Syk Inhibitors: New Computational Insights into Their Intraerythrocytic Action in Plasmodium falciparum Malaria


 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
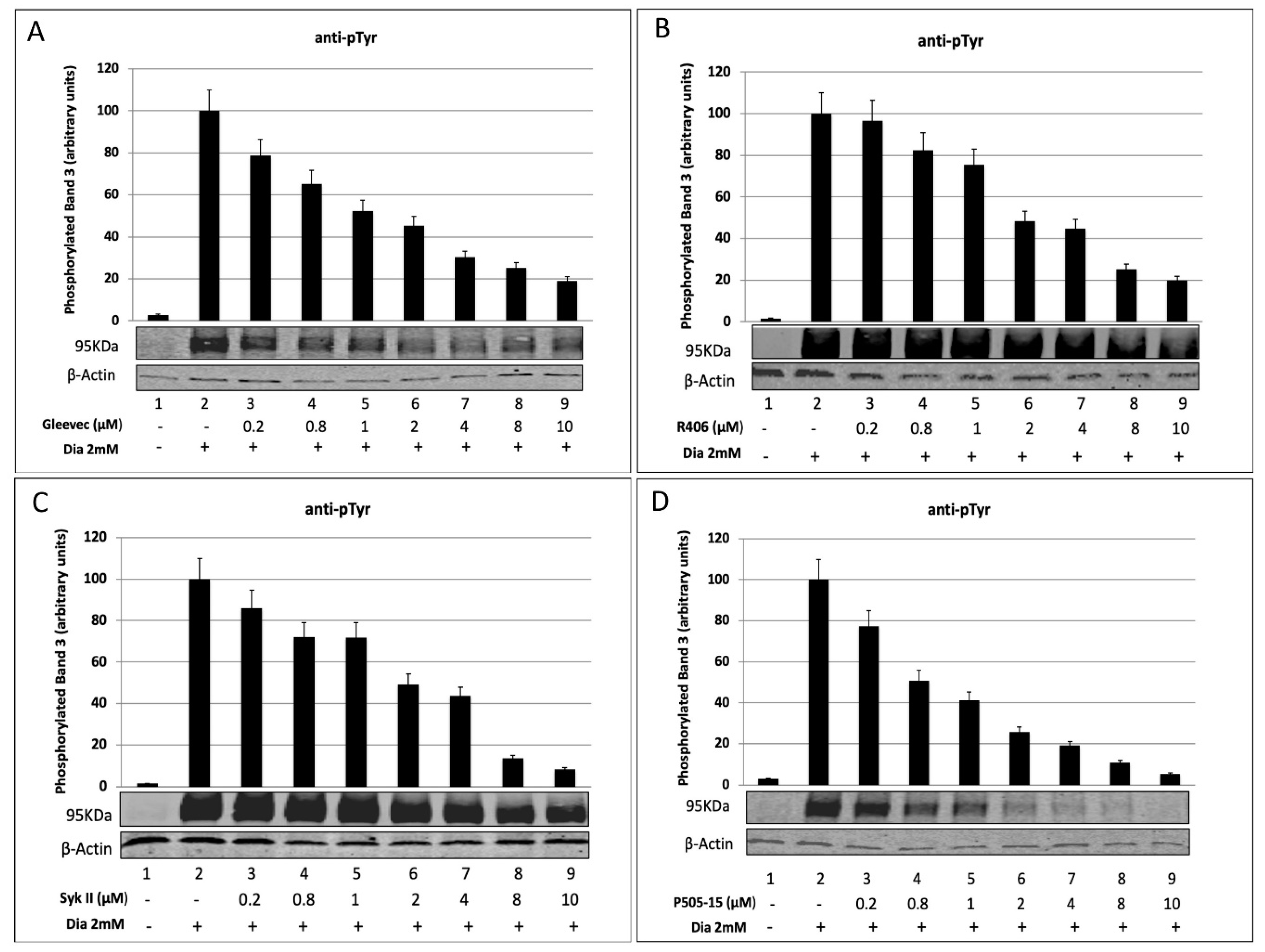
2.1. Densitometric Analysis of Band 3 Tyrosine Phosphorylation in Diamide-Treated Erythrocytes after Treatment with Syk Inhibitors
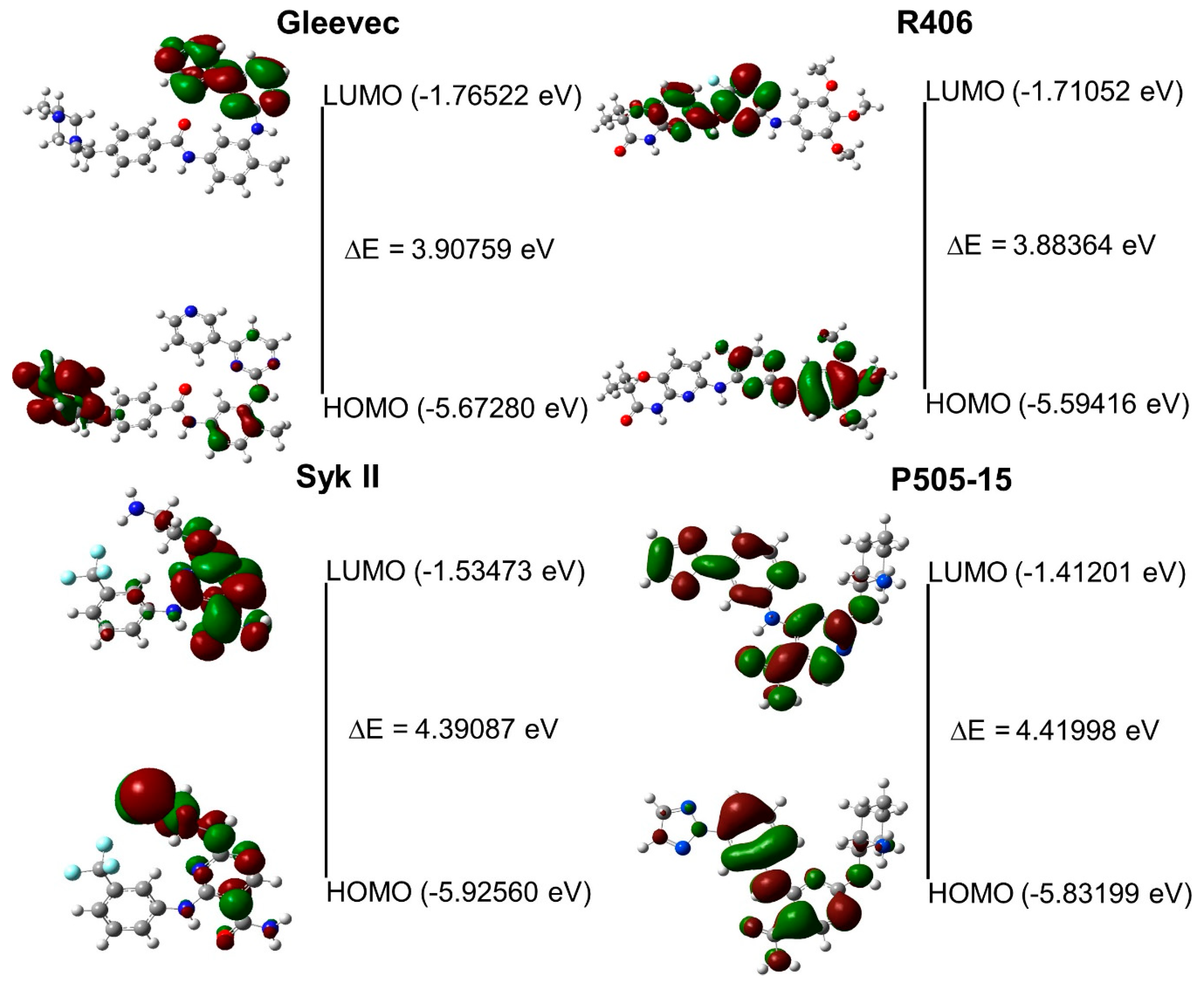
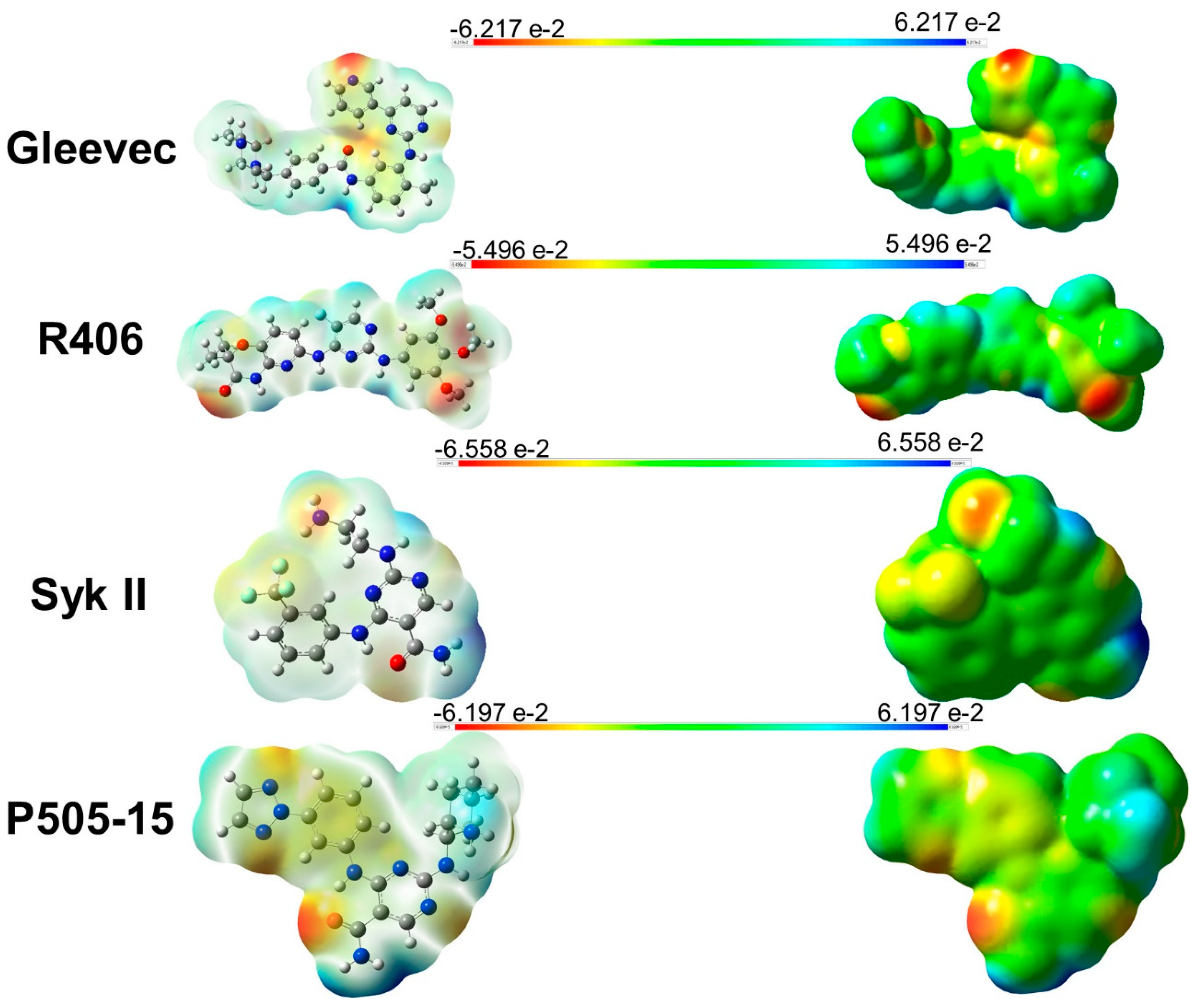
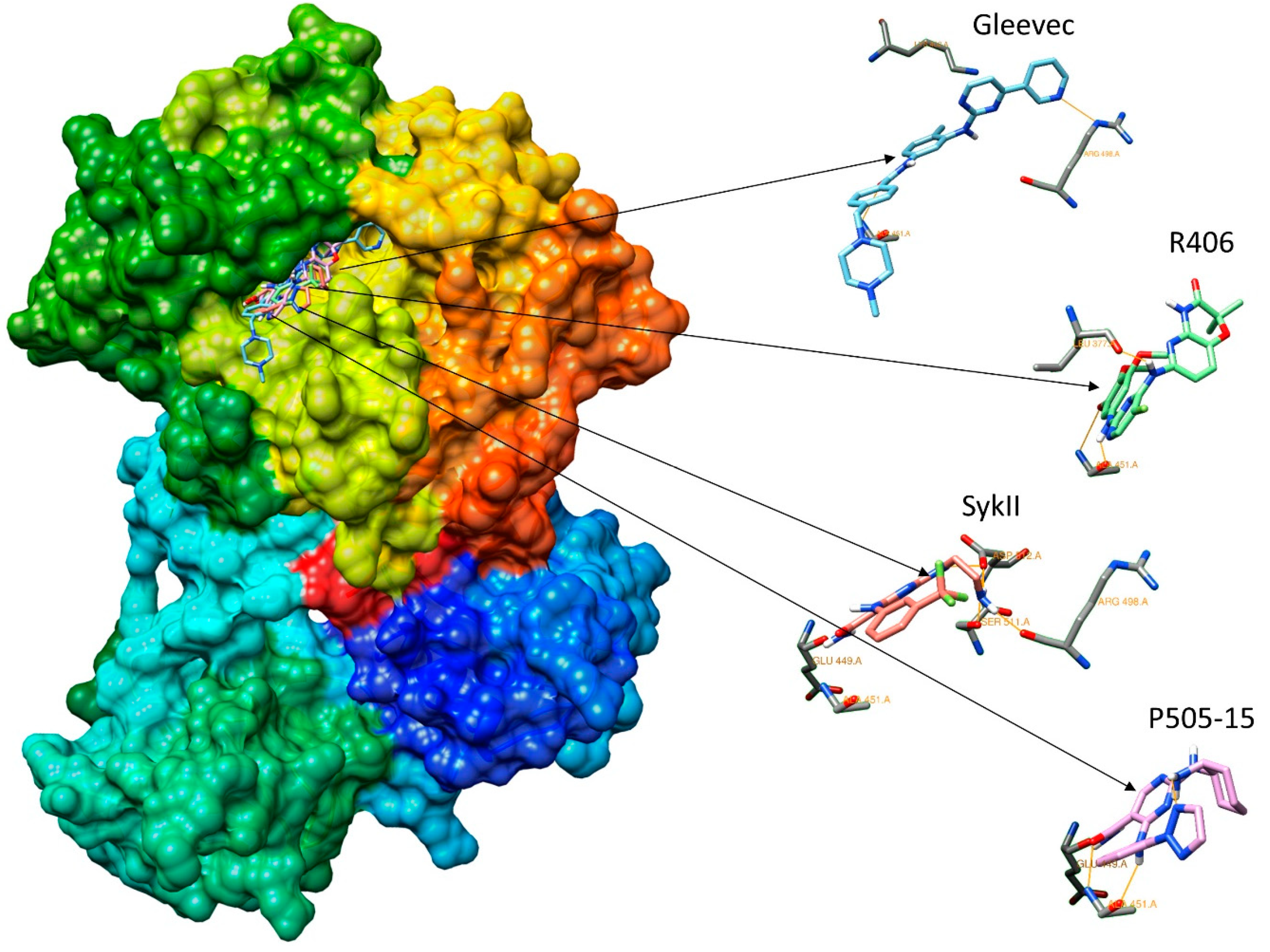
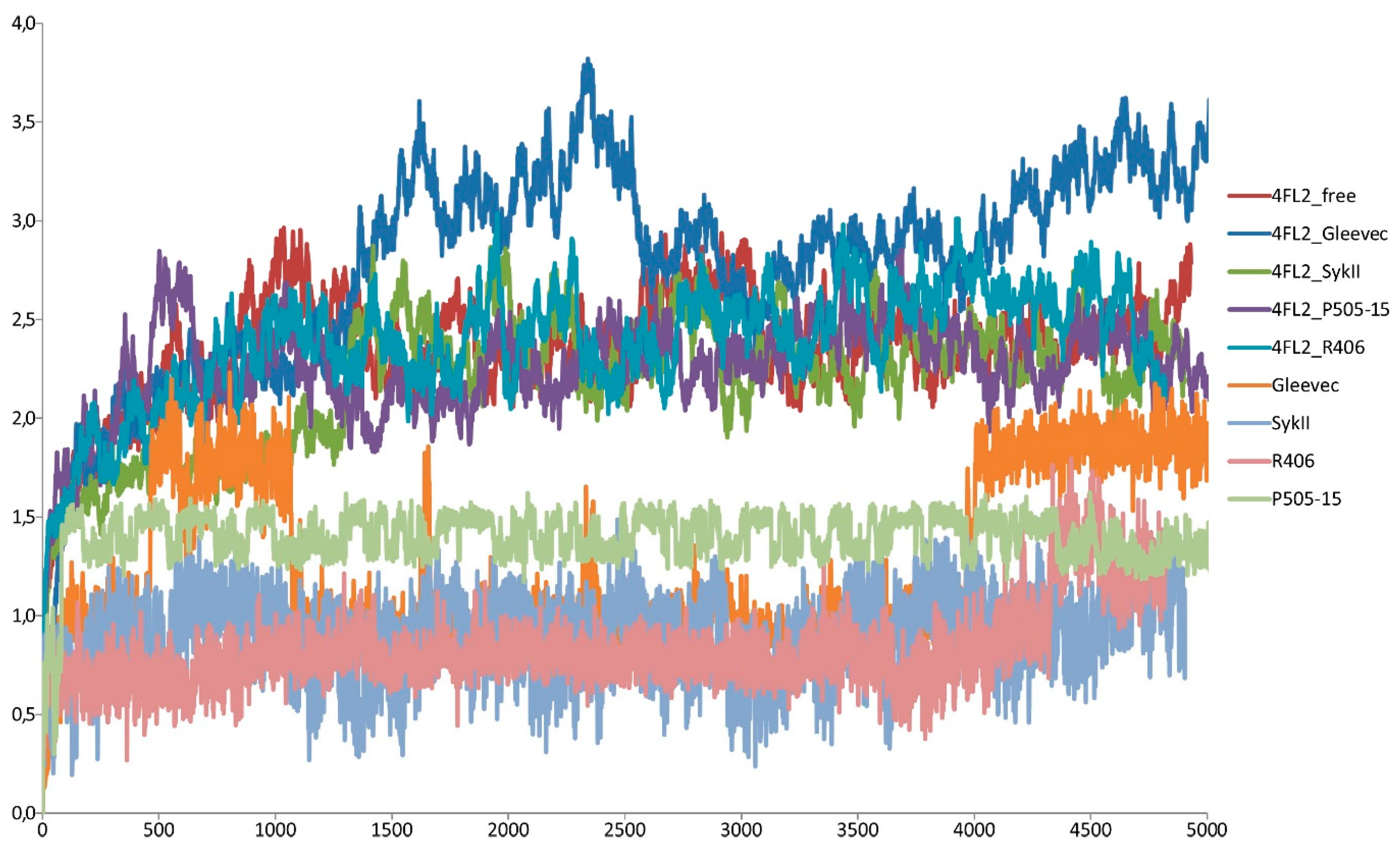
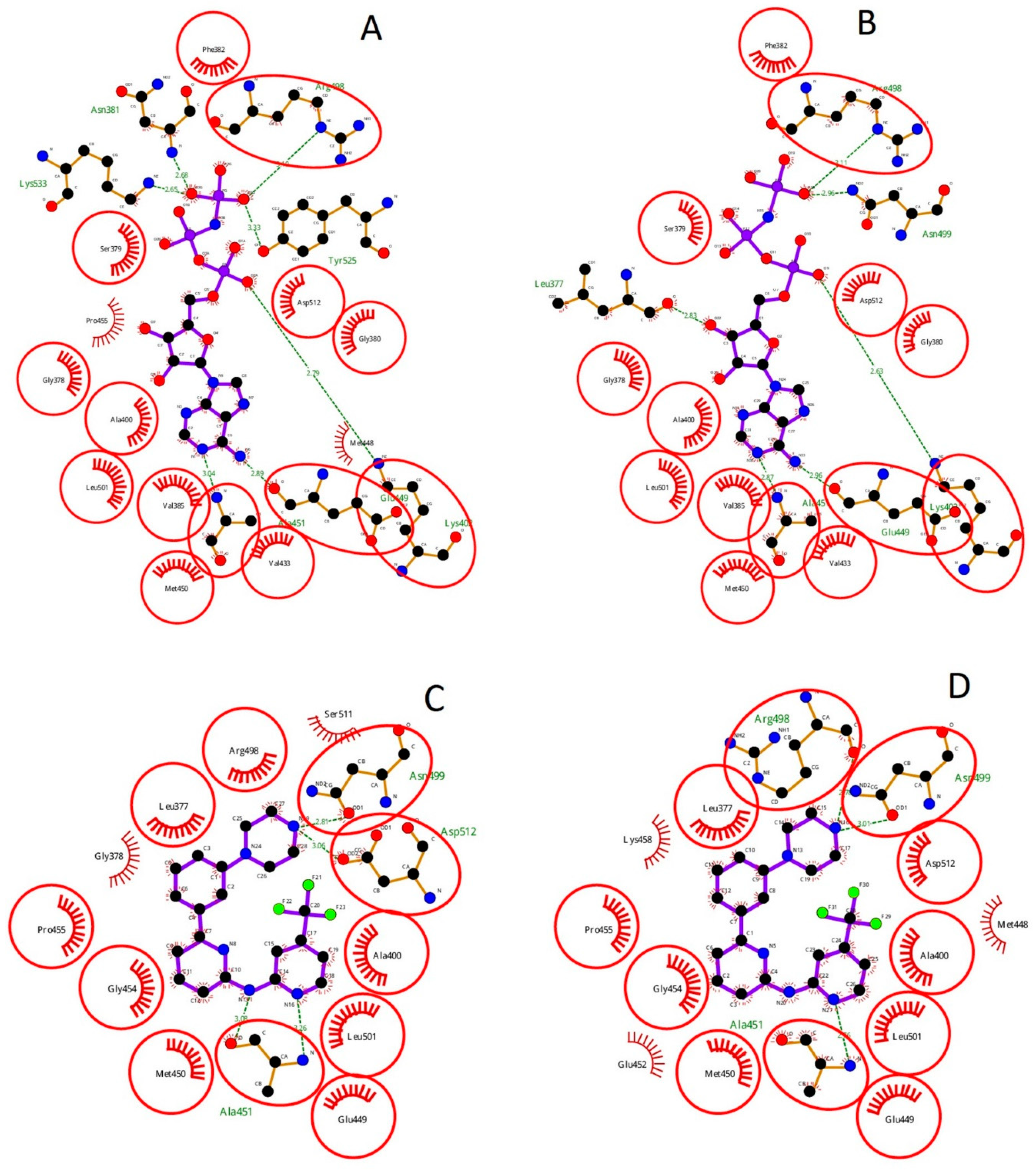
2.2. Assessment of Syk Inhibitor Efficacy through Computational Studies
3. Materials and Methods
3.1. In Vitro Experiments
3.2. Treatment of Red Blood Cells
3.3. RBC Membrane Preparation
3.4. SDS-PAGE
3.5. Western Blot Analysis
3.6. Molecular Mechanics (MM) and Quantum Chemicals (QC)
3.7. Molecular Electrostatic Potential (MEP)
3.8. Molecular Docking
3.9. Molecular Dynamics (MD)
3.10. Validation of Molecular Docking Protocol
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTs | Artemisinin-based combination therapies |
| Syk | Spleen tyrosine kinase |
| ANP | Phosphoaminophosphonic acid adenylate ester |
| OSB | N-{6-[3-(piperazin-1-yl)phenyl]pyridin-2-yl}-4-(trifluoromethyl)pyridin-2-amine |
| MD | Molecular dynamics |
| QM | Quantum mechanics |
| QC | Quantum chemistry |
| RMSD | Root mean square deviation |
| RBCs | Red blood cells |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| MEP | Molecular electrostatic potential |
References
- Berton, G.; Mocsai, A.; Lowell, C.A. Src and Syk kinases: Key regulators of phagocytic cell activation. Trends Immunol. 2005, 26, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Sada, K.; Takano, T.; Yanagi, S.; Yamamura, H. Structure and function of Syk protein-tyrosine kinase. J. Biochem. 2001, 130, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Siraganian, R.P.; Zhang, J.; Suzuki, K.; Sada, K. Protein tyrosine kinase Syk in mast cell signaling. Mol. Immunol. 2002, 38, 1229–1233. [Google Scholar] [CrossRef]
- Singh, R.; Masuda, E.S.; Payan, D.G. Discovery and development of spleen tyrosine kinase (SYK) inhibitors. J. Med. Chem. 2012, 55, 3614–3643. [Google Scholar] [CrossRef]
- Chan, A.C.; Shaw, A.S. Regulation of. antigen receptor signal transduction by protein tyrosine kinases. Curr. Opin. Immunol. 1995, 8, 394–401. [Google Scholar] [CrossRef]
- Van Oers, N.S.; Weiss, A. The Syk/ZAP-70 protein tyrosine kinase connection to antigen receptor signalling processes. Semin. Immunol. 1995, 7, 227–236. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Müller, S.; Knapp, S. SH2 domains: Modulators of nonreceptor tyrosine kinase activity. Curr. Opin. Struct. Biol. 2009, 19, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Bond, P.J.; Faraldo-Gómez, J.D. Molecular mechanism of selective recruitment of Syk kinases by the membrane antigen-receptor complex. J. Biol. Chem. 2011, 286, 25872–25881. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.; Aulabaugh, A.; Rajamohan, F.; Liu, S.; Kaila, N.; Wan, Z.K.; Ryan, M.; Magyar, R.; Qiu, X. Biophysical and mechanistic insights into novel allosteric inhibitor of spleen tyrosine kinase. J. Biol. Chem. 2012, 10, 7717–7727. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, A.; Ferru, E.; Carta, F.; Mannu, F.; Simula, L.F.; Khadjavi, A.; Turrini, F. Irreversible AE1 Tyrosine Phosphorylation Leads to Membrane Vesiculation in G6PD Deficient Red Cells. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, A.; Ferru, E.; Giribaldi, G.; Mannu, F.; Carta, F.; Matte, A.; De Franceschi, L.; Turrini, F. Oxidized and poorly glycosylated band 3 is selectively phosphorylated by Syk kinase to form large membrane clusters in normal and G6PD-deficient red blood cells. Biochem. J. 2009, 2, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, A.; Ferru, E.; Carta, F.; Valente, E.; Pippia, P.; Turrini, F. Effect of heterozygous beta thalassemia on the phosphorylative response to Plasmodium falciparum infection. J. Proteom. 2012, 76, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Ferru, E.; Giger, K.; Pantaleo, A.; Campanella, E.; Grey, J.; Ritchie, K.; Vono, R.; Turrini, F.; Low, P.S. Regulation of membrane-cytoskeletal interactions by tyrosine phosphorylation of erythrocyte band 3. Blood 2011, 22, 5998–6006. [Google Scholar] [CrossRef] [Green Version]
- Ferru, E.; Pantaleo, A.; Carta, F.; Mannu, F.; Khadjavi, A.; Gallo, V.; Turrini, F. Thalassemic erythrocytes release microparticles loaded with hemichromes by redox activation of p72Syk kinase. Haematologica 2014, 99, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Kesely, K.R.; Pantaleo, A.; Turrini, F.M.; Olupot-olupot, P.; Low, P.S. Inhibition of an Erythrocyte Tyrosine Kinase with Imatinib Prevents Plasmodium falciparum Egress and Terminates Parasitemia. PLoS ONE 2016, 11, e0164895. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, A.; Kesely, K.R.; Pau, M.C.; Tsamesidis, I.; Schwarzer, E.; Skorokhod, O.A.; Chien, H.D.; Ponzi, M.; Bertuccini, L.; Low, P.S.; et al. Syk inhibitors interfere with erythrocyte membrane modification during P falciparum growth and suppress parasite egress. Blood 2017, 130, 1031–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsamesidis, I.; Reybier, K.; Marchetti, G.; Pau, M.C.; Virdis, P.; Fozza, C.; Nepveu, F.; Low, P.S.; Turrini, F.M.; Pantaleo, A. Syk kinase inhibitors synergize with artemisinins by enhancing oxidative stress in Plasmodium falciparum-parasitized erythrocytes. Antioxidants 2020, 9, 753. [Google Scholar] [CrossRef]
- Sechi, M.; Derudas, M.; Dallocchio, R.; Dessì, A.; Bacchi, A.; Sannia, L.; Carta, F.; Palomba, M.; Ragab, O.; Chan, C.; et al. Design and Synthesis of Novel Indole -Diketo Acid Derivatives as HIV-1 Integrase Inhibitors. J. Med. Chem. 2004, 47, 5298–5310. [Google Scholar] [CrossRef]
- Pani, G.; Dessì, A.; Dallocchio, R.; Scherm, B.; Azara, E.; Delogu, G.; Migheli, Q. Natural Phenolic Inhibitors of Trichothecene Biosynthesis by the Wheat Fungal Pathogen Fusarium culmorum: A Computational Insight into the Structure-Activity Relationship. PLoS ONE 2016, 11, e0157316. [Google Scholar] [CrossRef]
- Marangon, E.; Citterio, M.; Sala, F.; Barisone, E.; Lippi, A.A.; Rizzari, C.; Biondi, A.; D’Incalci, M.; Zucchetti, M. Pharmacokinetic profile of imatinib mesylate and N-desmethyl-imatinib (CGP 74588) in children with newly diagnosed Ph+ acute leukemias. Cancer Chemother. Pharmacol. 2009, 3, 563–566. [Google Scholar] [CrossRef]
- Champagne, M.A.; Capdeville, R.; Krailo, M.; Qu, W.; Peng, B.; Rosamilia, M.; Therrien, M.; Zoellner, U.; Blaney, S.M.; Bernstein, M. Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: Results from a Children’s Oncology Group phase 1 study. Blood 2004, 9, 2655–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annesley, C.E.; Brown, P. Novel agents for the treatment of childhood acute leukemia. Ther. Adv. Hematol. 2015, 6, 61–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinblatt, M.E.; Genovese, M.C.; Ho, M.; Rosiak-Jedrychowicz, S.H.K.; Kavanaugh, A.; Millson, D.S.; Leon, G.; van der Heijde, D. Effects of Fostamatinib, an Oral Spleen Tyrosine Kinase Inhibitor, in Rheumatoid Arthritis Patients with an Inadequate Response to Methotrexate: Results from a Phase III, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study. Arthritis Rheumatol. 2014, 66, 3255–3264. [Google Scholar] [CrossRef] [PubMed]
- Podolanczuk, A.; Lazarus, A.H.; Crow, A.R.; Grossbard, E.; Bussel, J.B. Of mice and men: An open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood 2009, 113, 3154–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauzin, S.; Ding, H.; Burdevet, D.; Borisch, B.; Hoessli, D.C. Membrane-associated signaling in human B-lymphoma lines. Exp. Cell. Res. 2011, 2, 151–162. [Google Scholar] [CrossRef]
- Friedberg, J.W.; Sharman, J.; Sweetenham, J.; Johnston, P.B.; Vose, J.M.; LaCasce, A.; Schaefer-Cutillo, J.; De Vos, S.; Sinha, R.; Leonard, J.P.; et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin Lymphoma and chronic lymphocytic leukemia. Blood 2010, 115, 2578–2585. [Google Scholar] [CrossRef]
- Coffey, G.; DeGuzman, F.; Inagaki, M.; Pak, Y.; MDelaney, S.; Ives, D.; Betz, A.; Jia, Z.J.; Pandey, A.; Baker, D.; et al. Specific Inhibition of Spleen Tyrosine Kinase Suppresses Leukocyte Immune Function and Inflammation in Animal Models of Rheumatoid Arthritis. J. Pharmacol. Exp. Ther. 2011, 340, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Spurgeon, S.E.; Coffey, G.; Fletcher, L.B.; Burke, R.; Tyner, J.W.; Druker, B.J.; Loriaux, M.M. The Selective Syk Inhibitor P505-15 (PRT062607) Inhibits B Cell Signaling and Function In Vitro and In Vivo and Augments the Activity of Fludarabine in Chronic Lymphocytic Leukemia. J. Pharmacol. Exp. Ther. 2013, 344, 378–387. [Google Scholar] [CrossRef] [Green Version]
- Hisamichi, H.; Naito, R.; Toyoshima, A.; Kawano, N.; Ichikawa, A.; Orita, A.; Tsukamoto, S. Synthetic studies on novel Syk inhibitors. Part 1: Synthesis and structure-activity relationships of pyrimidine-5-carboxamide derivatives. Bioorg. Med. Chem. 2005, 13, 4936–4951. [Google Scholar] [CrossRef]
- Yi, Y.; Son, Y.; Ryou, C.; Sung, G.; Kim, J.; Cho, J.Y. Functional Roles of Syk in Macrophage-Mediated Inflammatory Responses. Mediat. Inflamm. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bordin, L.; Brunati, A.M.; Donella-Deana, A.; Baggio, B.; Toninello, A.; Clari, G. Band 3 is an anchor protein and a target for SHP-2 tyrosine phosphatase in human erythrocytes. Blood 2002, 100, 276–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, M.L.; Isaacson, C.C.; Burg, D.L.; Geahlen, R.L.; Low, P.S. Phosphorylation of human erythrocyte band 3 by endogenous p72syk. J. Biol. Chem. 1994, 269, 955–959. [Google Scholar] [PubMed]
- Bordin, L.; Zen, F.; Ion-Popa, F.; Barbetta, M.; Baggio, B.; Clari, G. Band 3 tyr-phosphorylation in normal and glucose-6-phospate dehydrogenase-deficient human erythrocytes. Mol. Membr. Biol. 2005, 5, 411–420. [Google Scholar] [CrossRef]
- Villaseñor, A.G.; Kondru, R.; Hoangdung, H.; Wang, S.; Papp, E.; Shaw, D.; Barnett, J.W.; Browner, M.F.; Kuglstatter, A. Structural Insights for Design of Potent Spleen Tyrosine Kinase Inhibitors from Crystallographic Analysis of Three Inhibitor Complexes. Chem. Biol. Drug Des. 2009, 73, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M.; Forns, P.; Erra, M.; Lopez, M.; Maldonado, M.; Orellana, A.; Carreno, C.; Ramis, I.; Miralpeix, M.; Vidal, B. Highly potent aminopyridines as Syk kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5419–5423. [Google Scholar] [CrossRef] [PubMed]
- Al-Sabagh, A.M.; Nasser, N.M.; Farag, A.A.; Migahed, M.A.; Eissa AM, F.; Mahmoud, T. Structure effect of some amine derivatives on corrosion inhibition efficiency for carbon steel in acidic media using electrochemical and Quantum Theory Methods. Egypt. J. Pet. 2013, 22, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Traxler, P.; Furet, P. Strategies toward the Design of Novel and Selective Protein Tyrosine Kinase Inhibitors. Pharmacol. Ther. 1999, 82, 195–206. [Google Scholar] [CrossRef]
- Thoma, G.; Smith, A.B.; Eis, M.J.; Van Vangrevelinghe, E.; Blanz, J.; Aichholz, R.; Zerwes, H. Discovery and Profiling of a Selective and Efficacious Syk Inhibitor. J. Med. Chem. 2015, 58, 1950–1963. [Google Scholar] [CrossRef]
- Miah SM, S.; Sada, K.; Tuazon, P.T.; Ling, J.; Maeno, K.; Kyo, S.; Yamamura, H. Activation of Syk Protein Tyrosine Kinase in Response to Osmotic Stress Requires Interaction with p21-Activated Protein Kinase Pak2/γ-PAK. Mol. Cell. Biol. 2004, 24, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Grädler, U.; Schwarz, D.; Dresing, V.; Musil, D.; Bomke, J.; Frech, M.; Wegener, A. Structural and biophysical characterization of the Syk activation switch. J. Mol. Biol. 2013, 425, 309–333. [Google Scholar] [CrossRef]
- Zeifman, A.A.; Titov, Y.; Svitanko, I.V.; Rakitina, T.V.; Lipkin, A.V.; Stroylov, V.S.; Chilov, G.G. Rational design and synthesis of novel Syk-kinase inhibitors. Mendeleev Commun. 2012, 22, 73–74. [Google Scholar] [CrossRef]
- Padilla, F.; Bhagirath, N.; Chen, S.; Chiao, E.; Goldstein, D.M.; Hermann, J.C.; Lucas, M.C. Pyrrolopyrazines as selective spleen tyrosine kinase inhibitors. J. Med. Chem. 2013, 56, 1677–1692. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, R.; Chen, Y.; Zheng, Q.; Fan, S.; Liu, P. A combined experimental and computational study of Vam3, a derivative of resveratrol, and syk interaction. Int. J. Mol. Sci. 2014, 15, 17188–17203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef] [PubMed]
- De Lucia, S.; Tsamesidis, I.; Pau, M.C.; Kesely, K.R.; Pantaleo, A.; Turrini, F. Induction of high tolerance to artemisinin by sub-lethal administration: A new in vitro model of P. falciparum. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fandeur, T.; Bonnefoy, S.; Mercereau-Puijalon, O. In vivo and in vitro derived Palo Alto lines of Plasmodium falciparum are genetically unrelated. Mol. Biochem. Parasitol. 1991, 47, 167–178. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef]
- Rivadeneira, E.M.; Wasserman, M.; Espinal, C.T. Separation and concentration of schizonts of Plasmodium falciparum by Percoll gradients. J. Protozool. 1983, 30, 367–370. [Google Scholar] [CrossRef]
- Le Nagard, H.; Vincent, C.; Mentré, F.; Le Bras, J. Online analysis of in vitro resistance to antimalarial drugs through nonlinear regression. Comput. Methods Programs Biomed. 2010, 104, 10–18. [Google Scholar] [CrossRef]
- Kaddouri, H.; Nakache, S.; Houzé, S.; Mentré, F.; Le Bras, J. Drug Susceptibility of Plasmodium falciparum Clinical Isolates from Africa using Plasmodium Lactate Dehydrogenase Immunodetection Assay and inhibitory Emax model for precise IC50 measurement. Antimicrob. Agents Chemother. 2006, 50, 3343–3349. [Google Scholar] [CrossRef] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel-an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Chang, G.; Guida, W.C.; Still, W.C. An internal-coordinate Monte Carlo method for searching conformational space. J. Am. Chem. Soc. 1989, 111, 4379–4386. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. Gauss View, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. 1999, 17, 57–61. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Kaus, J.W.; Pierce, L.T.; Walker, R.C.; McCammon, J.A. Improving the Efficiency of Free Energy Calculations in the Amber Molecular Dynamics Package. J. Chem. Theory Comput. 2013, 9, 4131–4139. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Dessì, A.; Peluso, P.; Dallocchio, R.; Weiss, R.; Andreotti, G.; Allocca, M.; Aubert, E.; Pale, P.; Mamane, V.; Cossu, S. Rational design, synthesis, characterization and evaluation of iodinated 4,4’-bipyridines as new transthyretin fibrillogenesis inhibitors. Molecules 2020, 25, 2213. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. Docking screens for novel ligands conferring new biology. J. Med. Chem. 2016, 59, 4103–4120. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Syk Inhibitors | IC50 (μΜ) |
|---|---|
| P505-15 | 0.64 |
| Gleevec | 0.77 |
| R406 | 0.83 |
| Syk II | 1.72 |
| Palo Alto Strain | 24 h | 48 h | ||
|---|---|---|---|---|
| IC50 (μΜ) | Range (μΜ) | IC50 (μΜ) | Range (μΜ) | |
| P505-15 | 0.83±0.06 | 0.78−0.90 | 0.49±0.07 | 0.42−0.61 |
| R406 | 2.62±0.83 | 1.42−3.85 | 0.55±0.19 | 0.25−0.92 |
| Gleevec | 3.81±0.55 | 3.24−4.95 | 1.55±0.13 | 1.32−1.74 |
| SYK II | 5.01±0.44 | 4.65−5.87 | 0.90±0.16 | 0.82−1.25 |
| H-Bond Interaction | ||||||||
|---|---|---|---|---|---|---|---|---|
| Ligands | Pose | % | H-Bond | Ligands Atom | Protein Atom | Distance (Å) | LogP | D. M. (Debye) |
| Gleevec | 1 | 73 | 4 | N33(NA) | Lys402:HZ1(HD) | 2.217 | 3.83 | 5.1101 |
| O10(OA) | Ala451:HN(HD) | 2.393 | ||||||
| N38(NA) | Arg498:HE(HD) | 2.147 | ||||||
| H27(HD) | Asp512:OD2(OA) | 1.995 | ||||||
| R406 | 11 | 16 | 3 | H9(HD) | Leu377:O(OA) | 1.875 | 3.37 | 3.4870 |
| H25(HD) | Ala451:O(OA) | 2.262 | ||||||
| O36(OA) | Ala451:HN(HD) | 2.317 | ||||||
| SykII | 1 | 56 | 7 | H23(HD) | Glu449:O(OA) | 2.075 | 1.22 | 4.4175 |
| H24(HD) | Glu449:O(OA) | 2.233 | ||||||
| O25(OA) | Ala451:HN(HD) | 2.299 | ||||||
| H29(HD) | Arg498:O(OA) | 2.063 | ||||||
| H30(HD) | Ser511:HG(HD) | 2.558 | ||||||
| H8(HD) | Asp512:OD2(OA) | 2.191 | ||||||
| H30(HD) | Asp512:OD2(OA) | 2.025 | ||||||
| P505-15 | 8 | 37 | 4 | H8(HD) | Ala451:O(OA) | 2.301 | 1.02 | 2.4285 |
| O21(OA) | Ala451:HN(HD) | 1.888 | ||||||
| H23(HD) | Glu449:O(OA) | 1.940 | ||||||
| H24(HD) | Glu449:O(OA) | 2.448 | ||||||
| R406 | Syk II | Gleevec | P505-15 |
|---|---|---|---|
| Leu377 * | Leu377 | Arg338 | Leu377 |
| Gly378 | Gly378 | Leu377 | Val385 |
| Ser379 | Val385 | Ser379 | Ala400 |
| Gly380 | Ala400 | Gly380 | Met448 |
| Val385 | Lys402 | Asn381 | Glu449 ** |
| Ala400 | Met448 | Phe382 | Met450 |
| Lys402 | Glu449 ** | Val385 | Ala451 ** |
| Val433 | Met450 | Ala400 | Glu452 |
| Met448 | Ala451 * | Lys402 * | Gly454 |
| Glu449 | Gly454 | Met448 | Pro455 |
| Met450 | Pro455 | Met450 | Lys458 |
| Ala451 ** | Lys458 | Ala451 * | Leu501 |
| Glu452 | Arg498 * | Leu453 | |
| Gly454 | Leu501 | Gly454 | |
| Pro455 | Ser511 * | Arg498 * | |
| Lys458 | Asp512 ** | Asn499 | |
| Leu501 | Leu501 | ||
| Asp512 | Ser511 | ||
| Asp512 * | |||
| Tyr525 | |||
| His531 |
| Ligand | M.B.E | E.F.E.B | E.I.C, Ki |
|---|---|---|---|
| Gleevec | −10.57 | −10.73 | 13.65 nM |
| P505-15 | −7.69 | −8.81 | 346.88 nM |
| R406 | −7.28 | −7.94 | 1.50 μM |
| SykII | −6.80 | −6.95 | 8.03 μM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchetti, G.; Dessì, A.; Dallocchio, R.; Tsamesidis, I.; Pau, M.C.; Turrini, F.M.; Pantaleo, A. Syk Inhibitors: New Computational Insights into Their Intraerythrocytic Action in Plasmodium falciparum Malaria. Int. J. Mol. Sci. 2020, 21, 7009. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197009
Marchetti G, Dessì A, Dallocchio R, Tsamesidis I, Pau MC, Turrini FM, Pantaleo A. Syk Inhibitors: New Computational Insights into Their Intraerythrocytic Action in Plasmodium falciparum Malaria. International Journal of Molecular Sciences. 2020; 21(19):7009. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197009
Chicago/Turabian StyleMarchetti, Giuseppe, Alessandro Dessì, Roberto Dallocchio, Ioannis Tsamesidis, Maria Carmina Pau, Francesco Michelangelo Turrini, and Antonella Pantaleo. 2020. "Syk Inhibitors: New Computational Insights into Their Intraerythrocytic Action in Plasmodium falciparum Malaria" International Journal of Molecular Sciences 21, no. 19: 7009. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197009