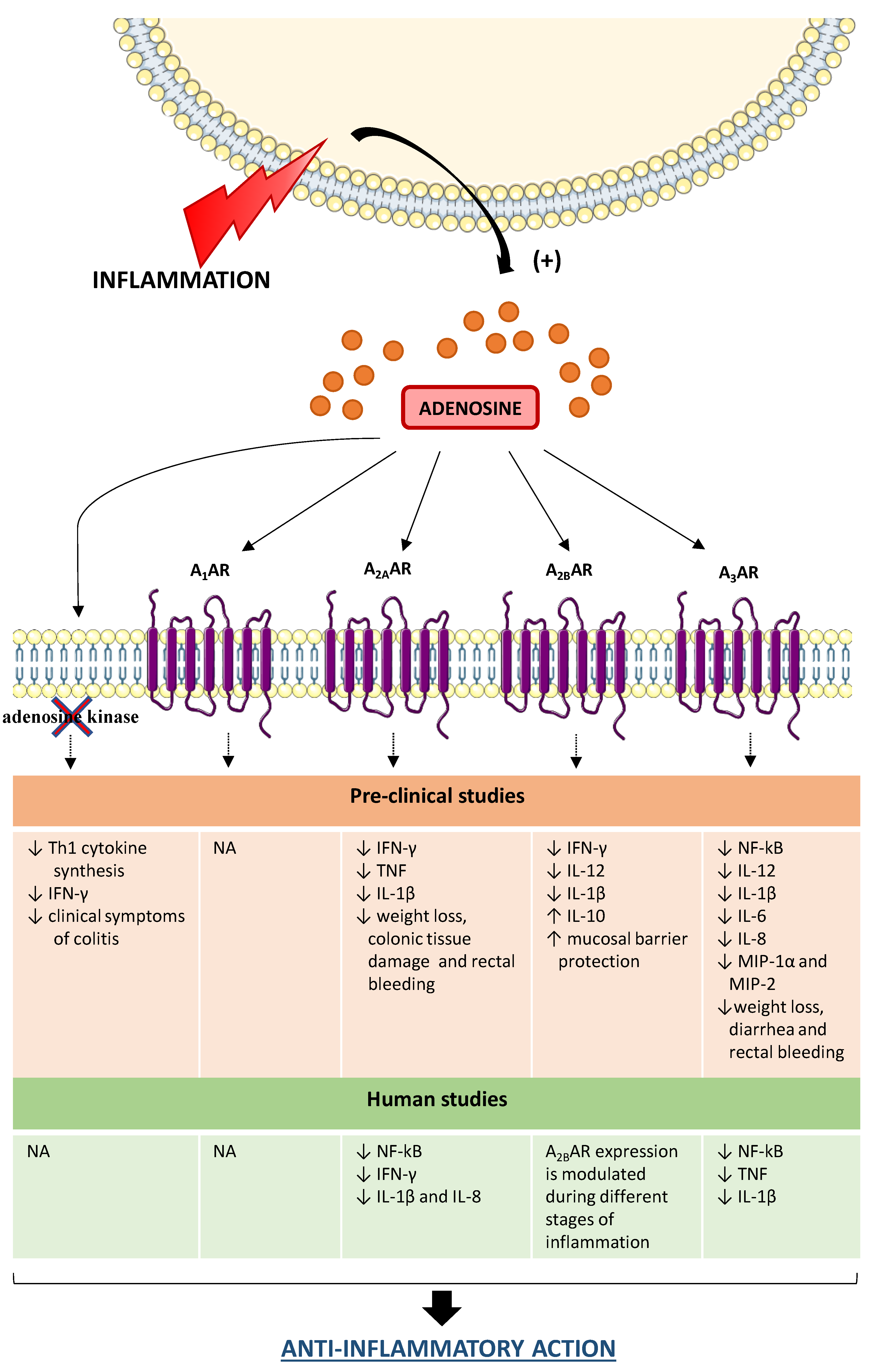
3. Adenosine System and Inflammatory Bowel Diseases (IBDs)
4. Adenosine System and Immune Cell Interaction in Colitis-Associated Cancer Pathogenesis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Marone, G.; Mantovani, A. Cancer Inflammation and Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuomisto, A.E.; Makinen, M.J.; Vayrynen, J.P. Systemic inflammation in colorectal cancer: Underlying factors, effects, and prognostic significance. World J. Gastroenterol. 2019, 25, 4383–4404. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L.; Svrcek, M.; Seksik, P.; Bouvier, A.M.; Simon, T.; Allez, M.; Brixi, H.; Gornet, J.M.; Altwegg, R.; Beau, P.; et al. Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease. Gastroenterology 2013, 145, 166–175.e168. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Jin, X.; Man, C.; Gao, Z.; Wang, X. Meta-analysis of the association between the inflammatory potential of diet and colorectal cancer risk. Oncotarget 2017, 8, 59592–59600. [Google Scholar] [CrossRef] [PubMed]
- De Waal, G.M.; De Villiers, W.J.S.; Forgan, T.; Roberts, T.; Pretorius, E. Colorectal cancer is associated with increased circulating lipopolysaccharide, inflammation and hypercoagulability. Sci. Rep. 2020, 10, 8777. [Google Scholar] [CrossRef] [PubMed]
- Keller, D.S.; Windsor, A.; Cohen, R.; Chand, M. Colorectal cancer in inflammatory bowel disease: Review of the evidence. Tech. Coloproctol. 2019, 23, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.J.; Qiu, X.Y.; Mao, X.Q.; Li, X.T.; Zhang, H.J. Systematic review with meta-analysis: Thiopurines decrease the risk of colorectal neoplasia in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2018, 47, 318–331. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Shen, Z.F.; Wu, B.S.; Xu, C.B.; He, Z.Q.; Chen, T.; Shang, H.T.; Xie, C.F.; Huang, S.Y.; Chen, Y.G.; et al. Risk of Colorectal Cancer in Ulcerative Colitis Patients: A Systematic Review and Meta-Analysis. Gastroenterol. Res. Pract. 2019, 2019, 5363261. [Google Scholar] [CrossRef] [Green Version]
- D’Antongiovanni, V.; Martinelli, S.; Richter, S.; Canu, L.; Guasti, D.; Mello, T.; Romagnoli, P.; Pacak, K.; Eisenhofer, G.; Mannelli, M.; et al. The microenvironment induces collective migration in SDHB-silenced mouse pheochromocytoma spheroids. Endocr. Relat. Cancer 2017, 24, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Richter, S.; D’Antongiovanni, V.; Martinelli, S.; Bechmann, N.; Riverso, M.; Poitz, D.M.; Pacak, K.; Eisenhofer, G.; Mannelli, M.; Rapizzi, E. Primary fibroblast co-culture stimulates growth and metabolism in Sdhb-impaired mouse pheochromocytoma MTT cells. Cell Tissue Res. 2018, 374, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, X.; Wu, F.; Zhou, Y.; Bao, Z.; Li, H.; Zheng, P.; Zhao, S. Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and EMT in gastric cancer. Cell Death Dis. 2019, 10, 918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umakoshi, M.; Takahashi, S.; Itoh, G.; Kuriyama, S.; Sasaki, Y.; Yanagihara, K.; Yashiro, M.; Maeda, D.; Goto, A.; Tanaka, M. Macrophage-mediated transfer of cancer-derived components to stromal cells contributes to establishment of a pro-tumor microenvironment. Oncogene 2019, 38, 2162–2176. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.; Sharma, N.; Gupta, K.B.; Dhiman, M. Role of immune system in tumor progression and carcinogenesis. J. Cell. Biochem. 2018, 119, 5028–5042. [Google Scholar] [CrossRef]
- Bonavita, E.; Galdiero, M.R.; Jaillon, S.; Mantovani, A. Phagocytes as Corrupted Policemen in Cancer-Related Inflammation. Adv. Cancer Res. 2015, 128, 141–171. [Google Scholar] [CrossRef] [Green Version]
- Nakatsumi, H.; Matsumoto, M.; Nakayama, K.I. Noncanonical Pathway for Regulation of CCL2 Expression by an mTORC1-FOXK1 Axis Promotes Recruitment of Tumor-Associated Macrophages. Cell Rep. 2017, 21, 2471–2486. [Google Scholar] [CrossRef] [Green Version]
- Seager, R.J.; Hajal, C.; Spill, F.; Kamm, R.D.; Zaman, M.H. Dynamic interplay between tumour, stroma and immune system can drive or prevent tumour progression. Converg. Sci. Phys. Oncol. 2017, 3, 034002. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Schetter, A.J.; Heegaard, N.H.; Harris, C.C. Inflammation and cancer: Interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2010, 31, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Blay, J.; White, T.D.; Hoskin, D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997, 57, 2602–2605. [Google Scholar]
- Arab, S.; Hadjati, J. Adenosine Blockage in Tumor Microenvironment and Improvement of Cancer Immunotherapy. Immune Netw. 2019, 19, e23. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, D.W.; Butler, J.J.; Drapeau, D.; Haeryfar, S.M.; Blay, J. Adenosine acts through an A3 receptor to prevent the induction of murine anti-CD3-activated killer T cells. Int. J. Cancer 2002, 99, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Novitskiy, S.V.; Sachsenmeier, K.F.; Fornai, M.; Blandizzi, C.; Hasko, G. Switching off CD73: A way to boost the activity of conventional and targeted antineoplastic therapies. Drug Discov. Today 2017, 22, 1686–1696. [Google Scholar] [CrossRef]
- Liu, H.; Xia, Y. Beneficial and detrimental role of adenosine signaling in diseases and therapy. J. Appl. Physiol. 2015, 119, 1173–1182. [Google Scholar] [CrossRef]
- Lofgren, L.; Pehrsson, S.; Hagglund, G.; Tjellstrom, H.; Nylander, S. Accurate measurement of endogenous adenosine in human blood. PLoS ONE 2018, 13, e0205707. [Google Scholar] [CrossRef] [Green Version]
- Deussen, A.; Stappert, M.; Schafer, S.; Kelm, M. Quantification of extracellular and intracellular adenosine production: Understanding the transmembranous concentration gradient. Circulation 1999, 99, 2041–2047. [Google Scholar] [CrossRef]
- Hasko, G.; Cronstein, B.N. Adenosine: An endogenous regulator of innate immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. The Purinergic System as a Pharmacological Target for the Treatment of Immune-Mediated Inflammatory Diseases. Pharmacol. Rev. 2019, 71, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.V.; Kjaergaard, J.; Lukashev, D.; Ohta, A. Hypoxia-adenosinergic immunosuppression: Tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5947–5952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spychala, J. Tumor-promoting functions of adenosine. Pharmacol. Ther. 2000, 87, 161–173. [Google Scholar] [CrossRef]
- Hasko, G.; Pacher, P. Regulation of macrophage function by adenosine. Arter. Thromb. Vasc. Biol. 2012, 32, 865–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, J. Adenosine metabolism and cancer. Focus on “Adenosine downregulates DPPIV on HT-29 colon cancer cells by stimulating protein tyrosine phosphatases and reducing ERK1/2 activity via a novel pathway”. Am. J. Physiol. Cell Physiol. 2006, 291, C405–C406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Zhang, J.; Zhang, Q.; Chen, P.; Song, J.; Yu, S.; Liu, H.; Liu, F.; Song, C.; Yang, D.; et al. Adenosine induces apoptosis in human liver cancer cells through ROS production and mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2014, 448, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Colucci, R.; La Motta, C.; Tuccori, M.; Awwad, O.; Da Settimo, F.; Blandizzi, C.; Fornai, M. Adenosine deaminase in the modulation of immune system and its potential as a novel target for treatment of inflammatory disorders. Curr. Drug Targets 2012, 13, 842–862. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K. Extracellular adenosine signaling in molecular medicine. J. Mol. Med. 2013, 91, 141–146. [Google Scholar] [CrossRef]
- Pastor-Anglada, M.; Urtasun, N.; Perez-Torras, S. Intestinal Nucleoside Transporters: Function, Expression, and Regulation. Compr. Physiol. 2018, 8, 1003–1017. [Google Scholar] [CrossRef]
- Antonioli, L.; Fornai, M.; Colucci, R.; Ghisu, N.; Tuccori, M.; Del Tacca, M.; Blandizzi, C. Regulation of enteric functions by adenosine: Pathophysiological and pharmacological implications. Pharmacol. Ther. 2008, 120, 233–253. [Google Scholar] [CrossRef]
- Archer, R.G.; Pitelka, V.; Hammond, J.R. Nucleoside transporter subtype expression and function in rat skeletal muscle microvascular endothelial cells. Br. J. Pharmacol. 2004, 143, 202–214. [Google Scholar] [CrossRef] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathological overproduction: The bad side of adenosine. Br. J. Pharmacol. 2017, 174, 1945–1960. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wainwright, D.A.; Wu, J.D.; Wan, Y.; Matei, D.E.; Zhang, Y.; Zhang, B. CD73: An emerging checkpoint for cancer immunotherapy. Immunotherapy 2019, 11, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yaguchi, T.; Lim, C.R.; Ishizawa, Y.; Nakano, T.; Nishizaki, T. Tuning of apoptosis-mediator gene transcription in HepG2 human hepatoma cells through an adenosine signal. Cancer Lett. 2010, 291, 225–229. [Google Scholar] [CrossRef]
- Sheth, S.; Brito, R.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Adenosine receptors: Expression, function and regulation. Int. J. Mol. Sci. 2014, 15, 2024–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol. 2008, 32, 527–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinger, M.; Freissmuth, M.; Nanoff, C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell. Signal. 2002, 14, 99–108. [Google Scholar] [CrossRef]
- Da Silva, C.G.; Jarzyna, R.; Specht, A.; Kaczmarek, E. Extracellular nucleotides and adenosine independently activate AMP-activated protein kinase in endothelial cells: Involvement of P2 receptors and adenosine transporters. Circ. Res. 2006, 98, e39–e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boison, D. Adenosine kinase: Exploitation for therapeutic gain. Pharmacol. Rev. 2013, 65, 906–943. [Google Scholar] [CrossRef] [Green Version]
- Mato, J.M.; Martinez-Chantar, M.L.; Lu, S.C. Methionine metabolism and liver disease. Annu. Rev. Nutr. 2008, 28, 273–293. [Google Scholar] [CrossRef] [Green Version]
- Nadeem, M.S.; Kumar, V.; Al-Abbasi, F.A.; Kamal, M.A.; Anwar, F. Risk of colorectal cancer in inflammatory bowel diseases. Semin. Cancer Biol. 2019, 64, 51–60. [Google Scholar] [CrossRef]
- Andersen, N.N.; Jess, T. Has the risk of colorectal cancer in inflammatory bowel disease decreased? World J. Gastroenterol. 2013, 19, 7561–7568. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Kareemi, H.; Parab, R.; Barkema, H.W.; Quan, H.; Myers, R.P.; Kaplan, G.G. Incidence of primary sclerosing cholangitis: A systematic review and meta-analysis. Hepatology 2011, 53, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Hernandez, V.; Myrelid, P.; Kariv, R.; Tsianos, E.; Toruner, M.; Marti-Gallostra, M.; Spinelli, A.; Van Der Meulen-de Jong, A.E.; Yuksel, E.S.; et al. Colorectal cancer in inflammatory bowel disease: Results of the 3rd ECCO pathogenesis scientific workshop (I). J. Crohn’s Colitis 2014, 8, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.11–15.25.14. [Google Scholar] [CrossRef] [PubMed]
- Alex, P.; Zachos, N.C.; Nguyen, T.; Gonzales, L.; Chen, T.E.; Conklin, L.S.; Centola, M.; Li, X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm. Bowel Dis. 2009, 15, 341–352. [Google Scholar] [CrossRef]
- Linden, J. Molecular approach to adenosine receptors: Receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 775–787. [Google Scholar] [CrossRef]
- Hasko, G.; Cronstein, B. Regulation of inflammation by adenosine. Front. Immunol. 2013, 4, 85. [Google Scholar] [CrossRef] [Green Version]
- Siegmund, B.; Rieder, F.; Albrich, S.; Wolf, K.; Bidlingmaier, C.; Firestein, G.S.; Boyle, D.; Lehr, H.A.; Loher, F.; Hartmann, G.; et al. Adenosine kinase inhibitor GP515 improves experimental colitis in mice. J. Pharmacol. Exp. Ther. 2001, 296, 99–105. [Google Scholar]
- Pallio, G.; Bitto, A.; Pizzino, G.; Galfo, F.; Irrera, N.; Squadrito, F.; Squadrito, G.; Pallio, S.; Anastasi, G.P.; Cutroneo, G.; et al. Adenosine Receptor Stimulation by Polydeoxyribonucleotide Improves Tissue Repair and Symptomology in Experimental Colitis. Front. Pharmacol. 2016, 7, 273. [Google Scholar] [CrossRef] [Green Version]
- Selmeczy, Z.; Csoka, B.; Pacher, P.; Vizi, E.S.; Hasko, G. The adenosine A2A receptor agonist CGS 21680 fails to ameliorate the course of dextran sulphate-induced colitis in mice. Inflamm. Res. 2007, 56, 204–209. [Google Scholar] [CrossRef] [Green Version]
- Kolachala, V.; Asamoah, V.; Wang, L.; Obertone, T.S.; Ziegler, T.R.; Merlin, D.; Sitaraman, S.V. TNF-alpha upregulates adenosine 2b (A2b) receptor expression and signaling in intestinal epithelial cells: A basis for A2bR overexpression in colitis. Cell. Mol. Life Sci. 2005, 62, 2647–2657. [Google Scholar] [CrossRef]
- Kolachala, V.; Ruble, B.; Vijay-Kumar, M.; Wang, L.; Mwangi, S.; Figler, H.; Figler, R.; Srinivasan, S.; Gewirtz, A.; Linden, J.; et al. Blockade of adenosine A2B receptors ameliorates murine colitis. Br. J. Pharmacol. 2008, 155, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Frick, J.S.; MacManus, C.F.; Scully, M.; Glover, L.E.; Eltzschig, H.K.; Colgan, S.P. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J. Immunol. 2009, 182, 4957–4964. [Google Scholar] [CrossRef] [Green Version]
- Aherne, C.M.; Saeedi, B.; Collins, C.B.; Masterson, J.C.; McNamee, E.N.; Perrenoud, L.; Rapp, C.R.; Curtis, V.F.; Bayless, A.; Fletcher, A.; et al. Epithelial-specific A2B adenosine receptor signaling protects the colonic epithelial barrier during acute colitis. Mucosal Immunol. 2015, 8, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Grants, I.; Alhaj, M.; McKiernan, M.; Jacobson, M.; Hassanain, H.H.; Frankel, W.; Wunderlich, J.; Christofi, F.L. Impact of disrupting adenosine A(3) receptors (A(3)(-)/(-) AR) on colonic motility or progression of colitis in the mouse. Inflamm. Bowel Dis. 2011, 17, 1698–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabley, J.; Soriano, F.; Pacher, P.; Hasko, G.; Marton, A.; Wallace, R.; Salzman, A.; Szabo, C. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide, is protective in two murine models of colitis. Eur. J. Pharmacol. 2003, 466, 323–329. [Google Scholar] [CrossRef]
- Ren, T.; Tian, T.; Feng, X.; Ye, S.; Wang, H.; Wu, W.; Qiu, Y.; Yu, C.; He, Y.; Zeng, J.; et al. An adenosine A3 receptor agonist inhibits DSS-induced colitis in mice through modulation of the NF-kappaB signaling pathway. Sci. Rep. 2015, 5, 9047. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; He, Y.; Feng, X.; Ye, S.; Wang, H.; Tan, W.; Yu, C.; Hu, J.; Zheng, R.; Zhou, Y. MicroRNA-206 is involved in the pathogenesis of ulcerative colitis via regulation of adenosine A3 receptor. Oncotarget 2017, 8, 705–721. [Google Scholar] [CrossRef] [Green Version]
- Ren, T.; Qiu, Y.; Wu, W.; Feng, X.; Ye, S.; Wang, Z.; Tian, T.; He, Y.; Yu, C.; Zhou, Y. Activation of adenosine A3 receptor alleviates TNF-alpha-induced inflammation through inhibition of the NF-kappaB signaling pathway in human colonic epithelial cells. Mediat. Inflamm. 2014, 2014, 818251. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Zhou, Y.; Feng, X.; Ye, S.; Wang, H.; Wu, W.; Tan, W.; Yu, C.; Hu, J.; Zheng, R.; et al. MicroRNA-16 is putatively involved in the NF-kappaB pathway regulation in ulcerative colitis through adenosine A2a receptor (A2aAR) mRNA targeting. Sci. Rep. 2016, 6, 30824. [Google Scholar] [CrossRef]
- Ren, T.H.; Lv, M.M.; An, X.M.; Leung, W.K.; Seto, W.K. Activation of adenosine A3 receptor inhibits inflammatory cytokine production in colonic mucosa of patients with ulcerative colitis by down-regulating the nuclear factor-kappa B signaling. J. Dig. Dis. 2020, 21, 38–45. [Google Scholar] [CrossRef]
- Danese, S.; Malesci, A.; Vetrano, S. Colitis-associated cancer: The dark side of inflammatory bowel disease. Gut 2011, 60, 1609–1610. [Google Scholar] [CrossRef] [PubMed]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef] [PubMed]
- Parang, B.; Barrett, C.W.; Williams, C.S. AOM/DSS Model of Colitis-Associated Cancer. Methods Mol. Biol. 2016, 1422, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Aghaei, M.; Karami-Tehrani, F.; Panjehpour, M.; Salami, S.; Fallahian, F. Adenosine induces cell-cycle arrest and apoptosis in androgen-dependent and -independent prostate cancer cell lines, LNcap-FGC-10, DU-145, and PC3. Prostate 2012, 72, 361–375. [Google Scholar] [CrossRef]
- Yang, D.; Yaguchi, T.; Yamamoto, H.; Nishizaki, T. Intracellularly transported adenosine induces apoptosis in HuH-7 human hepatoma cells by downregulating c-FLIP expression causing caspase-3/-8 activation. Biochem. Pharmacol. 2007, 73, 1665–1675. [Google Scholar] [CrossRef]
- Wu, X.R.; He, X.S.; Chen, Y.F.; Yuan, R.X.; Zeng, Y.; Lian, L.; Zou, Y.F.; Lan, N.; Wu, X.J.; Lan, P. High expression of CD73 as a poor prognostic biomarker in human colorectal cancer. J. Surg. Oncol. 2012, 106, 130–137. [Google Scholar] [CrossRef]
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rebe, C.; Derangere, V.; Chevriaux, A.; Boidot, R.; Vegran, F.; Bonnefoy, N.; Vincent, J.; et al. Accumulation of MDSC and Th17 Cells in Patients with Metastatic Colorectal Cancer Predicts the Efficacy of a FOLFOX-Bevacizumab Drug Treatment Regimen. Cancer Res. 2016, 76, 5241–5252. [Google Scholar] [CrossRef] [Green Version]
- Ahlmanner, F.; Sundstrom, P.; Akeus, P.; Eklof, J.; Borjesson, L.; Gustavsson, B.; Lindskog, E.B.; Raghavan, S.; Quiding-Jarbrink, M. CD39(+) regulatory T cells accumulate in colon adenocarcinomas and display markers of increased suppressive function. Oncotarget 2018, 9, 36993–37007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Cheng, B.; Li, F.S.; Ding, J.H.; Feng, Y.Y.; Zhuo, G.Z.; Wei, H.F.; Zhao, K. High expression of CD39/ENTPD1 in malignant epithelial cells of human rectal adenocarcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 9411–9419. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bowser, J.L.; Phan, L.H.; Eltzschig, H.K. The Hypoxia-Adenosine Link during Intestinal Inflammation. J. Immunol. 2018, 200, 897–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitkovsky, M.; Lukashev, D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 2005, 5, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, D.; Klebanov, B.; Kojima, H.; Grinberg, A.; Ohta, A.; Berenfeld, L.; Wenger, R.H.; Ohta, A.; Sitkovsky, M. Cutting edge: Hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J. Immunol. 2006, 177, 4962–4965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashev, D.; Ohta, A.; Sitkovsky, M. Hypoxia-dependent anti-inflammatory pathways in protection of cancerous tissues. Cancer Metastasis Rev. 2007, 26, 273–279. [Google Scholar] [CrossRef]
- Wu, R.; Chen, Y.; Li, F.; Li, W.; Zhou, H.; Yang, Y.; Pei, Z. Effects of CD73 on human colorectal cancer cell growth in vivo and in vitro. Oncol. Rep. 2016, 35, 1750–1756. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J.; et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 515. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [Green Version]
- Richard, E.; Alam, S.M.; Arredondo-Vega, F.X.; Patel, D.D.; Hershfield, M.S. Clustered charged amino acids of human adenosine deaminase comprise a functional epitope for binding the adenosine deaminase complexing protein CD26/dipeptidyl peptidase IV. J. Biol. Chem. 2002, 277, 19720–19726. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.Y.; Mujoomdar, M.; Blay, J. Adenosine down-regulates the surface expression of dipeptidyl peptidase IV on HT-29 human colorectal carcinoma cells: Implications for cancer cell behavior. Am. J. Pathol. 2004, 165, 319–330. [Google Scholar] [CrossRef]
- Tan, E.Y.; Richard, C.L.; Zhang, H.; Hoskin, D.W.; Blay, J. Adenosine downregulates DPPIV on HT-29 colon cancer cells by stimulating protein tyrosine phosphatase(s) and reducing ERK1/2 activity via a novel pathway. Am. J. Physiol. Cell Physiol. 2006, 291, C433–C444. [Google Scholar] [CrossRef]
- Decking, U.K.; Schlieper, G.; Kroll, K.; Schrader, J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ. Res. 1997, 81, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A. A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol. 2016, 7, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitkovsky, M.V.; Hatfield, S.; Abbott, R.; Belikoff, B.; Lukashev, D.; Ohta, A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol. Res. 2014, 2, 598–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Muller, C.E.; Murakami, T.; Robson, S.C. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030–1040. [Google Scholar] [CrossRef] [Green Version]
- Iannone, R.; Miele, L.; Maiolino, P.; Pinto, A.; Morello, S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia 2013, 15, 1400–1409. [Google Scholar] [CrossRef] [Green Version]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I.; et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008, 112, 1822–1831. [Google Scholar] [CrossRef]
- Romio, M.; Reinbeck, B.; Bongardt, S.; Huls, S.; Burghoff, S.; Schrader, J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. Am. J. Physiol. Cell Physiol. 2011, 301, C530–C539. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef] [Green Version]
- Cekic, C.; Day, Y.J.; Sag, D.; Linden, J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef] [Green Version]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [Green Version]
- Panther, E.; Corinti, S.; Idzko, M.; Herouy, Y.; Napp, M.; La Sala, A.; Girolomoni, G.; Norgauer, J. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood 2003, 101, 3985–3990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, M.; Shang, L.; Santaolalla, R.; Sotolongo, J.; Pastorini, C.; Espana, C.; Ungaro, R.; Harpaz, N.; Cooper, H.S.; Elson, G.; et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm. Bowel Dis. 2011, 17, 1464–1473. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Cytokines, IBD, and colitis-associated cancer. Inflamm. Bowel Dis. 2015, 21, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryzhov, S.; Novitskiy, S.V.; Goldstein, A.E.; Biktasova, A.; Blackburn, M.R.; Biaggioni, I.; Dikov, M.M.; Feoktistov, I. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J. Immunol. 2011, 187, 6120–6129. [Google Scholar] [CrossRef] [Green Version]
- Ehrentraut, H.; Westrich, J.A.; Eltzschig, H.K.; Clambey, E.T. Adora2b adenosine receptor engagement enhances regulatory T cell abundance during endotoxin-induced pulmonary inflammation. PLoS ONE 2012, 7, e32416. [Google Scholar] [CrossRef]
- Sorrentino, C.; Miele, L.; Porta, A.; Pinto, A.; Morello, S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget 2015, 6, 27478–27489. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Fan, J.; Thompson, L.F.; Zhang, Y.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J. Clin. Investig. 2011, 121, 2371–2382. [Google Scholar] [CrossRef]
- Ryzhov, S.V.; Pickup, M.W.; Chytil, A.; Gorska, A.E.; Zhang, Q.; Owens, P.; Feoktistov, I.; Moses, H.L.; Novitskiy, S.V. Role of TGF-beta signaling in generation of CD39+CD73+ myeloid cells in tumors. J. Immunol. 2014, 193, 3155–3164. [Google Scholar] [CrossRef] [Green Version]
- Harish, A.; Hohana, G.; Fishman, P.; Arnon, O.; Bar-Yehuda, S. A3 adenosine receptor agonist potentiates natural killer cell activity. Int. J. Oncol. 2003, 23, 1245–1249. [Google Scholar] [CrossRef]
- Wang, X.; Chen, D. Purinergic Regulation of Neutrophil Function. Front. Immunol. 2018, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Sundstrom, P.; Stenstad, H.; Langenes, V.; Ahlmanner, F.; Theander, L.; Ndah, T.G.; Fredin, K.; Borjesson, L.; Gustavsson, B.; Bastid, J.; et al. Regulatory T Cells from Colon Cancer Patients Inhibit Effector T-cell Migration through an Adenosine-Dependent Mechanism. Cancer Immunol. Res. 2016, 4, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.F.; Kondo, T.; Nakazawa, T.; Niu, D.F.; Mochizuki, K.; Kawasaki, T.; Yamane, T.; Katoh, R. Hypoxia-inducible adenosine A2B receptor modulates proliferation of colon carcinoma cells. Hum. Pathol. 2010, 41, 1550–1557. [Google Scholar] [CrossRef]
- Gessi, S.; Cattabriga, E.; Avitabile, A.; Gafa, R.; Lanza, G.; Cavazzini, L.; Bianchi, N.; Gambari, R.; Feo, C.; Liboni, A.; et al. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 5895–5901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyazit, Y.; Koklu, S.; Tas, A.; Purnak, T.; Sayilir, A.; Kurt, M.; Turhan, T.; Celik, T.; Suvak, B.; Torun, S.; et al. Serum adenosine deaminase activity as a predictor of disease severity in ulcerative colitis. J. Crohn’s Colitis 2012, 6, 102–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonioli, L.; Fornai, M.; Colucci, R.; Awwad, O.; Ghisu, N.; Tuccori, M.; Da Settimo, F.; La Motta, C.; Natale, G.; Duranti, E.; et al. The blockade of adenosine deaminase ameliorates chronic experimental colitis through the recruitment of adenosine A2A and A3 receptors. J. Pharmacol. Exp. Ther. 2010, 335, 434–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Ben, D.; Antonioli, L.; Lambertucci, C.; Spinaci, A.; Fornai, M.; D’Antongiovanni, V.; Pellegrini, C.; Blandizzi, C.; Volpini, R. Approaches for designing and discovering purinergic drugs for gastrointestinal diseases. Expert Opin. Drug Discov. 2020, 15, 687–703. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Braganhol, E.; Whiteside, T.L. Inhibition of the Adenosinergic Pathway in Cancer Rejuvenates Innate and Adaptive Immunity. Int. J. Mol. Sci. 2019, 20, 5698. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.; Al-Taei, S.; Webber, J.; Mason, M.D.; Tabi, Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J. Immunol. 2011, 187, 676–683. [Google Scholar] [CrossRef]
- Mannavola, F.; Salerno, T.; Passarelli, A.; Tucci, M.; Interno, V.; Silvestris, F. Revisiting the Role of Exosomes in Colorectal Cancer: Where Are We Now? Front. Oncol. 2019, 9, 521. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Harvey, J.B.; Phan, L.H.; Villarreal, O.E.; Bowser, J.L. CD73′s Potential as an Immunotherapy Target in Gastrointestinal Cancers. Front. Immunol. 2020, 11, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Antongiovanni, V.; Fornai, M.; Pellegrini, C.; Benvenuti, L.; Blandizzi, C.; Antonioli, L. The Adenosine System at the Crossroads of Intestinal Inflammation and Neoplasia. Int. J. Mol. Sci. 2020, 21, 5089. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145089
D’Antongiovanni V, Fornai M, Pellegrini C, Benvenuti L, Blandizzi C, Antonioli L. The Adenosine System at the Crossroads of Intestinal Inflammation and Neoplasia. International Journal of Molecular Sciences. 2020; 21(14):5089. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145089
Chicago/Turabian StyleD’Antongiovanni, Vanessa, Matteo Fornai, Carolina Pellegrini, Laura Benvenuti, Corrado Blandizzi, and Luca Antonioli. 2020. "The Adenosine System at the Crossroads of Intestinal Inflammation and Neoplasia" International Journal of Molecular Sciences 21, no. 14: 5089. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145089