Therapeutic Potential of Cathelicidin Peptide LL-37, an Antimicrobial Agent, in a Murine Sepsis Model


Abstract
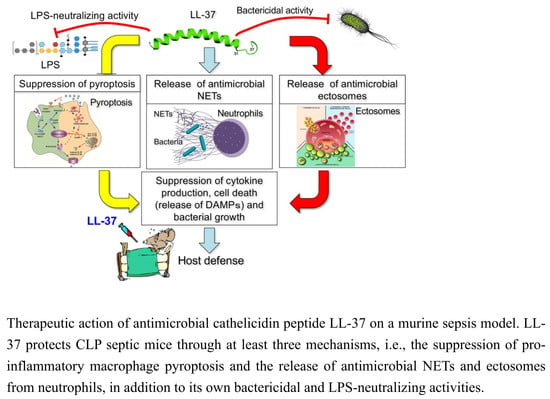
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
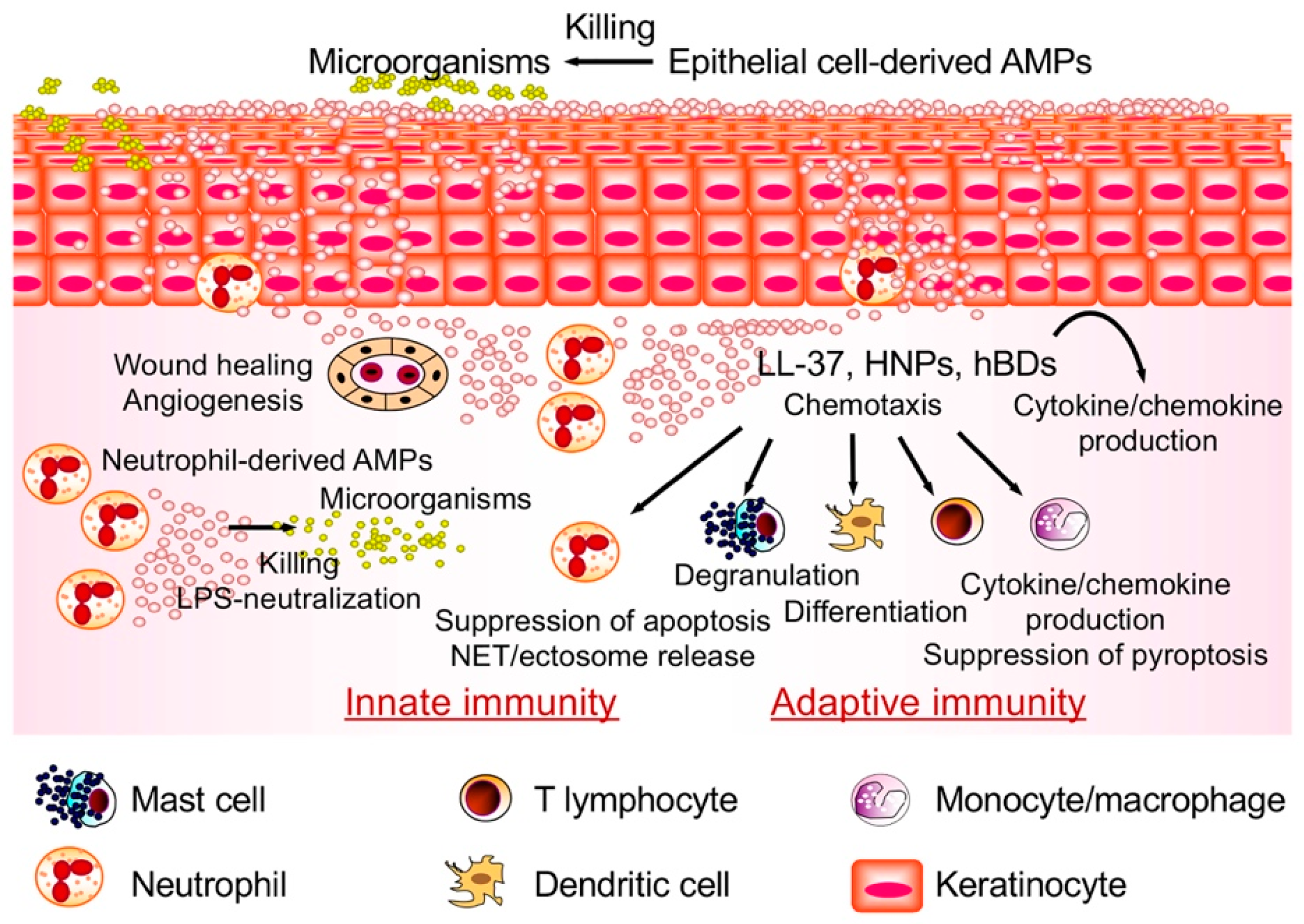
1. Introduction
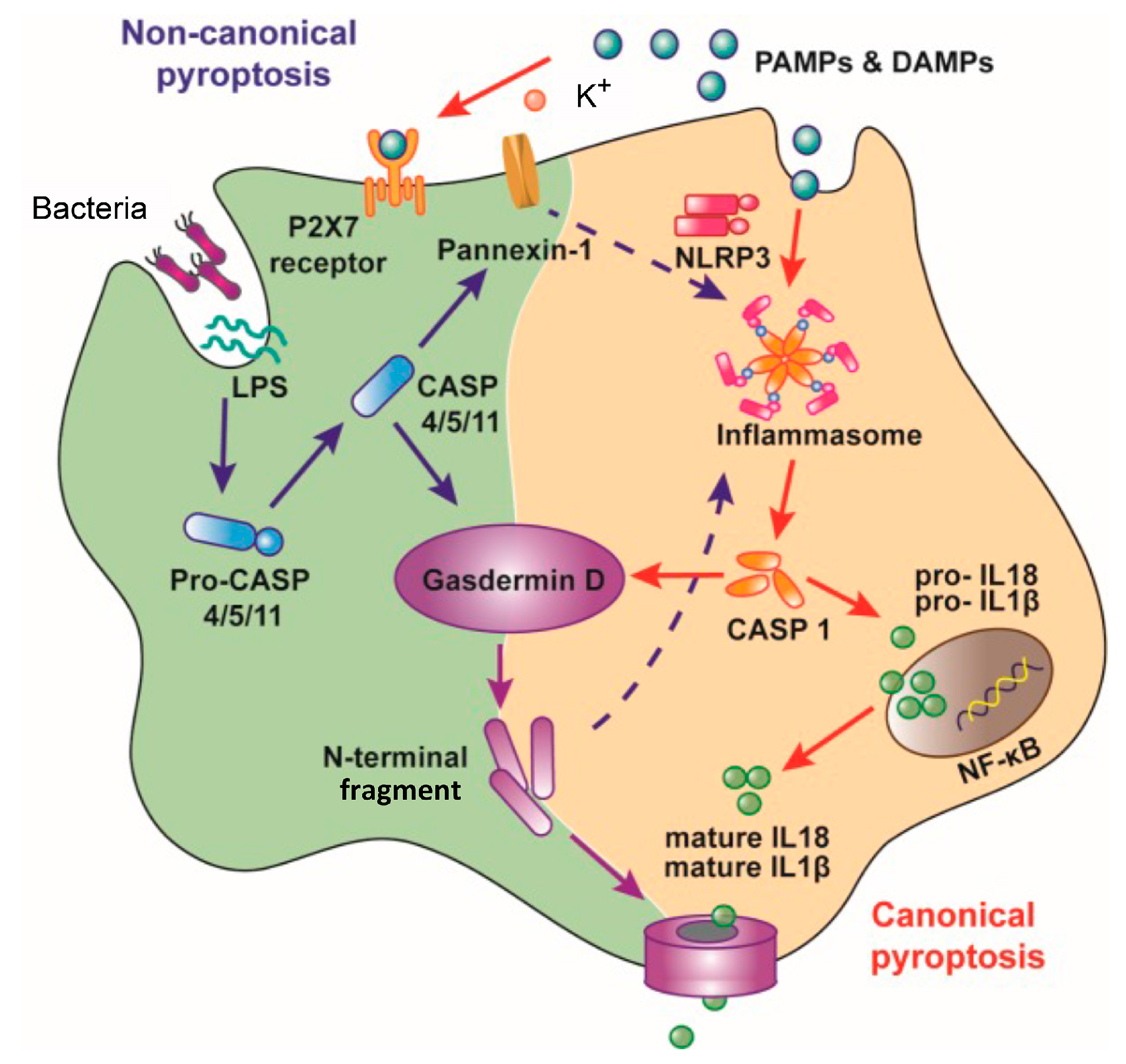
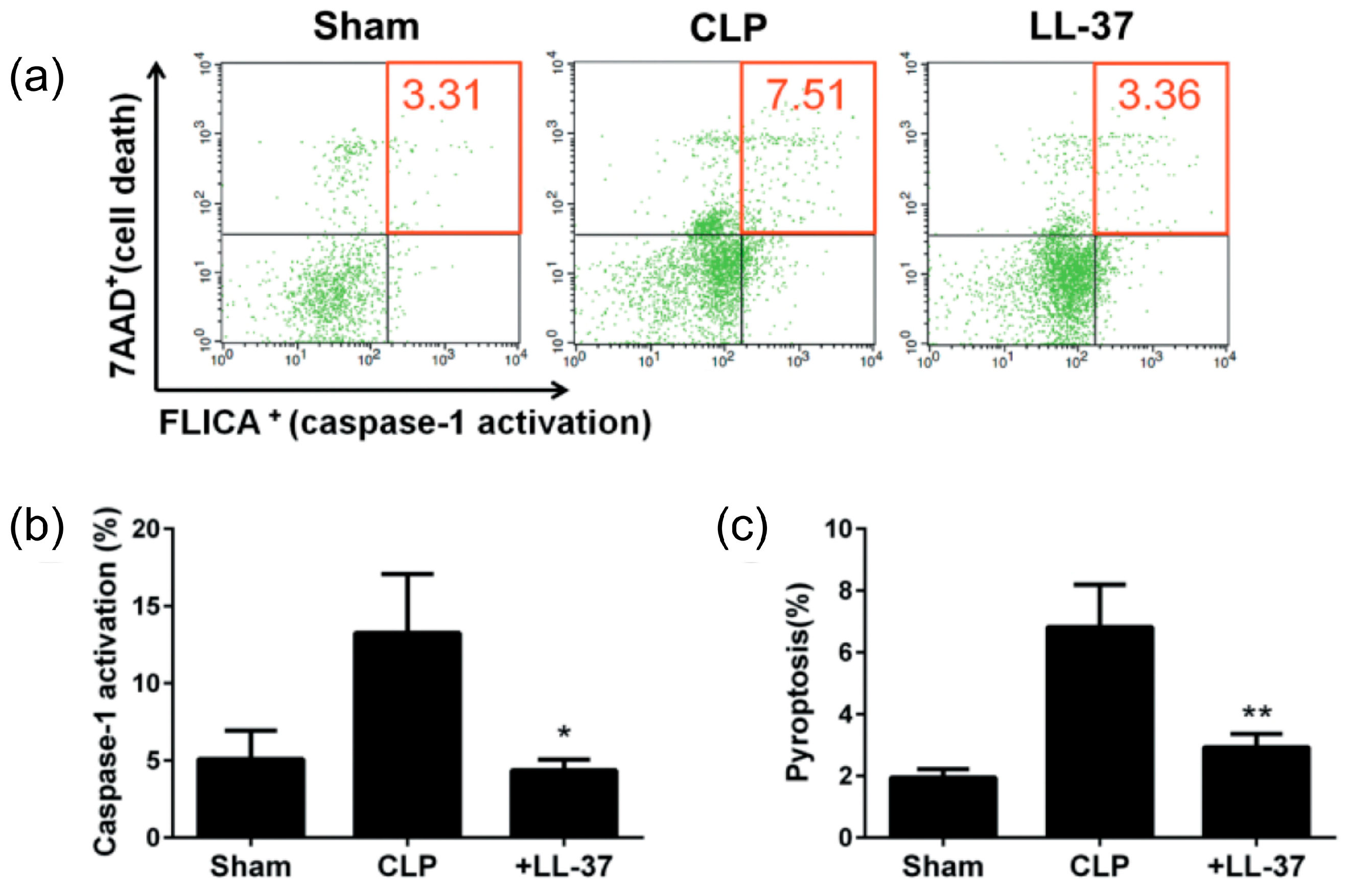
2. Inhibition of the Macrophage Pyroptosis and Improvement of the CLP Mouse Survival by LL-37
3. LL-37 induces NET Release from Neutrophils and Reduces the Inflammatory Response in CLP Mice
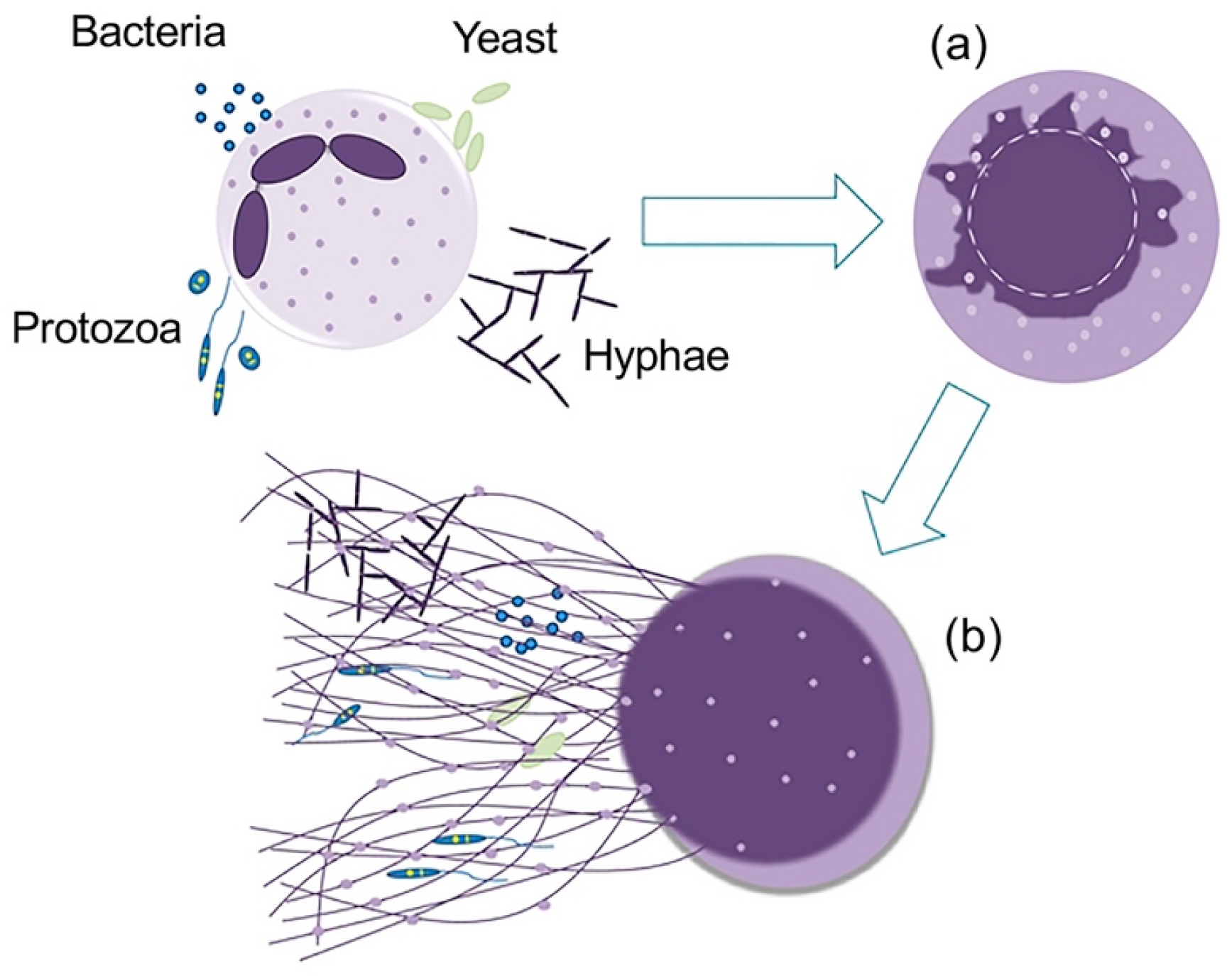
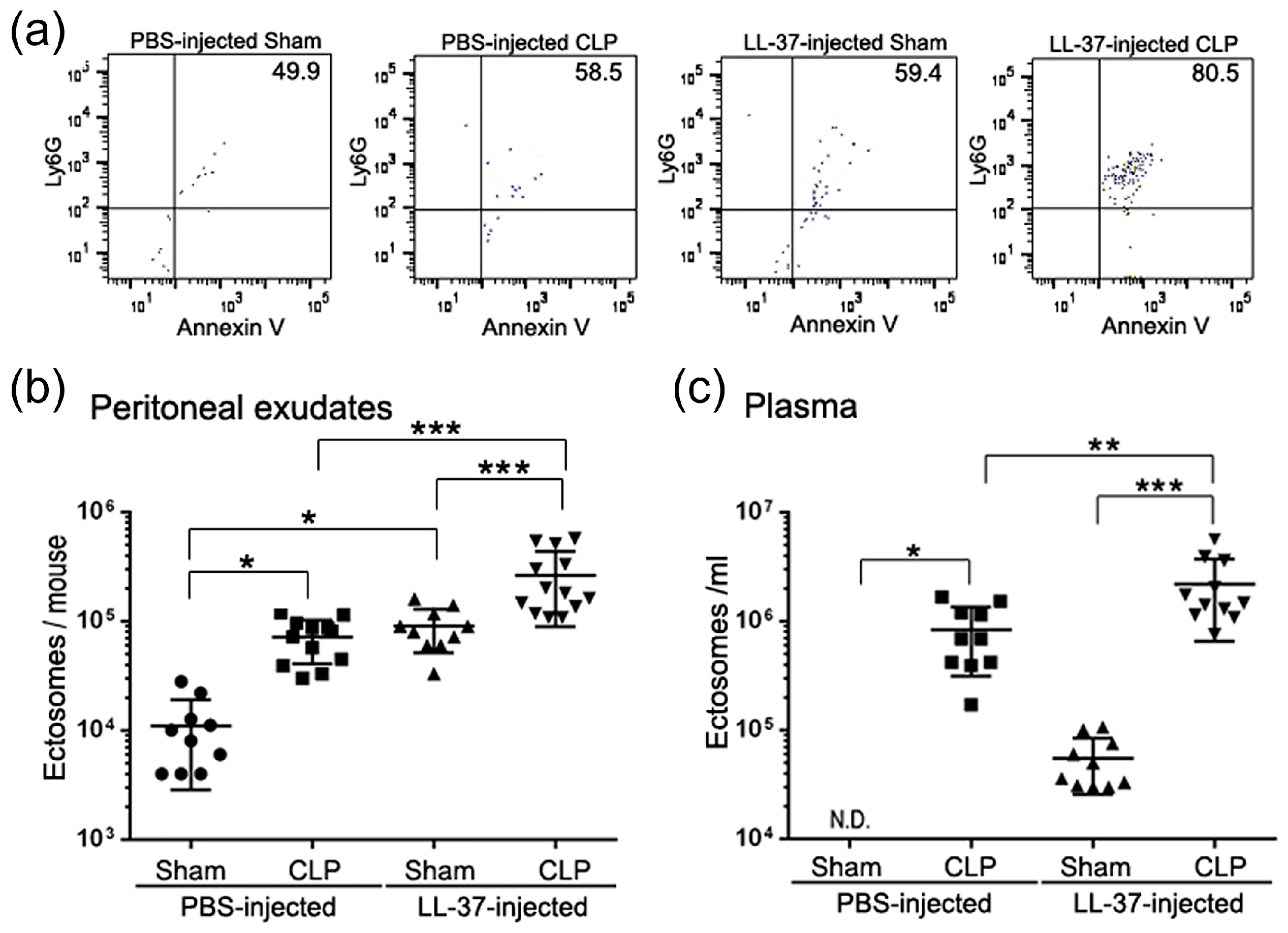
4. LL-37 Stimulates the Release of Antimicrobial Ectosomes from Neutrophils and Improves the Septic Condition
5. Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C. The search for effective therapy for sepsis: Back to the drawing board? JAMA 2011, 306, 2614–2615. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.M., Jr.; Ali, N.A.; Aberegg, S.K.; Abraham, E. Sepsis. Am. J. Med. 2007, 120, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, M.R. Dysregulation of the immune response in severe sepsis. Am. J. Med. Sci. 2004, 328, 220–229. [Google Scholar] [CrossRef]
- Wesche, D.E.; Lomas-Neira, J.L.; Perl, M.; Chung, C.S.; Ayala, A. Leukocyte apoptosis and its significance in sepsis and shock. J. Leukoc. Biol. 2005, 78, 325–337. [Google Scholar] [CrossRef]
- Pinheiro da Silva, F.; Nizet, V. Cell death during sepsis: Integration of disintegration in the inflammatory response to overwhelming infection. Apoptosis 2009, 14, 509–521. [Google Scholar] [CrossRef] [Green Version]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2011, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef]
- Casey, L.C.; Balk, R.A.; Bone, R.C. Plasma cytokine and endotoxin levels correlate with survival in patients with the sepsis syndrome. Ann. Intern. Med. 1993, 119, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Nathan, N. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Camicia, G.; Pozner, R.; de Larrañaga, G. Neutrophil extracellular traps in sepsis. Shock 2014, 42, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Kuijpers, T.W.; Wirawan, E.; Lippens, S.; Vandenabeele, P.; Vanden Berghe, T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011, 18, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [Green Version]
- Kalra, H.; Drummen, G.P.; Mathivanan, S. Focus on extracellular vesicles: Introducing the next small big thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef] [Green Version]
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat. Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef]
- Timár, C.I.; Lorincz, A.M.; Csépányi-Kömi, R.; Vályi-Nagy, A.; Nagy, G.; Buzás, E.I.; Iványi, Z.; Kittel, A.; Powell, D.W.; McLeish, K.R.; et al. Antibacterial effect of microvesicles released from human neutrophilic granulocytes. Blood 2013, 121, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Dalli, J.; Norling, L.V.; Montero-Melendez, T.; Canova, D.F.; Lashin, H.; Pavlov, A.M.; Sukhorukov, G.B.; Hinds, C.J.; Perretti, M. Microparticle alpha-2-macroglobulin enhances pro-resolving responses and promotes survival in sepsis. EMBO Mol. Med. 2014, 6, 27–42. [Google Scholar] [CrossRef]
- Lashin, H.M.S.; Nadkarni, S.; Oggero, S.; Jones, H.R.; Knight, J.C.; Hinds, C.J.; Perretti, M. Microvesicle subsets in sepsis due to community acquired pneumonia compared to faecal peritonitis. Shock 2018, 49, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, I. Have host defense peptides been acting in innate immunity since the trilobites of the Cambrian period 540 million years ago? Juntendo Med. J. 2016, 62, 96–97. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Nagaoka, I.; Ogawa, H.; Okumura, K. Multifunctional antimicrobial proteins and peptides: Natural activators of immune systems. Curr. Pharm. Des. 2009, 15, 2393–2413. [Google Scholar] [CrossRef] [PubMed]
- Larrick, J.W.; Hirata, M.; Balint, R.F.; Lee, J.; Zhong, J.; Wright, S. Human CAP18: A novel antimicrobial lipopolysaccharide-binding protein. Infec. Immun. 1995, 63, 1291–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudmundsson, G.H.; Agerberth, B.; Odeberg, J.; Bergman, T.; Olsson, B.; Salcedo, R. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur. J. Biochem. 1996, 238, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Dürr, U.H.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochem. Biophys. Acta 2006, 1758, 1408–1425. [Google Scholar] [CrossRef] [Green Version]
- Nagaoka, I.; Kuwahara-Arai, K.; Tamura, H.; Hiramatsu, K.; Hirata, M. Augmentation of the bactericidal activities of human cathelicidin CAP18/LL-37-derived antimicrobial peptides by amino acid substitutions. Inflamm. Res. 2005, 54, 66–73. [Google Scholar] [CrossRef]
- Nagaoka, I.; Hirota, S.; Niyonsaba, F.; Hirata, M.; Adachi, Y.; Tamura, H.; Heumann, D. Cathelicidin family of antibacterial peptides CAP18 and CAP11 inhibit the expression of TNF-α by blocking the binding of LPS to CD14+ cells. J. Immunol. 2001, 167, 3329–3338. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.E.; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8, 402–410. [Google Scholar] [CrossRef]
- Nagaoka, I.; Tamura, H.; Hirata, M. An antimicrobial cathelicidin peptide, human CAP18/LL-37, suppresses neutrophil apoptosis via the activation of formyl-peptide receptor-like 1 and P2X7. J. Immunol. 2006, 176, 3044–3052. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Murakami, T.; Kuwahara-Arai, K.; Tamura, H.; Hiramatsu, K.; Nagaoka, I. Human anti-microbial cathelicidin peptide LL-37 suppresses the LPS-induced apoptosis of endothelial cells. Int. Immunol. 2011, 23, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Murakami, T.; Suzuki, K.; Tamura, H.; Reich, J.; Kuwahara-Arai, K.; Iba, T.; Nagaoka, I. Antimicrobial cathelicidin peptide LL-37 inhibits the pyroptosis of macrophages and improves the survival of polybacterial septic mice. Int. Immunol. 2016, 28, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoda, H.; Nakamura, K.; Hu, Z.; Tamura, H.; Reich, J.; Kuwahara-Arai, K.; Iba, T.; Tabe, Y.; Nagaoaka, I. Antimicrobial cathelicidin peptide LL-37 induces NET formation and suppresses the inflammatory responses in a mouse septic model. Mol. Med. Rep. 2017, 16, 5618–5626. [Google Scholar] [CrossRef]
- Kumagai, Y.; Murakami, T.; Kuwahara-Arai, K.; Iba, T.; Reich, J.; Nagaoka, I. Antimicrobial peptide LL-37 ameliorates a murine sepsis model via the induction of microvesicle release from neutrophils. Innate Immun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; O’Donnell, J.A.; Gerlic, M. Pyroptotic death storms and cytopenia. Curr. Opin. Immunol. 2014, 26, 128–137. [Google Scholar] [CrossRef]
- Cinel, I.; Opal, S.M. Molecular biology of inflammation and sepsis: A primer. Crit. Care Med. 2009, 37, 291–304. [Google Scholar] [CrossRef]
- Latz, E. The inflammasomes: Mechanisms of activation and function. Curr. Opin. Immunol. 2010, 22, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J.; et al. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Faubel, S.; Ljubanovic, D.; Mitra, A.; Falk, S.A.; Kim, J.; Tao, Y.; Soloviev, A.; Reznikov, L.L.; Dinarello, C.A.; et al. Endotoxemic acute renal failure is attenuated in caspase-1-deficient mice. Am. J. Physiol. Renal Physiol. 2005, 288, 997–1004. [Google Scholar] [CrossRef]
- Hu, Z.; Murakami, T.; Suzuki, K.; Tamura, H.; Kuwahara-Arai, K.; Iba, T.; Nagaoka, I. Antimicrobial cathelicidin peptide LL-37 inhibits the LPS/ATP-induced pyroptosis of macrophages by dual mechanism. PLoS ONE 2014, 9, e85765. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Lin, S.; Wan, B.; Velani, B.; Zhu, Y. Pyroptosis in liver disease: New insights into disease mechanisms. Aging Dis. 2019, 10, 1094–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Yu, M.; Pan, X.; Ren, C.; Peng, W.; Li, X.; Jiang, W.; Zheng, J.; Zhou, H. Artesunate reduces serum lipopolysaccharide in cecal ligation/puncture mice via enhanced LPS internalization by macrophages through increased mRNA expression of scavenger receptors. Int. J. Mol. Sci. 2014, 15, 1143–1161. [Google Scholar] [CrossRef] [PubMed]
- Sumi, Y.; Woehrle, T.; Chen, Y.; Bao, Y.; Li, X.; Yao, Y.; Inoue, Y.; Tanaka, H.; Junger, W.G. Plasma ATP is required for neutrophil activation in a mouse sepsis model. Shock 2014, 42, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Wang, X.; Yan, C.; Gao, Q.; Li, S.A.; Liu, J.; Zhou, K.; Guo, X.; Lee, W.; Zhang, Y. Adenosine-5’-triphosphate (ATP) protects mice against bacterial infection by activation of the NLRP3 inflammasome. PLoS ONE 2013, 8, e63759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, R.J.; Stam, E.J.; Downs, J.T.; Otterness, I.G. ATP induces the release of IL-1 from LPS-primed cells in vivo. J. Immunol. 1995, 154, 2821–2828. [Google Scholar] [PubMed]
- Cauwels, A.; Rogge, E.; Vandendriessche, B.; Shiva, S.; Brouckaert, P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014, 5, e1102. [Google Scholar] [CrossRef] [Green Version]
- Guimarães-Costa, A.B.; Nascimento, M.T.; Wardini, A.B.; Pinto-da-Silva, L.H.; Saraiva, E.M. ETosis: A microbicidal mechanism beyond cell death. J. Parasitol. Res. 2012, 2012, 929743. [Google Scholar] [CrossRef]
- Neumann, A.; Berends, E.T.; Nerlich, A.; Molhoek, E.M.; Gallo, R.L.; Meerloo, T.; Nizet, V.; Naim, H.Y.; von Kockritz-Blickwede, M. The antimicrobial peptide LL-37 facilitates the formation of neutrophil extracellular traps. Biochem. J. 2014, 464, 3–11. [Google Scholar] [CrossRef]
- Bouchon, A.; Facchetti, F.; Weigand, M.A.; Colonna, M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001, 410, 1103–1107. [Google Scholar] [CrossRef]
- Gómez-Piña, V.; Soares-Schanoski, A.; Rodríguez-Rojas, A.; del Fresno, C.; García, F.; Vallejo-Cremades, M.T.; Fernández-Ruiz, I.; Arnalich, F.; Fuentes-Prior, P.; López-Collazo, E. Metalloproteinases shed TREM-1 ectodomain from lipopolysaccharide-stimulated human monocytes. J. Immunol. 2007, 179, 4065–4073. [Google Scholar] [CrossRef] [Green Version]
- Gibot, S.; Kolopp-Sarda, M.N.; Bene, M.C.; Cravoisy, A.; Levy, B.; Faure, G.C.; Bollaert, P.E. Plasma level of a triggering receptor expressed on myeloid cells-1: Its diagnostic accuracy in patients with suspected sepsis. Ann. Intern. Med. 2004, 141, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Matzinger P: The danger model: A renewed sense of self. Science 2002, 296, 301–305. [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo1, C.Y.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1999, 1462, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, α-helical antimicrobial peptides. Biopolymers 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Mookherjee, N.; Hancock, R.E. Cationic host defence peptides: Innate immune regulatory peptides as a novel approach for treating infections. Cell. Mol. Life Sci. 2007, 64, 922–933. [Google Scholar] [CrossRef]
- Schuerholz, T.; Doemming, S.; Hornef, M.; Martin, L.; Simon, T.P.; Heinbockel, L.; Brandenburg, K.; Marx, G. The anti-inflammatory effect of the synthetic antimicrobial peptide 19-2.5 in a murine sepsis model: A prospective randomized study. Crit. Care 2013, 17, R3. [Google Scholar] [CrossRef] [Green Version]
- Pfalzgraff, A.; Heinbockel, L.; Su, Q.; Brandenburg, K.; Weindl, G. Synthetic anti-endotoxin peptides inhibit cytoplasmic LPS-mediated responses. Biochem. Pharmacol. 2017, 140, 64–72. [Google Scholar] [CrossRef]
















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagaoka, I.; Tamura, H.; Reich, J. Therapeutic Potential of Cathelicidin Peptide LL-37, an Antimicrobial Agent, in a Murine Sepsis Model. Int. J. Mol. Sci. 2020, 21, 5973. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21175973
Nagaoka I, Tamura H, Reich J. Therapeutic Potential of Cathelicidin Peptide LL-37, an Antimicrobial Agent, in a Murine Sepsis Model. International Journal of Molecular Sciences. 2020; 21(17):5973. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21175973
Chicago/Turabian StyleNagaoka, Isao, Hiroshi Tamura, and Johannes Reich. 2020. "Therapeutic Potential of Cathelicidin Peptide LL-37, an Antimicrobial Agent, in a Murine Sepsis Model" International Journal of Molecular Sciences 21, no. 17: 5973. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21175973