Role of the Ubiquitin Proteasome System in the Regulation of Blood Pressure: A Review

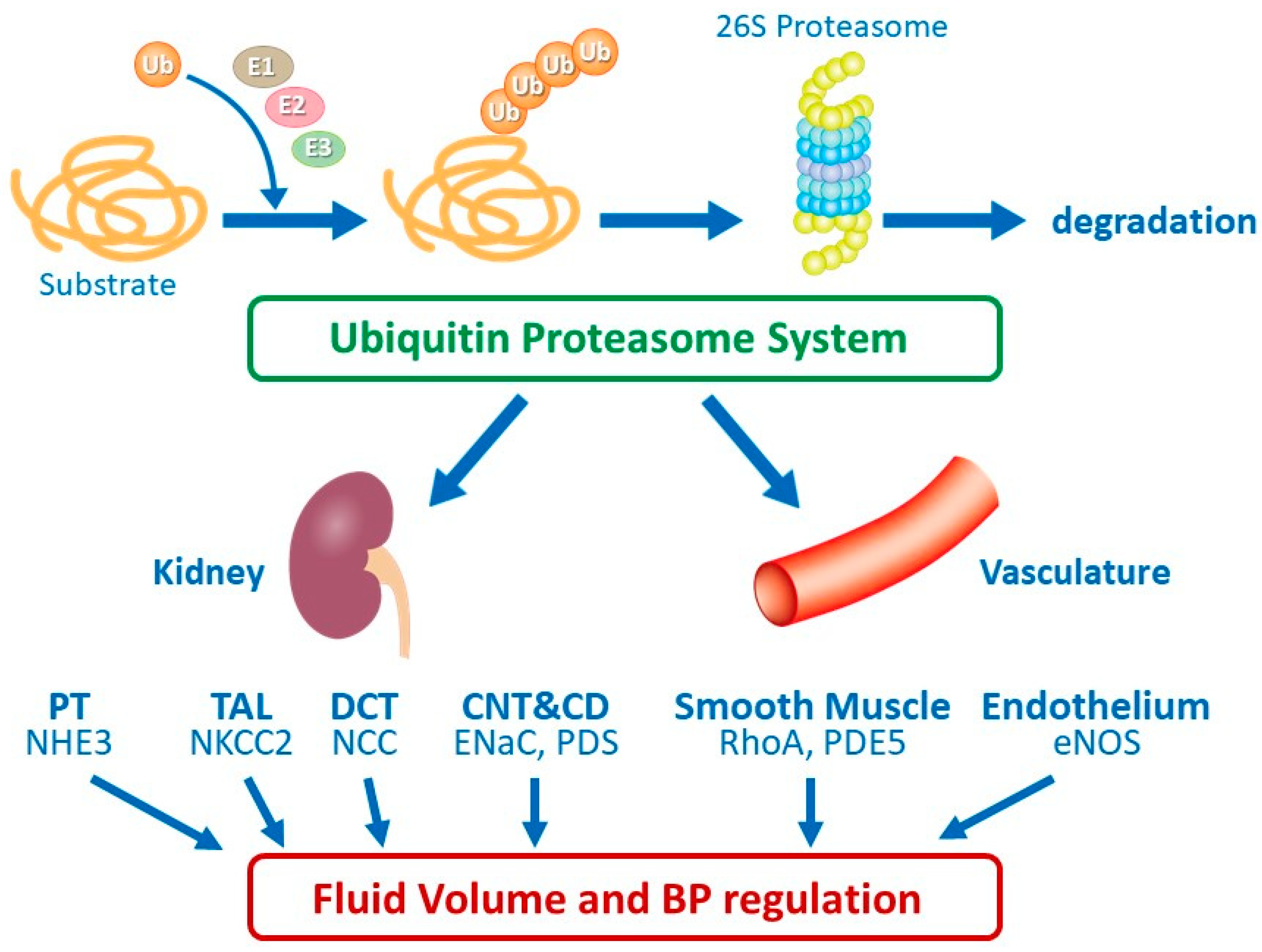{kind=link}
Abstract
:1. Introduction
2. Role of UPS in the Regulation of Tubular Function in the Kidney
2.1. Proximal Tubule
2.2. Thick Ascending Limb
2.3. Distal Convoluted Tubule
2.3.1. KLHL3-Mediated WNK4 Ubiquitylation and NCC
2.3.2. NEDD4-2-Mediated Ubiquitylation and NCC
2.4. Connecting Tubule and Collecting Duct
2.4.1. NEDD4-2-Mediated Ubiquitylation and ENaC
2.4.2. NEDD4-2 and Pendrin
3. Role of UPS in the Regulation of Vascular Function
3.1. Proteasome Inhibitors and Cardiovascular Disorders
3.2. Vascular Endothelial Cells
3.3. Vascular Smooth Muscle Cells
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 20-HETE | 20-Hydroxyeicosatetraenoic acid |
| AT1R | Angiotensin II type 1 receptor |
| BH4 | Tetrahydrobiopterin |
| BP | Blood pressure |
| CD | Collecting duct |
| CNT | Connecting tubule |
| CUL3 | Cullin 3 |
| DCT | Distal convoluted tubule |
| E1 | Ubiquitin-activating enzyme |
| E2 | Ubiquitin-conjugating enzyme |
| E3 | Ubiquitin protein ligase |
| ENaC | Epithelial sodium channel |
| eNOS | Endothelial NO synthase |
| GTPCH | GTP cyclohydrolase |
| IC | Intercalated cell |
| KLHL3 | Kelch-like 3 |
| NCC | Na+-Cl− cotransporter |
| NEDD4-2 | Neuronal precursor cell-expressed developmentally downregulated 4-2 |
| NHE3 | Na+/H+ exchanger isoform 3 |
| NKCC2 | Na+-K+-2Cl− cotransporter |
| NO | Nitric oxide |
| PDE | Phosphodiesterase |
| PHAII | Pseudohypoaldosteronism type II |
| PKA | Protein kinase A |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PT | Proximal tubule |
| RhoBTB1 | Rho-related BTB domain-containing protein 1 |
| RING | Really interesting new gene |
| Sgk1 | Serum/glucocorticoid-regulated kinase 1 |
| SPAK | STE20/SPS1-related proline-alanine-rich protein kinase |
| TAL | Thick ascending limb |
| UPS | Ubiquitin proteasome system |
| VEC | Vascular endothelial cell |
| VSMC | Vascular smooth muscle cell |
| WNK | With-no-lysine |
References
- Guyton, A.C. The surprising kidney-fluid mechanism for pressure control--its infinite gain! Hypertension 1990, 16, 725–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lifton, R.P.; Gharavi, A.G.; Geller, D.S. Molecular mechanisms of human hypertension. Cell 2001, 104, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Boyden, L.M.; Choi, M.; Choate, K.A.; Nelson-Williams, C.J.; Farhi, A.; Toka, H.R.; Tikhonova, I.R.; Bjornson, R.; Mane, S.M.; Colussi, G.; et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012, 482, 98–102. [Google Scholar] [CrossRef]
- Shibata, S.; Zhang, J.; Puthumana, J.; Stone, K.L.; Lifton, R.P. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc. Natl. Acad. Sci. USA 2013, 110, 7838–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, L.; Kiowski, W.; Bühler, F.R.; Lüscher, T.F. Indirect evidence for release of endothelium-derived relaxing factor in human forearm circulation in vivo. Blunted response in essential hypertension. Circulation 1990, 81, 1762–1767. [Google Scholar] [PubMed] [Green Version]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ashton, D.S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664–666. [Google Scholar] [CrossRef]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef]
- Leonard, A.M.; Chafe, L.L.; Montani, J.P.; Van Vliet, B.N. Increased salt-sensitivity in endothelial nitric oxide synthase-knockout mice. Am. J. Hypertens 2006, 19, 1264–1269. [Google Scholar] [CrossRef] [Green Version]
- Lahera, V.; Salom, M.G.; Miranda-Guardiola, F.; Moncada, S.; Romero, J.C. Effects of NG-nitro-L-arginine methyl ester on renal function and blood pressure. Am. J. Physiol. 1991, 261, F1033–F1037. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Haas, A.L.; Warms, J.V.; Rose, I.A. Ubiquitin adenylate: Structure and role in ubiquitin activation. Biochemistry 1983, 22, 4388–4394. [Google Scholar] [PubMed]
- Jentsch, S. The ubiquitin-conjugation system. Annu. Rev. Genet. 1992, 26, 179–207. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [PubMed]
- Clague, M.J.; Heride, C.; Urbé, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, S.J.; Timmers, H.T. The family of ubiquitin-conjugating enzymes (E2s): Deciding between life and death of proteins. FASEB J. 2010, 24, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Kocinsky, H.S.; Girardi, A.C.; Biemesderfer, D.; Nguyen, T.; Mentone, S.; Orlowski, J.; Aronson, P.S. Use of phospho-specific antibodies to determine the phosphorylation of endogenous Na+/H+ exchanger NHE3 at PKA consensus sites. Am. J. Physiol. Renal Physiol. 2005, 289, F249–F258. [Google Scholar]
- Thomson, R.B.; Wang, T.; Thomson, B.R.; Tarrats, L.; Girardi, A.; Mentone, S.; Soleimani, M.; Kocher, O.; Aronson, P.S. Role of PDZK1 in membrane expression of renal brush border ion exchangers. Proc. Natl. Acad. Sci. USA 2005, 102, 13331–13336. [Google Scholar]
- No, Y.R.; He, P.; Yoo, B.K.; Yun, C.C. Unique regulation of human Na+/H+ exchanger 3 (NHE3) by Nedd4-2 ligase that differs from non-primate NHE3s. J. Biol. Chem. 2014, 289, 18360–18372. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, M.; Kaimori, J.Y.; Yamamoto, S.; Matsui, I.; Hamano, T.; Takabatake, Y.; Ecelbarger, C.M.; Takahara, S.; Isaka, Y.; Rakugi, H. Azilsartan Improves Salt Sensitivity by Modulating the Proximal Tubular Na+-H+ Exchanger-3 in Mice. PLoS ONE 2016, 11, e0147786. [Google Scholar] [CrossRef]
- Russell, J.M. Sodium-potassium-chloride cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [PubMed]
- Wu, J.; Liu, X.; Lai, G.; Yang, X.; Wang, L.; Zhao, Y. Synergistical effect of 20-HETE and high salt on NKCC2 protein and blood pressure via ubiquitin-proteasome pathway. Hum. Genet. 2013, 132, 179–187. [Google Scholar] [PubMed]
- Ares, G.R. cGMP induces degradation of NKCC2 in the thick ascending limb via the ubiquitin-proteasomal system. Am. J. Physiol. Renal Physiol. 2019, 316, F838–F846. [Google Scholar] [PubMed]
- Gamba, G. The thiazide-sensitive Na+-Cl- cotransporter: Molecular biology, functional properties, and regulation by WNKs. Am. J. Physiol. Renal Physiol. 2009, 297, F838–F848. [Google Scholar]
- Mayan, H.; Munter, G.; Shaharabany, M.; Mouallem, M.; Pauzner, R.; Holtzman, E.J.; Farfel, Z. Hypercalciuria in familial hyperkalemia and hypertension accompanies hyperkalemia and precedes hypertension: Description of a large family with the Q565E WNK4 mutation. J. Clin. Endocrinol. Metab. 2004, 89, 4025–4030. [Google Scholar]
- Mayan, H.; Vered, I.; Mouallem, M.; Tzadok-Witkon, M.; Pauzner, R.; Farfel, Z. Pseudohypoaldosteronism type II: Marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J. Clin. Endocrinol. Metab. 2002, 87, 3248–3254. [Google Scholar]
- Wilson, F.H.; Disse-Nicodeme, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.M.; et al. Human hypertension caused by mutations in WNK kinases. Science 2001, 293, 1107–1112. [Google Scholar]
- Louis-Dit-Picard, H.; Barc, J.; Trujillano, D.; Miserey-Lenkei, S.; Bouatia-Naji, N.; Pylypenko, O.; Beaurain, G.; Bonnefond, A.; Sand, O.; Simian, C.; et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012, 44, 456–460, S451–S453. [Google Scholar]
- Vitari, A.C.; Deak, M.; Morrice, N.A.; Alessi, D.R. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J. 2005, 391, 17–24. [Google Scholar]
- Ohta, A.; Schumacher, F.R.; Mehellou, Y.; Johnson, C.; Knebel, A.; Macartney, T.J.; Wood, N.T.; Alessi, D.R.; Kurz, T. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: Disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem. J. 2013, 451, 111–122. [Google Scholar] [PubMed]
- Wakabayashi, M.; Mori, T.; Isobe, K.; Sohara, E.; Susa, K.; Araki, Y.; Chiga, M.; Kikuchi, E.; Nomura, N.; Mori, Y.; et al. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 2013, 3, 858–868. [Google Scholar] [PubMed] [Green Version]
- Wu, G.; Peng, J.B. Disease-causing mutations in KLHL3 impair its effect on WNK4 degradation. FEBS Lett. 2013, 587, 1717–1722. [Google Scholar]
- Shibata, S.; Arroyo, J.P.; Castaneda-Bueno, M.; Puthumana, J.; Zhang, J.; Uchida, S.; Stone, K.L.; Lam, T.T.; Lifton, R.P. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc. Natl. Acad. Sci. USA 2014, 111, 15556–15561. [Google Scholar]
- Yoshizaki, Y.; Mori, Y.; Tsuzaki, Y.; Mori, T.; Nomura, N.; Wakabayashi, M.; Takahashi, D.; Zeniya, M.; Kikuchi, E.; Araki, Y.; et al. Impaired degradation of WNK by Akt and PKA phosphorylation of KLHL3. Biochem. Biophys. Res. Commun. 2015, 467, 229–234. [Google Scholar]
- Castañeda-Bueno, M.; Arroyo, J.P.; Zhang, J.; Puthumana, J.; Yarborough, O., 3rd; Shibata, S.; Rojas-Vega, L.; Gamba, G.; Rinehart, J.; Lifton, R.P. Phosphorylation by PKC and PKA regulate the kinase activity and downstream signaling of WNK4. Proc. Natl. Acad. Sci. USA 2017, 114, E879–E886. [Google Scholar]
- Bazúa-Valenti, S.; Rojas-Vega, L.; Castañeda-Bueno, M.; Barrera-Chimal, J.; Bautista, R.; Cervantes-Pérez, L.G.; Vázquez, N.; Plata, C.; Murillo-de-Ozores, A.R.; González-Mariscal, L.; et al. The Calcium-Sensing Receptor Increases Activity of the Renal NCC through the WNK4-SPAK Pathway. J. Am. Soc. Nephrol. 2018, 29, 1838–1848. [Google Scholar] [PubMed] [Green Version]
- Ishizawa, K.; Wang, Q.; Li, J.; Yamazaki, O.; Tamura, Y.; Fujigaki, Y.; Uchida, S.; Lifton, R.P.; Shibata, S. Calcineurin dephosphorylates Kelch-like 3, reversing phosphorylation by angiotensin II and regulating renal electrolyte handling. Proc. Natl. Acad. Sci. USA 2019, 116, 3155–3160. [Google Scholar]
- Ishizawa, K.; Xu, N.; Loffing, J.; Lifton, R.P.; Fujita, T.; Uchida, S.; Shibata, S. Potassium depletion stimulates Na-Cl cotransporter via phosphorylation and inactivation of the ubiquitin ligase Kelch-like 3. Biochem. Biophys. Res. Commun. 2016, 480, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Ishizawa, K.; Wang, Q.; Li, J.; Xu, N.; Nemoto, Y.; Morimoto, C.; Fujii, W.; Tamura, Y.; Fujigaki, Y.; Tsukamoto, K.; et al. Inhibition of Sodium Glucose Cotransporter 2 Attenuates the Dysregulation of Kelch-Like 3 and NaCl Cotransporter in Obese Diabetic Mice. J. Am. Soc. Nephrol. 2019, 30, 782–794. [Google Scholar] [CrossRef]
- McCormick, J.A.; Yang, C.L.; Zhang, C.; Davidge, B.; Blankenstein, K.I.; Terker, A.S.; Yarbrough, B.; Meermeier, N.P.; Park, H.J.; McCully, B.; et al. Hyperkalemic hypertension-associated cullin 3 promotes WNK signaling by degrading KLHL3. J. Clin. Invest. 2014, 124, 4723–4736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelius, R.J.; Si, J.; Cuevas, C.A.; Nelson, J.W.; Gratreak, B.D.K.; Pardi, R.; Yang, C.L.; Ellison, D.H. Renal COP9 Signalosome Deficiency Alters CUL3-KLHL3-WNK Signaling Pathway. J. Am. Soc. Nephrol. 2018, 29, 2627–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, J.P.; Lagnaz, D.; Ronzaud, C.; Vazquez, N.; Ko, B.S.; Moddes, L.; Ruffieux-Daidie, D.; Hausel, P.; Koesters, R.; Yang, B.; et al. Nedd4-2 modulates renal Na+-Cl- cotransporter via the aldosterone-SGK1-Nedd4-2 pathway. J. Am. Soc. Nephrol. 2011, 22, 1707–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faresse, N.; Lagnaz, D.; Debonneville, A.; Ismailji, A.; Maillard, M.; Fejes-Toth, G.; Náray-Fejes-Tóth, A.; Staub, O. Inducible kidney-specific Sgk1 knockout mice show a salt-losing phenotype. Am. J. Physiol. Renal Physiol. 2012, 302, F977–F985. [Google Scholar] [CrossRef] [PubMed]
- Ronzaud, C.; Loffing-Cueni, D.; Hausel, P.; Debonneville, A.; Malsure, S.R.; Fowler-Jaeger, N.; Boase, N.A.; Perrier, R.; Maillard, M.; Yang, B.; et al. Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J. Clin. Invest. 2013, 123, 657–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Al-Qusairi, L.; Donnelly, B.F.; Ronzaud, C.; Marciszyn, A.L.; Gong, F.; Chang, Y.P.; Butterworth, M.B.; Pastor-Soler, N.M.; Hallows, K.R.; et al. Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action. J. Clin. Invest. 2015, 125, 3433–3448. [Google Scholar] [CrossRef] [Green Version]
- Furusho, T.; Sohara, E.; Mandai, S.; Kikuchi, H.; Takahashi, N.; Fujimaru, T.; Hashimoto, H.; Arai, Y.; Ando, F.; Zeniya, M.; et al. Renal TNFα activates the WNK phosphorylation cascade and contributes to salt-sensitive hypertension in chronic kidney disease. Kidney Int. 2020, 97, 713–727. [Google Scholar] [CrossRef]
- Wu, P.; Su, X.T.; Gao, Z.X.; Zhang, D.D.; Duan, X.P.; Xiao, Y.; Staub, O.; Wang, W.H.; Lin, D.H. Renal Tubule Nedd4-2 Deficiency Stimulates Kir4.1/Kir5.1 and Thiazide-Sensitive NaCl Cotransporter in Distal Convoluted Tubule. J. Am. Soc. Nephrol. 2020, 31, 1226–1242. [Google Scholar] [CrossRef]
- Rossier, B.C. Epithelial sodium channel (ENaC) and the control of blood pressure. Curr. Opin. Pharmacol. 2014, 15, 33–46. [Google Scholar] [CrossRef]
- Pearce, D.; Soundararajan, R.; Trimpert, C.; Kashlan, O.B.; Deen, P.M.; Kohan, D.E. Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Schild, L.; Canessa, C.M.; Shimkets, R.A.; Gautschi, I.; Lifton, R.P.; Rossier, B.C. A mutation in the epithelial sodium channel causing Liddle disease increases channel activity in the Xenopus laevis oocyte expression system. Proc. Natl. Acad. Sci. USA 1995, 92, 5699–5703. [Google Scholar] [CrossRef] [Green Version]
- Hansson, J.H.; Nelson-Williams, C.; Suzuki, H.; Schild, L.; Shimkets, R.; Lu, Y.; Canessa, C.; Iwasaki, T.; Rossier, B.; Lifton, R.P. Hypertension caused by a truncated epithelial sodium channel gamma subunit: Genetic heterogeneity of Liddle syndrome. Nat. Genet. 1995, 11, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Schild, L.; Enomoto, N.; Matsui, N.; Marumo, F.; Rossier, B.C. Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J. Clin. Invest. 1996, 97, 1780–1784. [Google Scholar] [CrossRef] [PubMed]
- Staub, O.; Dho, S.; Henry, P.; Correa, J.; Ishikawa, T.; McGlade, J.; Rotin, D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 1996, 15, 2371–2380. [Google Scholar] [CrossRef]
- Staub, O.; Gautschi, I.; Ishikawa, T.; Breitschopf, K.; Ciechanover, A.; Schild, L.; Rotin, D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997, 16, 6325–6336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Pribanic, S.; Debonneville, A.; Jiang, C.; Rotin, D. The PY motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic 2007, 8, 1246–1264. [Google Scholar] [CrossRef]
- Wiemuth, D.; Ke, Y.; Rohlfs, M.; McDonald, F.J. Epithelial sodium channel (ENaC) is multi-ubiquitinated at the cell surface. Biochem. J. 2007, 405, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Patel, S.V.; Snyder, P.M. Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J. Biol. Chem. 2007, 282, 20207–20212. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Pearce, D. Regulation of the epithelial Na+ channel by the mTORC2/SGK1 pathway. Nephrol. Dial. Transplant. 2016, 31, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Bhalla, V.; Daidie, D.; Li, H.; Pao, A.C.; LaGrange, L.P.; Wang, J.; Vandewalle, A.; Stockand, J.D.; Staub, O.; Pearce, D. Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol. Endocrinol. 2005, 19, 3073–3084. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.P.; Cao, X.R.; Sweezer, E.M.; Kinney, T.S.; Williams, N.R.; Husted, R.F.; Nair, R.; Weiss, R.M.; Williamson, R.A.; Sigmund, C.D.; et al. Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am. J. Physiol. Renal Physiol. 2008, 295, F462–F470. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, S.; Ishigami, T.; Kino, T.; Chen, L.; Nakashima-Sasaki, R.; Araki, N.; Yatsu, K.; Fujita, M.; Umemura, S. An isoform of Nedd4-2 is critically involved in the renal adaptation to high salt intake in mice. Sci. Rep. 2016, 6, 27137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, D.M.; Ishigami, T.; Pankow, J.; von Niederhausern, A.; Alder, J.; Hunt, S.C.; Leppert, M.F.; Lalouel, J.M.; Weiss, R.B. Common variant of human NEDD4L activates a cryptic splice site to form a frameshifted transcript. J. Hum. Genet. 2002, 47, 665–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouladkou, F.; Alikhani-Koopaei, R.; Vogt, B.; Flores, S.Y.; Malbert-Colas, L.; Lecomte, M.C.; Loffing, J.; Frey, F.J.; Frey, B.M.; Staub, O. A naturally occurring human Nedd4-2 variant displays impaired ENaC regulation in Xenopus laevis oocytes. Am. J. Physiol. Renal Physiol. 2004, 287, F550–F561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, N.; Umemura, M.; Miyagi, Y.; Yabana, M.; Miki, Y.; Tamura, K.; Uchino, K.; Aoki, R.; Goshima, Y.; Umemura, S.; et al. Expression, transcription, and possible antagonistic interaction of the human Nedd4L gene variant: Implications for essential hypertension. Hypertension 2008, 51, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Wang, Y.; Wang, X.; Sun, K.; Zhou, X.; Hui, R. A functional variant of NEDD4L is associated with hypertension, antihypertensive response, and orthostatic hypotension. Hypertension 2009, 54, 796–801. [Google Scholar] [CrossRef] [Green Version]
- Nanami, M.; Pham, T.D.; Kim, Y.H.; Yang, B.; Sutliff, R.L.; Staub, O.; Klein, J.D.; Lopez-Cayuqueo, K.I.; Chambrey, R.; Park, A.Y.; et al. The Role of Intercalated Cell Nedd4-2 in BP Regulation, Ion Transport, and Transporter Expression. J. Am. Soc. Nephrol. 2018, 29, 1706–1719. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [Green Version]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hájek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Waxman, A.J.; Clasen, S.; Hwang, W.T.; Garfall, A.; Vogl, D.T.; Carver, J.; O’Quinn, R.; Cohen, A.D.; Stadtmauer, E.A.; Ky, B.; et al. Carfilzomib-Associated Cardiovascular Adverse Events: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, e174519. [Google Scholar] [CrossRef] [PubMed]
- Gavazzoni, M.; Vizzardi, E.; Gorga, E.; Bonadei, I.; Rossi, L.; Belotti, A.; Rossi, G.; Ribolla, R.; Metra, M.; Raddino, R. Mechanism of cardiovascular toxicity by proteasome inhibitors: New paradigm derived from clinical and pre-clinical evidence. Eur. J. Pharmacol. 2018, 828, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Chen-Scarabelli, C.; Corsetti, G.; Pasini, E.; Dioguardi, F.S.; Sahni, G.; Narula, J.; Gavazzoni, M.; Patel, H.; Saravolatz, L.; Knight, R.; et al. Spasmogenic Effects of the Proteasome Inhibitor Carfilzomib on Coronary Resistance, Vascular Tone and Reactivity. EBioMedicine 2017, 21, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, A.; Luthi, J.; Belohlavek, M.; Kortüm, K.M.; Mookadam, F.; Mayo, A.; Fonseca, R.; Bergsagel, P.L.; Reeder, C.B.; Mikhael, J.R.; et al. Carfilzomib and the cardiorenal system in myeloma: An endothelial effect? Blood Cancer J. 2016, 6, e384. [Google Scholar] [CrossRef]
- Yui, J.C.; Van Keer, J.; Weiss, B.M.; Waxman, A.J.; Palmer, M.B.; D’Agati, V.D.; Kastritis, E.; Dimopoulos, M.A.; Vij, R.; Bansal, D.; et al. Proteasome inhibitor associated thrombotic microangiopathy. Am. J. Hematol. 2016, 91, E348–E352. [Google Scholar] [CrossRef]
- Hobeika, L.; Self, S.E.; Velez, J.C. Renal thrombotic microangiopathy and podocytopathy associated with the use of carfilzomib in a patient with multiple myeloma. BMC Nephrol. 2014, 15, 156. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ Res. 2016, 119, 375–396. [Google Scholar] [CrossRef] [Green Version]
- Alp, N.J.; Channon, K.M. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef]
- Abdelghany, T.M.; Ismail, R.S.; Mansoor, F.A.; Zweier, J.R.; Lowe, F.; Zweier, J.L. Cigarette smoke constituents cause endothelial nitric oxide synthase dysfunction and uncoupling due to depletion of tetrahydrobiopterin with degradation of GTP cyclohydrolase. Nitric. Oxide 2018, 76, 113–121. [Google Scholar] [CrossRef]
- Xu, J.; Wang, S.; Wu, Y.; Song, P.; Zou, M.H. Tyrosine nitration of PA700 activates the 26S proteasome to induce endothelial dysfunction in mice with angiotensin II-induced hypertension. Hypertension 2009, 54, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, Y.; Song, P.; Zhang, M.; Wang, S.; Zou, M.H. Proteasome-dependent degradation of guanosine 5′-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation 2007, 116, 944–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, M.; Wilck, N.; Meiners, S.; Ludwig, A.; Baumann, G.; Stangl, K.; Stangl, V. Proteasome inhibition prevents experimentally-induced endothelial dysfunction. Life Sci. 2009, 84, 929–934. [Google Scholar] [CrossRef] [PubMed]
- De Martin, R.; Hoeth, M.; Hofer-Warbinek, R.; Schmid, J.A. The transcription factor NF-kappa B and the regulation of vascular cell function. Arterioscler Thromb. Vasc. Biol. 2000, 20, E83–E88. [Google Scholar] [PubMed] [Green Version]
- Takaoka, M.; Ohkita, M.; Matsumura, Y. Pathophysiological role of proteasome-dependent proteolytic pathway in endothelin-1-related cardiovascular diseases. Curr. Vasc Pharmacol. 2003, 1, 19–26. [Google Scholar] [CrossRef]
- Barroso, I.; Gurnell, M.; Crowley, V.E.; Agostini, M.; Schwabe, J.W.; Soos, M.A.; Maslen, G.L.; Williams, T.D.; Lewis, H.; Schafer, A.J.; et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999, 402, 880–883. [Google Scholar] [CrossRef]
- Savage, D.B.; Tan, G.D.; Acerini, C.L.; Jebb, S.A.; Agostini, M.; Gurnell, M.; Williams, R.L.; Umpleby, A.M.; Thomas, E.L.; Bell, J.D.; et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-gamma. Diabetes 2003, 52, 910–917. [Google Scholar] [CrossRef] [Green Version]
- Rahim, N.G.; Harismendy, O.; Topol, E.J.; Frazer, K.A. Genetic determinants of phenotypic diversity in humans. Genome Biol. 2008, 9, 215. [Google Scholar] [CrossRef] [Green Version]
- Mukohda, M.; Fang, S.; Wu, J.; Agbor, L.N.; Nair, A.R.; Ibeawuchi, S.C.; Hu, C.; Liu, X.; Lu, K.T.; Guo, D.F.; et al. RhoBTB1 protects against hypertension and arterial stiffness by restraining phosphodiesterase 5 activity. J. Clin. Invest. 2019, 129, 2318–2332. [Google Scholar] [CrossRef] [Green Version]
- Pelham, C.J.; Ketsawatsomkron, P.; Groh, S.; Grobe, J.L.; de Lange, W.J.; Ibeawuchi, S.R.; Keen, H.L.; Weatherford, E.T.; Faraci, F.M.; Sigmund, C.D. Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARγ and RhoA/Rho-kinase. Cell Metab. 2012, 16, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Rivero, F.; Dislich, H.; Glockner, G.; Noegel, A.A. The Dictyostelium discoideum family of Rho-related proteins. Nucleic Acids Res. 2001, 29, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Berthold, J.; Schenkova, K.; Ramos, S.; Miura, Y.; Furukawa, M.; Aspenstrom, P.; Rivero, F. Characterization of RhoBTB-depedent Cul3 ubiquitin ligase complexes—evidence for an autoregulatory mechanism. Exp. Cell Res. 2008, 314, 3453–3465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton-Cheh, C.; Johnson, T.; Gateva, V.; Tobin, M.D.; Bochud, M.; Coin, L.; Najjar, S.S.; Zhao, J.H.; Heath, S.C.; Eyheramendy, S.; et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet. 2009, 41, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, E.; Warren, H.R.; Mosen-Ansorena, D.; Mifsud, B.; Pazoki, R.; Gao, H.; Ntritsos, G.; Dimou, N.; Cabrera, C.P.; Karaman, I.; et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018, 50, 1412–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, S.; Khademi, F.; Somesh, B.P.; Rivero, F. Genomic organization and expression profile of the small GTPases of the RhoBTB family in human and mouse. Gene 2002, 298, 147–157. [Google Scholar] [CrossRef]
- Maass, P.G.; Aydin, A.; Luft, F.C.; Schächterle, C.; Weise, A.; Stricker, S.; Lindschau, C.; Vaegler, M.; Qadri, F.; Toka, H.R.; et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet. 2015, 47, 647–653. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell 2009, 35, 841–855. [Google Scholar] [CrossRef]
- Ibeawuchi, S.R.; Agbor, L.N.; Quelle, F.W.; Sigmund, C.D. Hypertension-causing Mutations in Cullin3 Protein Impair RhoA Protein Ubiquitination and Augment the Association with Substrate Adaptors. J. Biol. Chem. 2015, 290, 19208–19217. [Google Scholar] [CrossRef] [Green Version]
- Agbor, L.N.; Ibeawuchi, S.C.; Hu, C.; Wu, J.; Davis, D.R.; Keen, H.L.; Quelle, F.W.; Sigmund, C.D. Cullin-3 mutation causes arterial stiffness and hypertension through a vascular smooth muscle mechanism. JCI Insight. 2016, 1, e91015. [Google Scholar] [CrossRef] [Green Version]
- Abdel Khalek, W.; Rafael, C.; Loisel-Ferreira, I.; Kouranti, I.; Clauser, E.; Hadchouel, J.; Jeunemaitre, X. Severe Arterial Hypertension from Cullin 3 Mutations Is Caused by Both Renal and Vascular Effects. J. Am. Soc. Nephrol. 2019, 30, 811–823. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamazaki, O.; Hirohama, D.; Ishizawa, K.; Shibata, S. Role of the Ubiquitin Proteasome System in the Regulation of Blood Pressure: A Review. Int. J. Mol. Sci. 2020, 21, 5358. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155358
Yamazaki O, Hirohama D, Ishizawa K, Shibata S. Role of the Ubiquitin Proteasome System in the Regulation of Blood Pressure: A Review. International Journal of Molecular Sciences. 2020; 21(15):5358. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155358
Chicago/Turabian StyleYamazaki, Osamu, Daigoro Hirohama, Kenichi Ishizawa, and Shigeru Shibata. 2020. "Role of the Ubiquitin Proteasome System in the Regulation of Blood Pressure: A Review" International Journal of Molecular Sciences 21, no. 15: 5358. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155358