Metabolite Patterns in Human Myeloid Hematopoiesis Result from Lineage-Dependent Active Metabolic Pathways

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
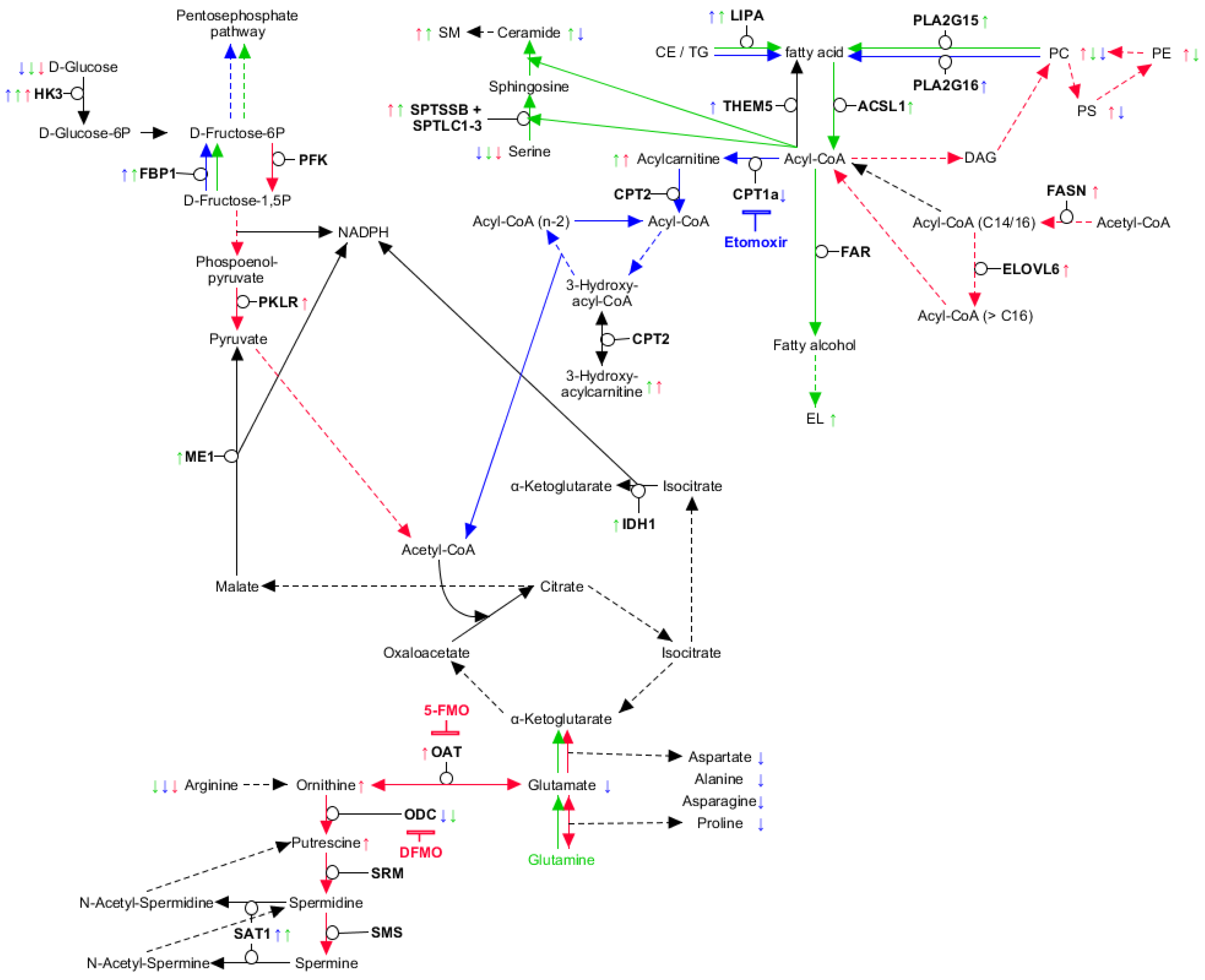
2. Results
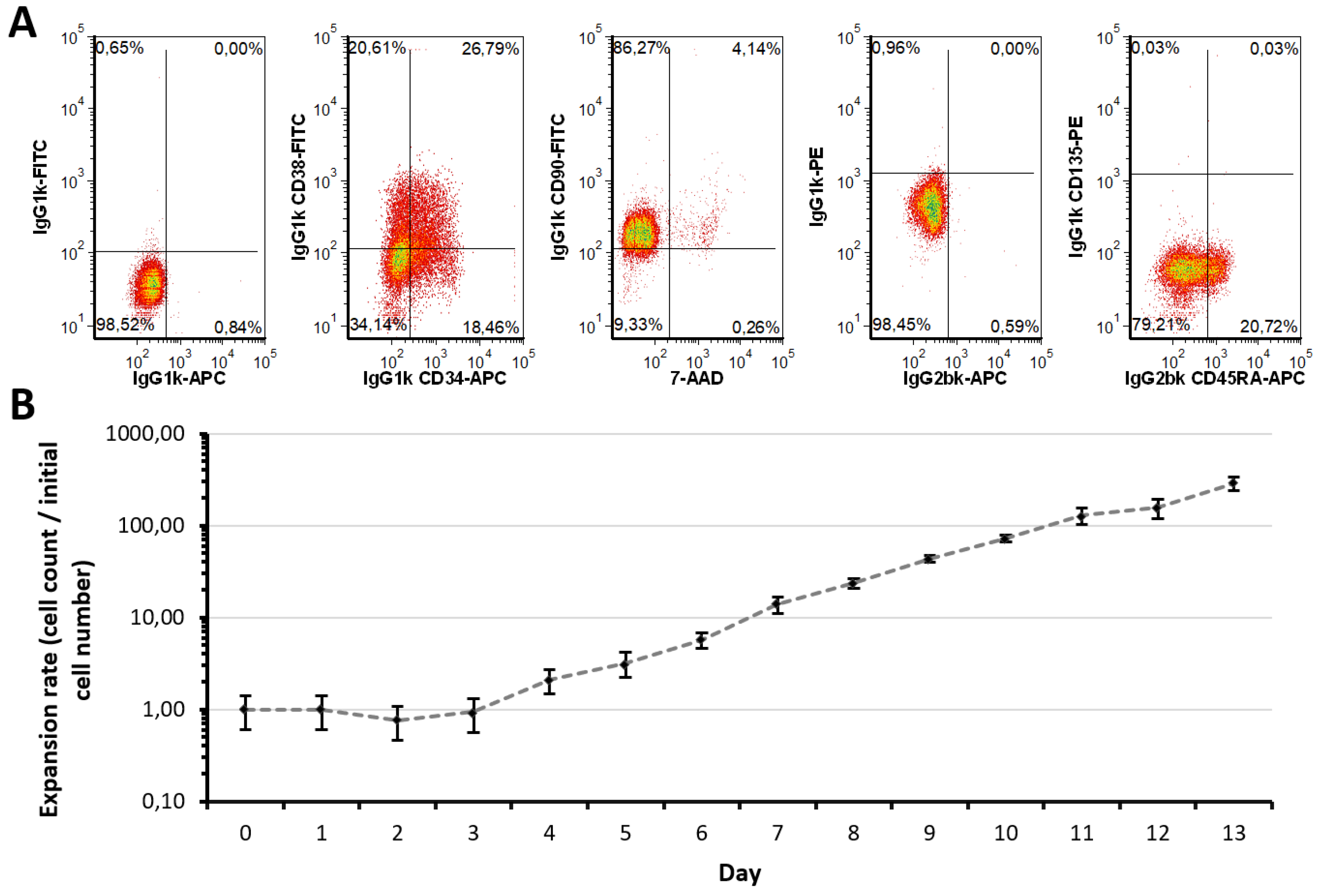
2.1. Expansion Approach Preserves Restricted Myeloid-Lineage Potential
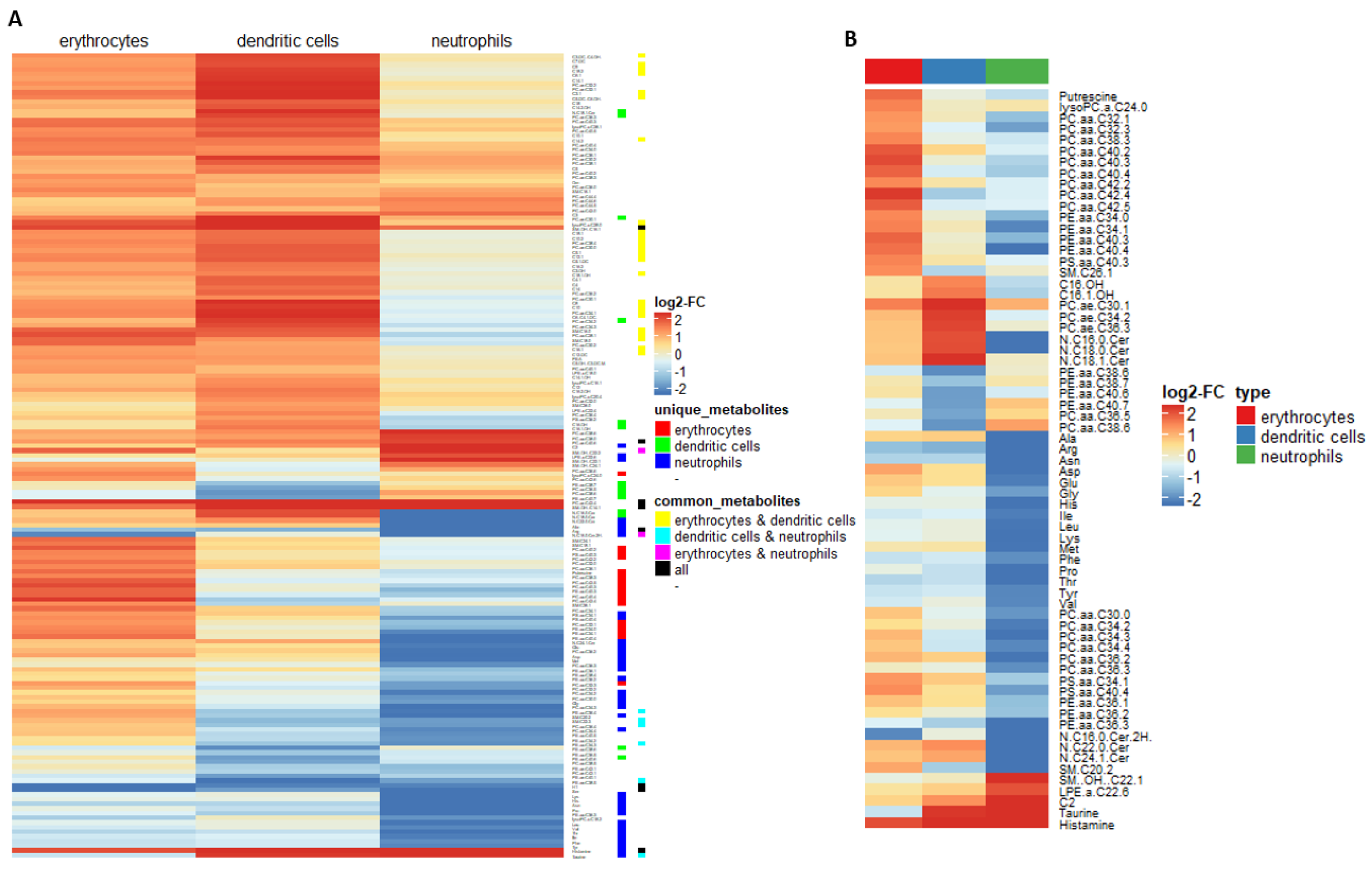
2.2. Myeloid Lineages Display Several Unique and Common Metabolic Patterns during Differentiation Accompanied by Changes in Pathway-Related Genes
2.3. Myeloid Lineages Own a Higher Hexose Consumption with Different Fate
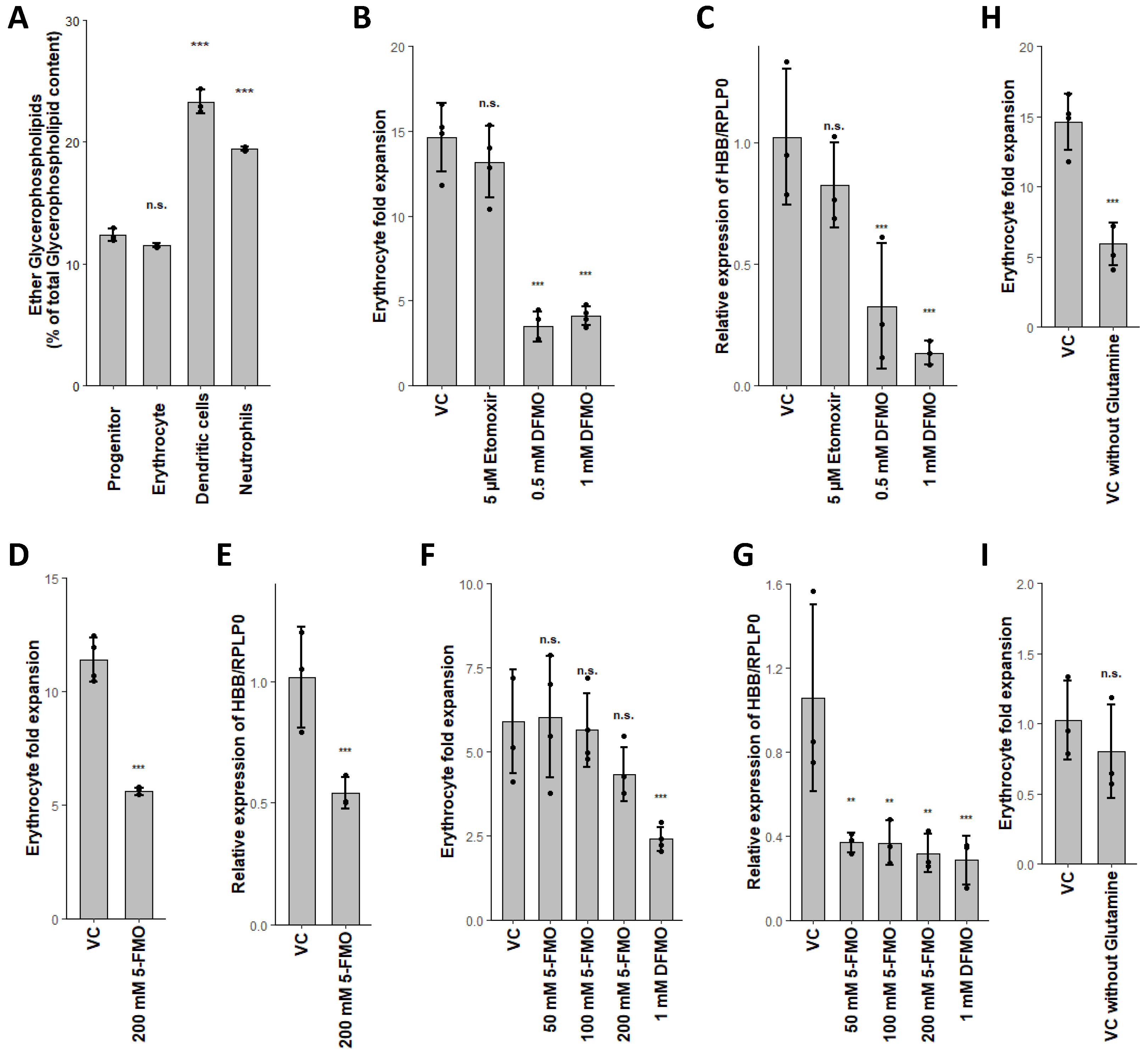
2.4. Combined Fatty Acid Generation and Respective Fate Is Unique for Each Myeloid Lineage
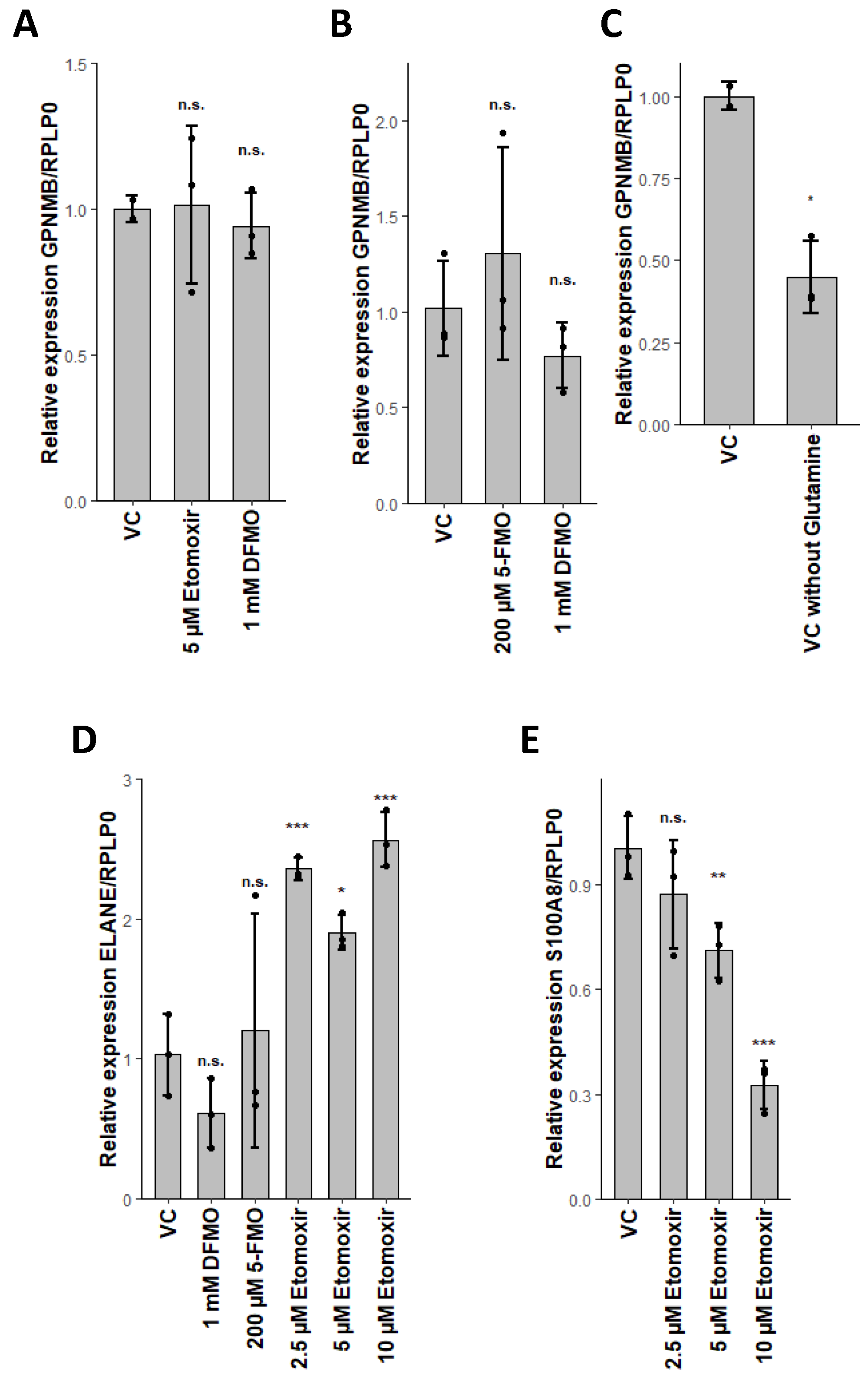
2.5. Glutaminolysis Crosstalk with FAO and Its Interconnection to Polyamine Synthesis in Erythrocytes
2.6. PLA2G15, PLA2G16 and Fatty Acid Levels Are Three Major Regulators of Unique Lipidomic Changes
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Flow Cytometry Analyses and Antibodies
4.3. Human CD34+ Cell Culture
4.4. Total RNA Isolation and RNA-Seq
4.5. Quantitative PCR Primer
4.6. LC/MS-based Metabolomics
4.7. Bioinformatic Analysis
4.8. Data Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-FMO | 5-fluoromethylornithine |
| aGPL | Acyl Glycerophospholipids |
| CD | Cluster of differentiation |
| CE | Cholesteryl esters |
| CFU | Colony forming unit |
| DC | Dendritic cells |
| DFMO | Difluormethyl-ornithine |
| EL | Ether lipids |
| EPO | Erythropoietin |
| F6P | Fructose-6-phosphate |
| FAO | Fatty acid oxidation |
| FA | Fatty acids |
| FDR | False Discovery Rate |
| G6P | Glucose-6-phosphate |
| GPL | Glycerophospholipids |
| HSC | Hematopoietic stem cell |
| HSPC | Hematopoietic stem and progenitor cells |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PS | Phosphatidylserine |
| PPP | Pentose phosphate pathway |
| ROS | Reactive oxygen species |
| TG | Triglycerides |
References
- Hunt, P.R. The C. elegans model in toxicity testing. J. Appl. Toxicol. 2017, 37, 50–59. [Google Scholar] [CrossRef]
- Miranda, J.P.; Leite, S.B.; Muller-Vieira, U.; Rodrigues, A.; Carrondo, M.J.T.; Alves, P.M. Towards an extended functional hepatocyte in vitro culture. Tissue Eng. Part C Methods 2009, 15, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Zhang, Q.; Carmichael, P.L.; Boekelheide, K.; Andersen, M.E. Toxicity testing in the 21st century: Defining new risk assessment approaches based on perturbation of intracellular toxicity pathways. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.L.; Honkalampi-Hämäläinen, U.; Weber, A.; Andersson, M.A.; Bertaud, F.; Castle, L.; Dahlman, O.; Hakulinen, P.; Hoornstra, D.; Lhuguenot, J.C.; et al. The BIOSAFEPAPER project for in vitro toxicity assessments: Preparation, detailed chemical characterisation and testing of extracts from paper and board samples. Food Chem. Toxicol. 2008, 46, 2498–2509. [Google Scholar] [CrossRef] [PubMed]
- Lipman, J.; Flint, O.; Bradlaw, J.; Frazier, J. Cell culture systems and in vitro toxicity testing. In Animals and Alternatives in Testing: History, Science and Ethics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1990; Volume 8, pp. 129–176. [Google Scholar]
- Moya, M.L.; Hsu, Y.H.; Lee, A.P.; Christopher, C.W.H.; George, S.C. In vitro perfused human capillary networks. Tissue Eng. Part C Methods 2013, 19, 730–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrod, M.; Chau, D.Y.S. An overview of tissue engineering as an alternative for toxicity assessment. J. Pharm. Pharm. Sci. 2016, 19, 31–71. [Google Scholar] [CrossRef]
- Pupovac, A.; Senturk, B.; Griffoni, C.; Maniura-Weber, K.; Rottmar, M.; McArthur, S.L. Toward Immunocompetent 3D Skin Models. Adv. Healthc. Mater. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Mahler, G.J.; Esch, M.B.; Stokol, T.; Hickman, J.J.; Shuler, M.L. Body-on-A-Chip systems for Animal-free toxicity testing. ATLA Altern. to Lab. Anim. 2016, 44, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Luz, A.L.; Tokar, E.J. Pluripotent Stem Cells in Developmental Toxicity Testing: A Review of Methodological Advances. Toxicol. Sci. 2018, 165, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Anson, B.D.; Kolaja, K.L.; Kamp, T.J. Opportunities for use of human iPS cells in predictive toxicology. Clin. Pharmacol. Ther. 2011, 89, 754–758. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.W.; Peters, M.F.; Dragan, Y.P. Human induced pluripotent stem cells and their use in drug discovery for toxicity testing. Toxicol. Lett. 2013, 219, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Balls, M.; Combes, R.D.; Bhogal, N. (Eds.) New Technologies for Toxicity Testing; Advances in Experimental Medicine and Biology; Springer US: New York, NY, USA, 2012; Volume 745, ISBN 978-1-4614-3054-4. [Google Scholar]
- Laiosa, M.D.; Tate, E.R. Fetal hematopoietic stem cells are the canaries in the coal mine that portend later life immune deficiency. Endocrinology 2015, 156, 3458–3465. [Google Scholar] [CrossRef] [PubMed]
- Van Den Heuvel, R.L.; Leppens, H.; Schoeters, G.E.R. Use of in vitro assays to assess hematotoxic effects of environmental compounds. Cell Biol. Toxicol. 2001, 17, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Mahalingaiah, P.K.; Palenski, T.; Van Vleet, T.R. An In Vitro Model of Hematotoxicity: Differentiation of Bone Marrow–Derived Stem/Progenitor Cells into Hematopoietic Lineages and Evaluation of Lineage-Specific Hematotoxicity. Curr. Protoc. Toxicol. 2018, 76, e45. [Google Scholar] [CrossRef] [PubMed]
- Owen, L.; Laird, K.; Wilson, P.B. Structure-activity modelling of essential oils, their components, and key molecular parameters and descriptors. Mol. Cell. Probes 2018, 38, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Hirode, M.; Ono, A.; Kiyosawa, N.; Omura, K.; Shimizu, T.; Mizukawa, Y.; Miyagishima, T.; Nagao, T.; Urushidani, T. A toxicogenomics approach for early assessment of potential non-genotoxic hepatocarcinogenicity of chemicals in rats. Toxicology 2008, 250, 15–26. [Google Scholar] [CrossRef]
- Duke, S.O.; Bajsa, J.; Pan, Z. Omics Methods for Probing the Mode of Action of Natural and Synthetic Phytotoxins. J. Chem. Ecol. 2013, 39, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, T.; Strigun, A.; Verlohner, A.; Huener, H.A.; Peter, E.; Herold, M.; Bordag, N.; Mellert, W.; Walk, T.; Spitzer, M.; et al. Prediction of liver toxicity and mode of action using metabolomics in vitro in HepG2 cells. Arch. Toxicol. 2018, 92, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Labine, L.M.; Simpson, M.J. The use of nuclear magnetic resonance (NMR) and mass spectrometry (MS)–based metabolomics in environmental exposure assessment. Curr. Opin. Environ. Sci. Heal. 2020, 15, 7–15. [Google Scholar] [CrossRef]
- Emwas, A.H.M. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. 2015, 1277, 161–193. [Google Scholar] [CrossRef]
- Potratz, S.; Tarnow, P.; Jungnickel, H.; Baumann, S.; Von Bergen, M.; Tralau, T.; Luch, A. Combination of Metabolomics with Cellular Assays Reveals New Biomarkers and Mechanistic Insights on Xenoestrogenic Exposures in MCF-7 Cells. Chem. Res. Toxicol. 2017, 30, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.A.; Burant, C.F.; Kennedy, R.T. Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal. Chem. 2011, 83, 3406–3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.; Evans, K.J.; Sexton, A.E.; Schofield, L.; Creek, D.J. Metabolomics-Based Elucidation of Active Metabolic Pathways in Erythrocytes and HSC-Derived Reticulocytes. J. Proteome Res. 2017, 16, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Sapcariu, S.C.; Kanashova, T.; Weindl, D.; Ghelfi, J.; Dittmar, G.; Hiller, K. Simultaneous extraction of proteins and metabolites from cells in culture. MethodsX 2014, 1, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Muschet, C.; Möller, G.; Prehn, C.; de Angelis, M.H.; Adamski, J.; Tokarz, J. Removing the bottlenecks of cell culture metabolomics: Fast normalization procedure, correlation of metabolites to cell number, and impact of the cell harvesting method. Metabolomics 2016, 12, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubyshkin, V.; Budisa, N. Amide rotation trajectories probed by symmetry. Org. Biomol. Chem. 2017, 15, 6764–6772. [Google Scholar] [CrossRef] [Green Version]
- Grootveld, M.; Percival, B.; Gibson, M.; Osman, Y.; Edgar, M.; Molinari, M.; Mather, M.L.; Casanova, F.; Wilson, P.B. Progress in low-field benchtop NMR spectroscopy in chemical and biochemical analysis. Anal. Chim. Acta 2019, 1067, 11–30. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Fan, Z.; Xu, H.; Tan, X.; Zhu, M. Metabolomics-based parallel discovery of xenobiotics and induced endogenous metabolic dysregulation in clinical toxicology. Biomed. Chromatogr. 2019, 33, e4413. [Google Scholar] [CrossRef]
- Ortmayr, K.; Dubuis, S.; Zampieri, M. Metabolic profiling of cancer cells reveals genome-wide crosstalk between transcriptional regulators and metabolism. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Leenders, J.; Grootveld, M.; Percival, B.; Gibson, M.; Casanova, F.; Wilson, P.B. Benchtop low-frequency 60 MHz NMR analysis of urine: A comparative metabolomics investigation. Metabolites 2020, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.H.; Leone, R.D.; Horton, M.R.; Powell, J.D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 2019, 18, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Carriere, J.; Lin, X.; Xie, N.; Feng, P. Interplay between cellular metabolism and cytokine responses during viral infection. Viruses 2018, 10, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef] [PubMed]
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016, 8, 28–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheding, S.; Loeffler, M.; Schmitz, S.; Seidel, H.J.; -Erich Wichmann, H. Hematotoxic effects of benzene analyzed by mathematical modeling. Toxicology 1992, 72, 265–279. [Google Scholar] [CrossRef]
- Nagata, M.; Arimitsu, N.; Ito, T.; Sekimizu, K. Antioxidant N-acetyl-l-cysteine inhibits erythropoietin-induced differentiation of erythroid progenitors derived from mouse fetal liver. Cell Biol. Int. 2007, 31, 252–256. [Google Scholar] [CrossRef]
- Zembron-Lacny, A.; Slowinska-Lisowska, M.; Szygula, Z.; Witkowski, Z.; Szyszka, K. Modulatory effect of N-acetylcysteine on pro-antioxidant status and haematological response in healthy men. J. Physiol. Biochem. 2010, 66, 15–21. [Google Scholar] [CrossRef]
- Scheding, S.; Media, J.E.; Nakeff, A. Acute toxic effects of 3′-azido-3′-deoxythymidine (AZT) on normal and regenerating murine hematopoiesis. Exp. Hematol. 1994, 22, 60–65. [Google Scholar]
- Luo, S.T.; Zhang, D.M.; Qin, Q.; Lu, L.; Luo, M.; Guo, F.C.; Shi, H.S.; Jiang, L.; Shao, B.; Li, M.; et al. The Promotion of Erythropoiesis via the Regulation of Reactive Oxygen Species by Lactic Acid. Sci. Rep. 2017, 7, 38105. [Google Scholar] [CrossRef]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oburoglu, L.; Tardito, S.; Fritz, V.; De Barros, S.C.; Merida, P.; Craveiro, M.; Mamede, J.; Cretenet, G.; Mongellaz, C.; An, X.; et al. Glucose and Glutamine Metabolism Regulate Human Hematopoietic Stem Cell Lineage Specification. Cell Stem Cell 2014, 15, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.J.; Lin, Y.C.; Lin, C.Y.; Pishesha, N.; Lewis, C.A.; Freinkman, E.; Farquharson, C.; Millán, J.L.; Lodish, H. Enhanced phosphocholine metabolism is essential for terminal erythropoiesis. Blood 2018, 131, 2955–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niskanen, E.; Kallio, A.; McCann, P.P.; Baker, D.G. The role of polyamine biosynthesis in hematopoietic precursor cell proliferation in mice. Blood 1983, 61, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Segal, G.M.; Stueve, T.; Adamson, J.W. Spermine and spermidine are non-specific inhibitors of in vitro hematopoiesis. Kidney Int. 1987, 31, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J. Mol. Biol. 2015, 427, 3389–3406. [Google Scholar] [CrossRef]
- Kaiser, L.; Weinschrott, H.; Quint, I.; Csuk, R.; Jung, M.; Deigner, H.-P. Fatty acid oxidation makes the difference: Lineage-selective disturbance by DEHP via ROS in myeloid hematopoiesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, L.; Guan, X.; Wang, H.; Shen, B.; Zhang, Y.; Ren, Z.; Ma, Y.; Ding, X.; Jiang, Y. A small-molecule/cytokine combination enhances hematopoietic stem cell proliferation via inhibition of cell differentiation. Stem Cell Res. Ther. 2017, 8, 169. [Google Scholar] [CrossRef] [Green Version]
- Manz, M.G.; Miyamoto, T.; Akashi, K.; Weissman, I.L. Prospective isolation of human clonogenic common myeloid progenitors. Proc. Natl. Acad. Sci. USA 2002, 99, 11872–11877. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, C.; Wang, L.; Shen, B.; Guan, X.; Tian, J.; Ren, Z.; Ding, X.; Ma, Y.; Dai, W.; et al. Large-Scale Ex Vivo Generation of Human Red Blood Cells from Cord Blood CD34 + Cells. Stem Cells Transl. Med. 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, R.; Ogata, H.; Iguchi, T.; Sogo, S.; Kushida, T.; Ito, T.; Inaba, M.; Ikehara, S.; Kobayashi, Y. Comparative analyses of megakaryocytes derived from cord blood and bone marrow. Br. J. Haematol. 2000, 108, 602–609. [Google Scholar] [CrossRef]
- Woo, S.-Y.; Jung, Y.-J.; Ryu, K.-H.; Park, H.-Y.; Kie, J.-H.; Im, S.-A.; Chung, W.-S.; Han, H.-S.; Seoh, J.-Y. In vitro differentiation of natural killer T cells from human cord blood CD34+ cells. Br. J. Haematol. 2003, 121, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Gutknecht, M.; Geiger, J.; Joas, S.; Dörfel, D.; Salih, H.R.; Müller, M.R.; Grünebach, F.; Rittig, S.M. The transcription factor MITF is a critical regulator of GPNMB expression in dendritic cells. Cell Commun. Signal. 2015, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Van Schaarenburg, R.A.; Suurmond, J.; Habets, K.L.L.; Brouwer, M.C.; Wouters, D.; Kurreeman, F.A.S.; Huizinga, T.W.J.; Toes, R.E.M.; Trouw, L.A. The production and secretion of complement component C1q by human mast cells. Mol. Immunol. 2016, 78, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Gliddon, D.R.; Howard, C.J. CD26 is expressed on a restricted subpopulation of dendritic cells in vivo. Eur. J. Immunol. 2002, 32, 1472. [Google Scholar] [CrossRef]
- Zimmerman, A.W.; Joosten, B.; Torensma, R.; Parnes, J.R.; van Leeuwen, F.N.; Figdor, C.G. Long-term engagement of CD6 and ALCAM is essential for T-cell proliferation induced by dendritic cells. Blood 2006, 107, 3212–3220. [Google Scholar] [CrossRef] [Green Version]
- Tessier Carle Ryckman, P.A.; Vandal, K.; Rouleau, P. Neutrophil Chemotaxis and Adhesion S100A8, S100A9, and S100A8/A9 Induce Proinflammatory Activities of S100: Proteins. J Immunol Ref. 2003, 170, 3233–3242. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. The Glycolytic Pathway Is Tightly Controlled. In Biochemistry, 5th ed.; W H Freeman: New York, NY, USA, 2002; ISBN 978-0716746843. [Google Scholar]
- Lieberman, M.D.; Cunningham, W.A. Type I and Type II error concerns in fMRI research: Re-balancing the scale. Soc. Cogn. Affect. Neurosci. 2009, 4, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Oburoglu, L.; Romano, M.; Taylor, N.; Kinet, S. Metabolic regulation of hematopoietic stem cell commitment and erythroid differentiation. Curr. Opin. Hematol. 2016, 23, 198–205. [Google Scholar] [CrossRef]
- Borregaard, N.; Herlin, T. Energy metabolism of human neutrophils during phagocytosis. J. Clin. Investig. 1982, 70, 550–557. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiamoncini, J.; Lima, T.M.; Hirabara, S.M.; Ecker, J.; Gorjão, R.; Romanatto, T.; Elolimy, A.; Worsch, S.; Laumen, H.; Bader, B.; et al. Medium-chain dicarboxylic acylcarnitines as markers of n-3 PUFA-induced peroxisomal oxidation of fatty acids. Mol. Nutr. Food Res. 2015, 59, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines--old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, S.E.; Evans, A.M. Carnitine and Acylcarnitines. Clin. Pharmacokinet. 2012, 51, 553–572. [Google Scholar] [CrossRef]
- Su, X.; Han, X.; Mancuso, D.J.; Abendschein, D.R.; Gross, R.W. Accumulation of long-chain acylcarnitine and 3-hydroxy acylcarnitine molecular species in diabetic myocardium: Identification of alterations in mitochondrial fatty acid processing in diabetic myocardium by shotgun lipidomics. Biochemistry 2005, 44, 5234–5245. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Komen, J.; Kemp, S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011, 278, 182–194. [Google Scholar] [CrossRef]
- Jansen, G.A.; Wanders, R.J.A. Alpha-Oxidation. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 1403–1412. [Google Scholar] [CrossRef] [Green Version]
- Grassi, L.; Pourfarzad, F.; Ullrich, S.; Merkel, A.; Were, F.; Carrillo-de-Santa-Pau, E.; Yi, G.; Hiemstra, I.H.; Tool, A.T.J.; Mul, E.; et al. Dynamics of Transcription Regulation in Human Bone Marrow Myeloid Differentiation to Mature Blood Neutrophils. Cell Rep. 2018, 24, 2784–2794. [Google Scholar] [CrossRef] [Green Version]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e8. [Google Scholar] [CrossRef]
- Kim, M.H.; Yang, D.; Kim, M.; Kim, S.Y.; Kim, D.; Kang, S.J. A late-lineage murine neutrophil precursor population exhibits dynamic changes during demand-adapted granulopoiesis. Sci. Rep. 2017, 7, 39804. [Google Scholar] [CrossRef]
- Cassago, A.; Ferreira, A.P.S.; Ferreira, I.M.; Fornezari, C.; Gomes, E.R.M.; Greene, K.S.; Pereira, H.M.; Garratt, R.C.; Dias, S.M.G.; Ambrosio, A.L.B. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Zhu, X.; Shi, Z.; Wu, T.; Wu, L. Metabolic Regulation of Dendritic Cell Differentiation. Front. Immunol. 2019, 10, 410. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Wakasawa, T.; Shima, Y.; Tsuboi, I.; Aizawa, S.; Tamai, I. Role of Polyamines Derived from Arginine in Differentiation and Proliferation of Human Blood Cells. Biol. Pharm. Bull. 2006, 29, 234–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballas, S.K.; Mohandas, N.; Marton, L.J.; Shohet, S.B. Stabilization of erythrocyte membranes by polyamines. Proc. Natl. Acad. Sci. USA 2006, 80, 1942–1946. [Google Scholar] [CrossRef] [Green Version]
- Bratton, D.L. Polyamine inhibition of transbilayer movement of plasma membrane phospholipids in the erythrocyte ghost. J. Biol. Chem. 1994, 269, 22517–22523. [Google Scholar]
- Farriol, M.; Segovia-Silvestre, T.; Venereo, Y.; Orta, X. Antioxidant effect of polyamines on erythrocyte cell membrane lipoperoxidation after free-radical damage. Phytother. Res. 2003, 17, 44–47. [Google Scholar] [CrossRef]
- Shima, Y.; Maeda, T.; Aizawa, S.; Tsuboi, I.; Kobayashi, D.; Kato, R.; Tamai, I. L-arginine import via cationic amino acid transporter CAT1 is essential for both differentiation and proliferation of erythrocytes. Blood 2006, 107, 1352–1356. [Google Scholar] [CrossRef] [Green Version]
- Ginguay, A.; Cynober, L.; Curis, E.; Nicolis, I. Ornithine Aminotransferase, an Important Glutamate-Metabolizing Enzyme at the Crossroads of Multiple Metabolic Pathways. Biology 2017, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Daune, G.; Gerhart, F.; Seiler, N. 5-Fluoromethylornithine, an irreversible and specific inhibitor of l-ornithine:2-oxo-acid aminotransferase. Biochem. J. 1988, 253, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Gavin, A.L.; Huang, D.; Huber, C.; Mårtensson, A.; Tardif, V.; Skog, P.D.; Blane, T.R.; Thinnes, T.C.; Osborn, K.; Chong, H.S.; et al. PLD3 and PLD4 are single-stranded acid exonucleases that regulate endosomal nucleic-acid sensing. Nat. Immunol. 2018, 19, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhao, L.; Larsson, A.; Venge, P. The identification of a phospholipase B precursor in human neutrophils. FEBS J. 2009, 276, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Voelker, D.R. Phosphatidylserine decarboxylase. Biochim. Biophys. Acta Lipids Lipid Metab. 1997, 1348, 236–244. [Google Scholar] [CrossRef]
- McMaster, C.R.; Bell, R.M. CDP-ethanolamine:1,2-diacylglycerol ethanolaminephosphotransferase. Biochim. Biophys. Acta Lipids Lipid Metab. 1997, 1348, 117–123. [Google Scholar] [CrossRef]
- Glukhova, A.; Hinkovska-Galcheva, V.; Kelly, R.; Abe, A.; Shayman, J.A.; Tesmer, J.J.G.G. Structure and function of lysosomal phospholipase A2 and lecithin:cholesterol acyltransferase. Nat. Commun. 2015, 6, 6250. [Google Scholar] [CrossRef] [Green Version]
- Uyama, T.; Ichi, I.; Kono, N.; Inoue, A.; Tsuboi, K.; Jin, X.-H.H.; Araki, N.; Aoki, J.; Arai, H.; Ueda, N. Regulation of peroxisomal lipid metabolism by catalytic activity of tumor suppressor H-rev107. J. Biol. Chem. 2012, 287, 2706–2718. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.B.; Russell, D.W. Mammalian Wax Biosynthesis I. Identification of two fatty acyl-Coenzyme A reductases with different substrate specificities and tissue distributions. J. Biol. Chem. 2004, 279, 37789–37797. [Google Scholar] [CrossRef] [Green Version]
- Honsho, M.; Asaoku, S.; Fujiki, Y. Posttranslational regulation of fatty acyl-CoA reductase 1, Far1, controls ether glycerophospholipid synthesis. J. Biol. Chem. 2010, 285, 8537–8542. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Gupta, S.D.; Gable, K.; Niranjanakumari, S.; Moitra, P.; Eichler, F.; Brown, R.H.; Harmon, J.M.; Dunn, T.M. Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proc. Natl. Acad. Sci. USA 2009, 106, 8186–8191. [Google Scholar] [CrossRef] [Green Version]
- Karigane, D.; Takubo, K. Metabolic regulation of hematopoietic and leukemic stem/progenitor cells under homeostatic and stress conditions. Int. J. Hematol. 2017, 106, 18–26. [Google Scholar] [CrossRef]
- Du, X.; Chapman, N.M.; Chi, H. Emerging roles of cellular metabolism in regulating dendritic cell subsets and function. Front. Cell Dev. Biol. 2018, 6, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiciński, M.; Wȩclewicz, M.M. Clozapine-induced agranulocytosis/granulocytopenia: Mechanisms and monitoring. Curr. Opin. Hematol. 2018, 25, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Ohtoyo, M.; Machinaga, N.; Inoue, R.; Hagihara, K.; Yuita, H.; Tamura, M.; Hashimoto, R.; Chiba, J.; Muro, F.; Watanabe, J.; et al. Component of Caramel Food Coloring, THI, Causes Lymphopenia Indirectly via a Key Metabolic Intermediate. Cell Chem. Biol. 2016, 23, 555–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demur, C.; Métais, B.; Canlet, C.; Tremblay-Franco, M.; Gautier, R.; Blas-Y-Estrada, F.; Sommer, C.; Gamet-Payrastre, L. Dietary exposure to a low dose of pesticides alone or as a mixture: The biological metabolic fingerprint and impact on hematopoiesis. Toxicology 2013, 308, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Rivedal, E.; Leithe, E. The benzene metabolite trans, trans-muconaldehyde blocks gap junction intercellular communication by cross-linking connexin43. Toxicol. Appl. Pharmacol. 2008, 232, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Menale, C.; Mita, D.G.; Diano, N.; Diano, S. Adverse Effects of Bisphenol A Exposure on Glucose Metabolism Regulation. Open Biotechnol. J. 2016, 10, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.C.; Aivazidis, S.; Kuzyk, C.L.; Jain, A.; Roede, J.R. Acute Maneb Exposure Significantly Alters Both Glycolysis and Mitochondrial Function in Neuroblastoma Cells. Toxicol. Sci. 2018, 165, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Burri, L.; Bjørndal, B.; Wergedahl, H.; Berge, K.; Bohov, P.; Svardal, A.; Berge, R.K. Tetradecylthioacetic Acid Increases Hepatic Mitochondrial β-Oxidation and Alters Fatty Acid Composition in a Mouse Model of Chronic Inflammation. Lipids 2011, 46, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Maćczak, A.; Cyrkler, M.; Bukowska, B.; Michałowicz, J. Eryptosis-inducing activity of bisphenol A and its analogs in human red blood cells (in vitro study). J. Hazard. Mater. 2016, 307, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Olchowik-Grabarek, E.; Makarova, K.; Mavlyanov, S.; Abdullajanova, N.; Zamaraeva, M. Comparative analysis of BPA and HQ toxic impacts on human erythrocytes, protective effect mechanism of tannins (Rhus typhina). Environ. Sci. Pollut. Res. 2018, 25, 1200–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanherwegen, A.S.; Eelen, G.; Ferreira, G.B.; Ghesquière, B.; Cook, D.P.; Nikolic, T.; Roep, B.; Carmeliet, P.; Telang, S.; Mathieu, C.; et al. Vitamin D controls the capacity of human dendritic cells to induce functional regulatory T cells by regulation of glucose metabolism. J. Steroid Biochem. Mol. Biol. 2019, 187, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Feng, Z.; Yi, X. A general introduction to adjustment for multiple comparisons. J. Thorac. Dis. 2017, 9, 1725–1729. [Google Scholar] [CrossRef] [Green Version]
- Kutmon, M.; van Iersel, M.P.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.R.; Evelo, C.T. PathVisio 3: An Extendable Pathway Analysis Toolbox. PLOS Comput. Biol. 2015, 11, e1004085. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Process | Fold Enrichment | Holm Adjusted p Value |
|---|---|---|
| Erythrocytes | ||
| Immune system process | 2.19 | 0.00009 |
| Cytokine-mediated signaling pathway | 3.50 | 0.00174 |
| Response to chemical | 1.81 | 0.00282 |
| Erythrocyte differentiation | 11.25 | 0.00355 |
| Erythrocyte homeostasis | 10.12 | 0.00771 |
| Dendritic Cells | ||
| Immune response | 2.76 | < 10−10 |
| Immune-system process | 2.35 | < 10−10 |
| Leukocyte activation involved in immune response | 4.34 | < 10−9 |
| Cell activation involved in immune response | 4.31 | < 10−9 |
| Leukocyte activation | 3.59 | < 10−9 |
| Neutrophil Granulocytes | ||
| Defense response | 4.09 | < 10−27 |
| Immune response | 3.47 | < 10−26 |
| Immune system process | 2.77 | < 10−24 |
| Immune effector process | 4.20 | < 10−22 |
| Myeloid cell activation involved in immune response | 5.98 | < 10−20 |
| Gene | Metabolic Pathway | Erythrocytes | Dendritic Cells | Neutrophils |
|---|---|---|---|---|
| SLC2A1 | Glycolysis | 1.69 | n.s. | n.s. |
| SLC2A5 | n.s. | n.s. | 1.94 | |
| SLC2A6 | n.s. | n.s. | 2.28 | |
| HK3 | 1.77 | 2.65 | 3.76 | |
| MINPP1 | 0.96 | −1.47 | n.s. | |
| PKLR | 4.42 | n.s. | n.s. | |
| FBP1 | n.s. | 4.44 | 5.36 | |
| OAT | Polyamine synthesis | 0.95 | n.s. | n.s. |
| ODC | n.s. | −1.89 | −1.47 | |
| SAT1 | n.s. | 1.28 | 1.16 | |
| MYC | 1.19 | n.s. | n.s. | |
| ME1 | NADPH production | n.s. | 2.88 | n.s. |
| IDH1 | n.s. | 1.97 | n.s. | |
| CPT1a | FA oxidation | n.s. | n.s. | −1.25 |
| FASN | FA biosynthesis | 1.10 | n.s. | n.s. |
| ACSL1 | n.s. | 1.14 | n.s. | |
| ELOVL6 | FA elongation | 1.41 | n.s. | n.s. |
| THEM5 | n.s. | n.s. | Inf | |
| SCD | Biosynthesis of unsaturated FA | 1.13 | 1.36 | n.s. |
| LIPA | Lipolysis | n.s. | 2.86 | 2.75 |
| PLD3 | Glycero-phospholipid metabolism | 0.96 | 2.54 | 2.33 |
| PLBD1 | 1.84 | n.s. | 3.118 | |
| PLA2G15 | n.s. | 1.24 | n.s. | |
| PLA2G16 | n.s. | n.s. | 2.73 | |
| LPCAT3 | 1.09 | n.s. | n.s. | |
| PISD | 1.03 | n.s. | n.s. | |
| PHOSPHO1 | 3.91 | n.s. | n.s. | |
| PPAP2B | −2.28 | 1.73 | 2.54 | |
| LPIN1 | n.s. | n.s. | -Inf | |
| PCYT1B | n.s. | −2.02 | −2.40 | |
| PLA2G7 | Ether lipid metabolism | 1.41 | 4.18 | 3.34 |
| SPTSSB | Sphingolipid metabolism | 3.11 | 5.07 | n.s. |
| ACER3 | n.s. | 1.14 | n.s. | |
| SMPD3 | Inf | n.s. | Inf | |
| PPAP2B | −2.28 | 1.73 | 2.54 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaiser, L.; Weinschrott, H.; Quint, I.; Blaess, M.; Csuk, R.; Jung, M.; Kohl, M.; Deigner, H.-P. Metabolite Patterns in Human Myeloid Hematopoiesis Result from Lineage-Dependent Active Metabolic Pathways. Int. J. Mol. Sci. 2020, 21, 6092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176092
Kaiser L, Weinschrott H, Quint I, Blaess M, Csuk R, Jung M, Kohl M, Deigner H-P. Metabolite Patterns in Human Myeloid Hematopoiesis Result from Lineage-Dependent Active Metabolic Pathways. International Journal of Molecular Sciences. 2020; 21(17):6092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176092
Chicago/Turabian StyleKaiser, Lars, Helga Weinschrott, Isabel Quint, Markus Blaess, René Csuk, Manfred Jung, Matthias Kohl, and Hans-Peter Deigner. 2020. "Metabolite Patterns in Human Myeloid Hematopoiesis Result from Lineage-Dependent Active Metabolic Pathways" International Journal of Molecular Sciences 21, no. 17: 6092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176092