Impaired Glycolysis Promotes Alcohol Exposure-Induced Apoptosis in HEI-OC1 Cells via Inhibition of EGFR Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
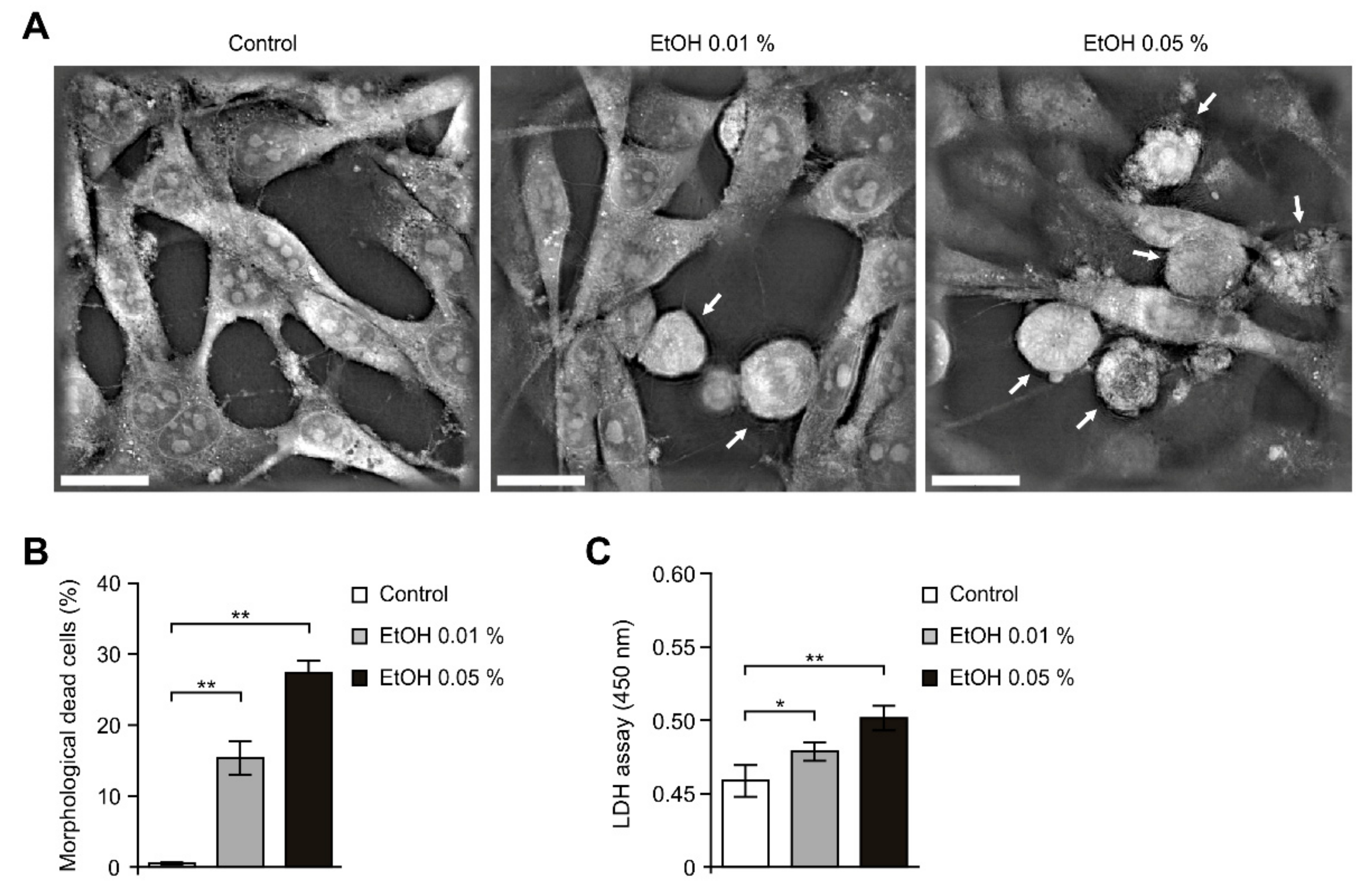
2.1. Alcohol Exposure Induced Cytotoxicity in HEI-OC1 Cells
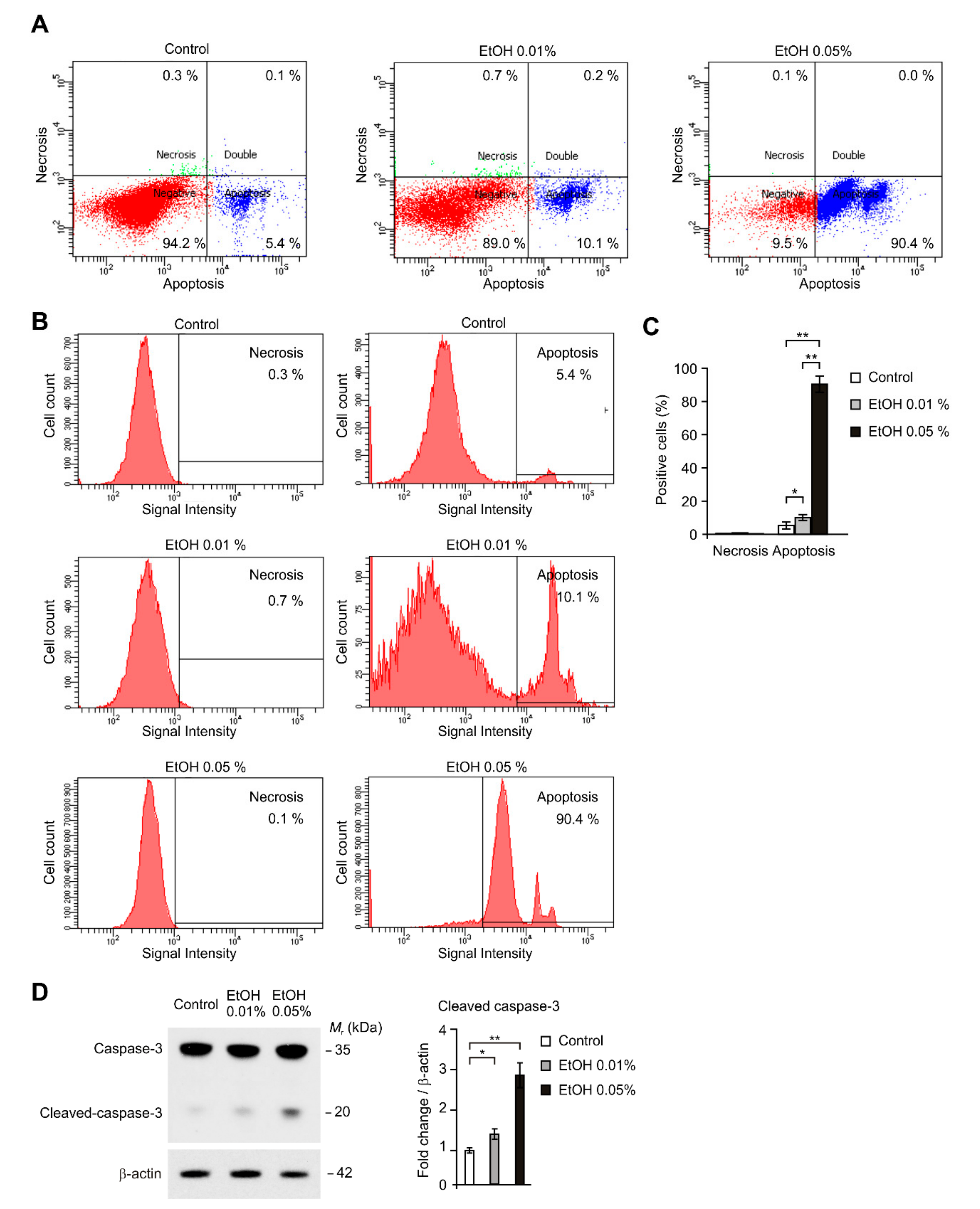
2.2. Alcohol Exposure Induced Apoptosis in HEI-OC1 Cells
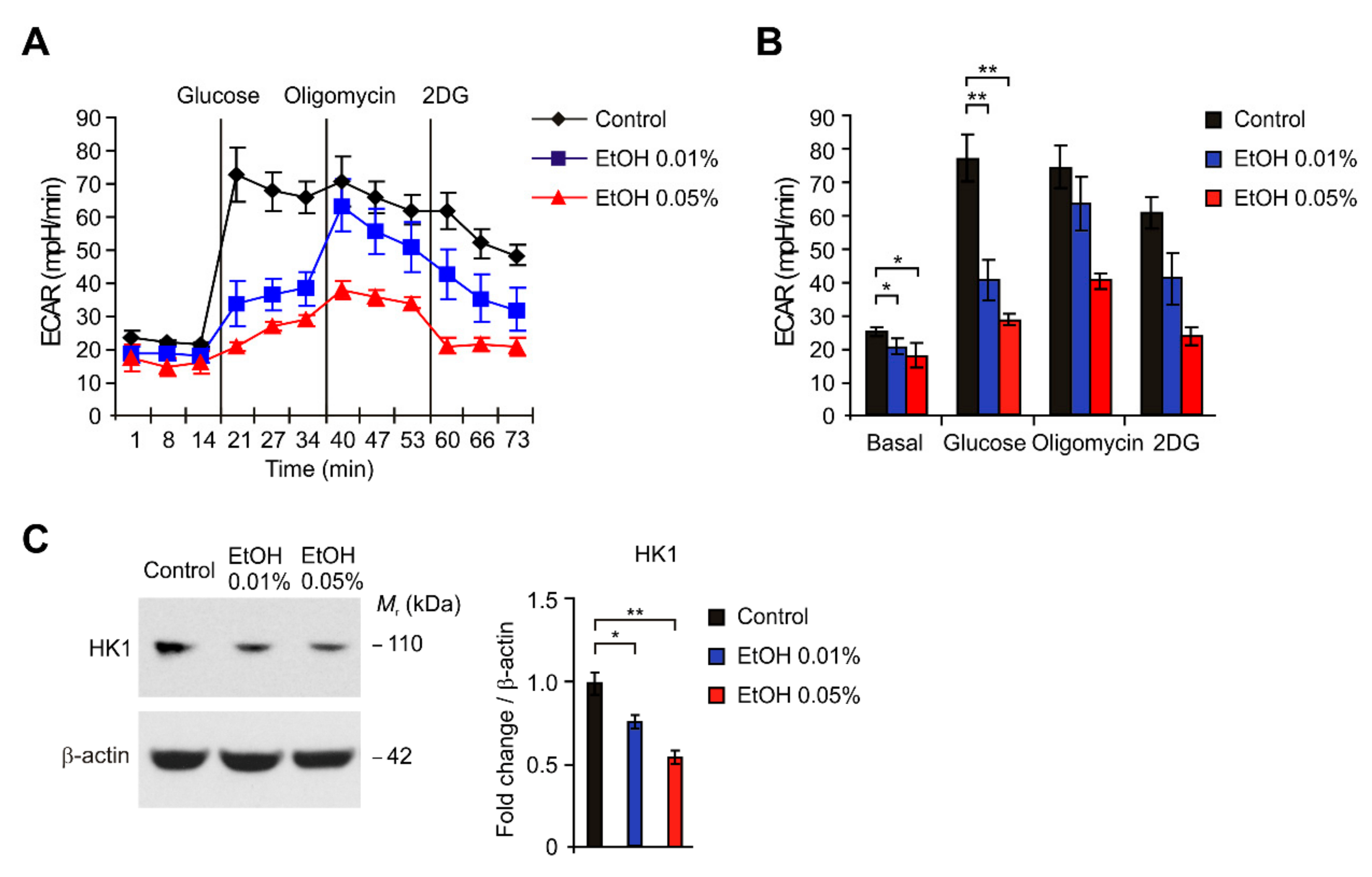
2.3. HK1-Dependent Glycolysis Is Suppressed by Alcohol Exposure in HEI-OC1 Cells
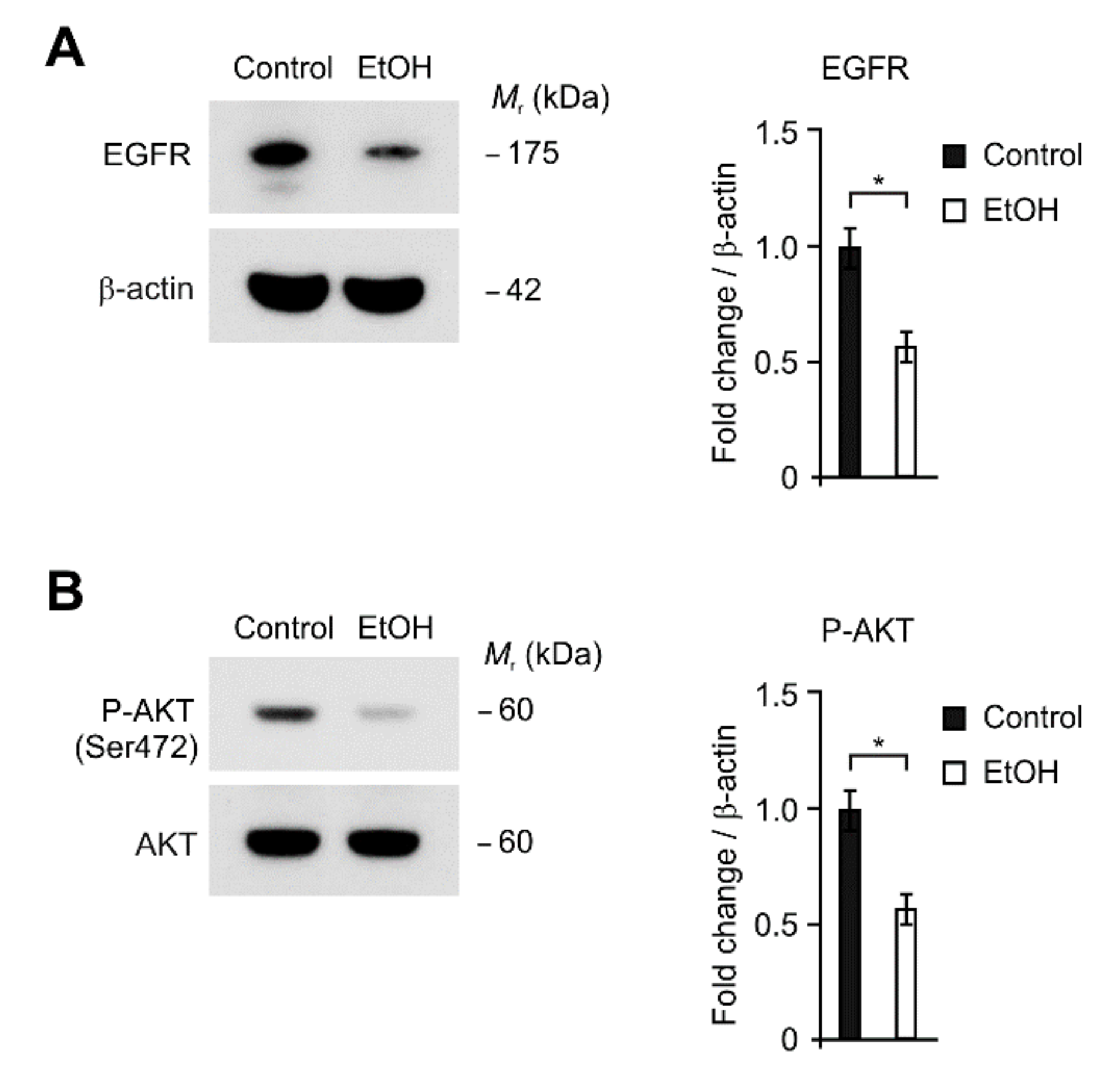
2.4. The Levels of EGFR and AKT Phosphorylation Were Reduced by Alcohol Exposure in HEI-OC1 Cells
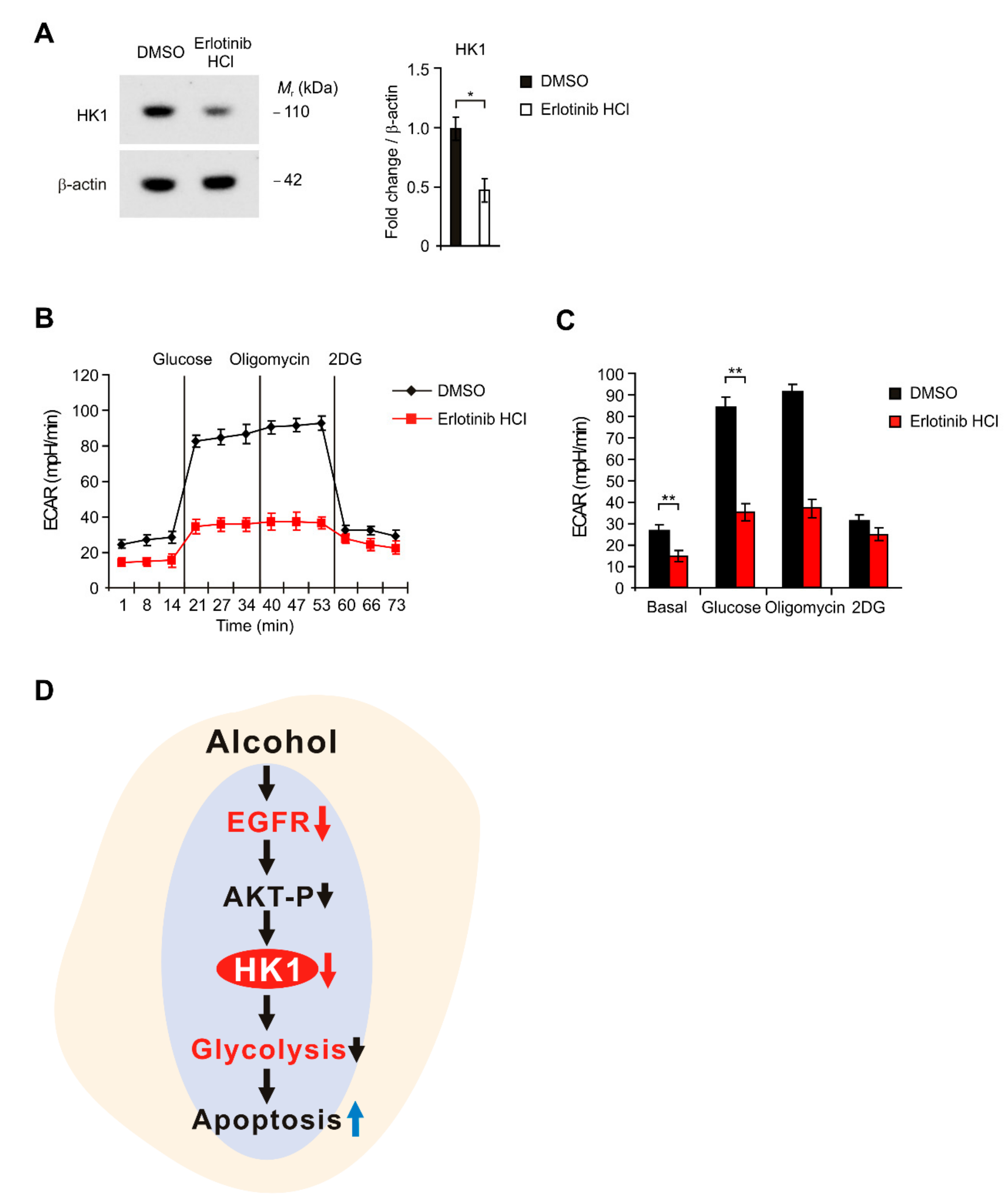
2.5. Inhibition of EGFR Suppressed HK1-Dependent Glycolysis in HEI-OC1 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. 3D Images
4.4. Cell Cytotoxicity Assay
4.5. Apoptosis/Necrosis Assay
4.6. Glycolysis Activity Assay
4.7. Immunoblot Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HK1 | hexokinase 1 |
| EGFR | epidermal growth factor receptor |
| ECAR | extracellular acidification rate |
| HEI-OC | house ear institute-organ of corti-1 |
| HEI-OC1 | phosphatidylinositol 3-kinase |
| LDH | lactate dehydrogenase |
References
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, E.; Morris, H.P.; Pedersen, P.L. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. R J. Biol. Chem. 1981, 256, 8699–8704. [Google Scholar]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Arora, K.K.; Fanciulli, M.; Pedersen, P.L. Glucose phosphorylation in tumor cells. Cloning, sequencing, and overexpression in active form of a fulllength cDNA encoding a mitochondrial bindable form of hexokinase. J. Biol. Chem. 1990, 265, 6481–6488. [Google Scholar]
- Azoulay-Zohar, H.; Aflalo, C. Binding of rat brain hexokinase to recombinant yeast mitochondria: Identification of necessary molecular determinants. J. Bioenerg. Biomembr. 1999, 31, 569–579. [Google Scholar] [CrossRef]
- Azoulay-Zohar, H.; Aflalo, C. Binding of rat brain hexokinase to recombinant yeast mitochondria: Identification of necessary physico-chemical determinants. Eur. J. Biochem. 2000, 267, 2973–2980. [Google Scholar] [CrossRef]
- Baijal, M.; Wilson, J.E. Residues putatively involved in binding of ATP and glucose 6-phosphate to a mammalian hexokinase: Site-directed mutation at analogous positions in the N- and C-terminal halves of the type I isozyme. Arch. Biochem. Biophys. 1995, 321, 413–420. [Google Scholar] [CrossRef]
- Albernaz, P.L. Hearing Loss, Dizziness, and Carbohydrate Metabolism. Int. Arch. Otorhinolaryngol. 2016, 20, 261–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, O.H.; Choi, M.G.; Park, C.H.; Kim, D.K.; Lee, J.S.; Lee, J.H. Hyperglycemia as a potential prognostic factor of idiopathic sudden sensorineural hearing loss. Otolaryngol. Head Neck Surg. 2014, 150, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, A.L.; Oesterle, E.C. Transforming growth factor alpha with insulin stimulates cell proliferation in vivo in adult rat vestibular sensory epithelium. J. Comp. Neurol. 1998, 393, 413–423. [Google Scholar] [CrossRef]
- Hume, C.R.; Kirkegaard, M.; Oesterle, E.C. ErbB expression: The mouse inner ear and maturation of the mitogenic response to heregulin. J. Assoc. Res. Otolaryngol. 2003, 4, 422–443. [Google Scholar] [CrossRef] [PubMed]
- Doetzlhofer, A.; White, P.M.; Johnson, J.E.; Segil, N.; Groves, A.K. In vitro growth and differentiation of mammalian sensory hair cell progenitors: A requirement for EGF and periotic mesenchyme. Dev. Biol. 2004, 272, 432–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, I.J.; Bailey, L.M.; Aghakhani, M.R.; Moss, S.E.; Futter, C.E. EGF stimulates annexin 1-dependent inward vesiculation in a multivesicular endosome subpopulation. EMBO J. 2006, 25, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montcouquiol, M.; Corwin, J.T. Intracellular signals that control cell proliferation in mammalian balance epithelia: Key roles for phosphatidylinositol-3 kinase, mammalian target of rapamycin, and S6 kinases in preference to calcium, protein kinase C, and mitogen-activated protein kinase. J. Neurosci. 2001, 21, 570–580. [Google Scholar]
- Wang, X.; McCullough, K.D.; Franke, T.F.; Holbrook, N.J. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J. Biol. Chem. 2000, 275, 14624–14631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humtsoe, J.O.; Kramer, R.H. Differential epidermal growth factor receptor signaling regulates anchorage-independent growth by modulation of the PI3K/AKT pathway. Oncogene 2010, 29, 1214–1226. [Google Scholar] [CrossRef] [Green Version]
- George, A.; Figueredo, V.M. Alcohol and arrhythmias: A comprehensive review. J. Cardiov. Med. 2010, 11, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef]
- Seitz, H.K.; Becker, P. Alcohol metabolism and cancer risk. Alcohol Res. Health 2007, 30, 38–47. [Google Scholar]
- Bagnardi, V.; Blanglardo, M.; La Vecchia, C.; Corrao, G. Alcohol consumption and the risk of cancer: A meta-analysis. Alcohol Res. Health 2001, 25, 263–270. [Google Scholar] [PubMed]
- Whitcomb, D.C. Genetics and alcohol: A lethal combination in pancreatic disease? Alcohol Clin. Exp. Res. 2011, 35, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.P. Genetic polymorphisms of alcohol metabolizing enzymes. Pathol. Biol. 2001, 49, 703–709. [Google Scholar] [CrossRef]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar]
- Homann, N.; Stickel, F.; König, I.R.; Jacobs, A.; Junghanns, K.; Benesova, M.; Schuppan, D.; Himsel, S.; Zuber-Jerger, I.; Hellerbrand, C.; et al. Alcohol dehydrogenase 1C*1 allele is a genetic marker for alcohol-associated cancer in heavy drinkers. Int. J. Cancer 2006, 118, 1998–2002. [Google Scholar] [CrossRef]
- Pöschl, G.; Seitz, H.K. Alcohol and cancer. Alcohol Alcohol. 2004, 39, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Hörster, F.; Tenkova, T.; et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef]
- Light, K.E.; Belcher, S.M.; Pierce, D.R. Time course and manner of purkinje neuron death following a single ethanol exposure on postnatal day 4 in the developing rat. Neuroscience 2002, 114, 327–337. [Google Scholar] [CrossRef]
- Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Nemmers, B.; Olney, J.W. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Dev. Brain Res. 2005, 155, 1–13. [Google Scholar] [CrossRef]
- Ting, J.W.; Lautt, W.W. The effect of acute, chronic, and prenatal ethanol exposure on insulin sensitivity. Pharm. Ther. 2006, 111, 346–373. [Google Scholar] [CrossRef]
- Manzo-Avalos, S.; Saavedra-Molina, A. Cellular and mitochondrial effects of alcohol consumption. Int. J. Environ. Res. Public Health 2010, 7, 4281–4304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaghef, L.; Farajdokht, F.; Erfani, M.; Majdi, A.; Sadigh-Eteghad, S.; Karimi, P.; Sandoghchian Shotorbani, S.; Seyedi Vafaee, M.; Mahmoudi, J. Cerebrolysin attenuates ethanol-induced spatial memory impairments through inhibition of hippocampal oxidative stress and apoptotic cell death in rats. Alcohol 2019, 79, 30251–30259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.A.; Suh, M.J. Hazardous Alcohol Consumption and the Risk of Hearing Impairment in Adults Based on the Korean National Health and Nutrition Survey: A Retrospective Study. J. Audiol. Otol. 2019, 23, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kalinec, G.M.; Webster, P.; Lim, D.J.; Kalinec, F. A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol. Neurootol. 2003, 8, 177–189. [Google Scholar] [CrossRef]
- Kalinec, G.M.; Park, C.; Thein, P.; Kalinec, F. Working with Auditory HEI-OC1 Cells. J. Vis. Exp. 2016, 115, e54425. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Sun, G.; Yang, Q.; Chen, C.; Qi, Q.; Wang, H.; Li, J. NLRX1 accelerates cisplatin-induced ototoxity in HEI-OC1 cells via promoting generation of ROS and activation of JNK signaling pathway. Sci. Rep. 2017, 7, 44311. [Google Scholar] [CrossRef] [Green Version]
- Butts, M.; Singh Paulraj, R.; Haynes, J.; Arthur, S.; Singh, S.; Sundaram, U. Moderate Alcohol Consumption Inhibits Sodium-Dependent Glutamine Co-Transport in Rat Intestinal Epithelial Cells in Vitro and Ex Vivo. Nutrients 2019, 11, 2516. [Google Scholar] [CrossRef] [Green Version]
- TeSlaa, T.; Teitell, M.A. Techniques to monitor glycolysis. Methods Enzymol. 2014, 542, 91–114. [Google Scholar]
- Moon, J.S.; Hisata, S.; Park, M.A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M.K. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Makinoshima, H.; Takita, M.; Matsumoto, S.; Yagishita, A.; Owada, S.; Esumi, H.; Tsuchihara, K. Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. J. Biol. Chem. 2014, 282, 20813–20823. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Nam, B.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Kim, W.S.; Choi, C.M.; Chang, E.J.; Koh, J.S.; et al. Enhanced Glycolysis Supports Cell Survival in EGFR-Mutant Lung Adenocarcinoma by Inhibiting Autophagy-Mediated EGFR Degradation. Cancer Res. 2018, 78, 4482–4496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felício, J.S.; de Souza d’Albuquerque Silva, L.; Martins, C.L.E.L.P.; Neto, J.F.A.; de Lemos, M.N.; de Souza Resende, F.; da Silva, W.M.; de Alcântara, A.L.; de Oliveira, M.C.N.I.; de Souza Neto, N.J.K.; et al. Cochlear dysfunction and microvascular complications in patients with type 1 diabetes mellitus. Diabetol. Metab. Syndr. 2018, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, C.; Kodama, S.; Tanaka, S.; Fujihara, K.; Hirasawa, R.; Yachi, Y.; Shimano, H.; Yamada, N.; Saito, K.; Sone, H. Diabetes and risk of hearing impairment in adults: A meta-analysis. J. Clin. Endocrinol. Metab. 2013, 98, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bainbridge, K.E.; Hoffman, H.J.; Cowie, C.C. Diabetes and hearing impairment in the united states: Audiometric evidence from the national health and nutrition examination survey, 1999 to 2004. Ann. Intern. Med. 2008, 149, 1–10. [Google Scholar] [CrossRef]
- Sugimoto, S.; Teranishi, M.; Fukunaga, Y.; Yoshida, T.; Sugiura, S.; Uchida, Y.; Oiso, Y.; Nakashima, T. Contributing factors to hearing of diabetic patients in an in-hospital education program. Acta Otolaryngol. 2013, 133, 1165–1172. [Google Scholar] [CrossRef]
- Lisowska, G.; Namysowski, G.; Morawski, K.; Strojek, K. Early identification of hearing impairment in patients with type 1 diabetes mellitus. Otol. Neurotol. 2001, 22, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Wilunda, C.; Kimura, T.; Takeuchi, M.; Kawakami, K. Prenatal Alcohol Exposure and Suspected Hearing Impairment Among Children: A Population-based Retrospective Cohort Study. Alcohol Alcohol. 2018, 53, 221–227. [Google Scholar] [CrossRef]
- Kahkonen, S.; Marttinen Rossi, E.; Yamashita, H. Alcohol impairs auditory processing of frequency changes and novel sounds: A combined MEG and EEG study. Psychopharmacology 2005, 177, 366–372. [Google Scholar] [CrossRef]
- Upile, T.; Sipaul, F.; Jerjes, W.; Singh, S.; Nouraei, S.A.; El Maaytah, M.; Andrews, P.; Graham, J.; Hopper, C.; Wright, A. The acute effects of alcohol on auditory thresholds. BMC Ear Nose Throat Disord. 2007, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Rosenhall, U.; Sixt, E.; Sundh, V.; Svanborg, A. Correlations between presbyacusis and extrinsic noxious factors. Audiology 1993, 32, 234–243. [Google Scholar] [CrossRef]
- Ribeiro, S.B.; Jacob, L.C.; de Alvarenga, K.F.; Marques, J.M.; Campelo, R.M.; Tschoeke, S.N. Auditory assessment of alcoholics in abstinence. Braz. J. Otorhinolaryngol. 2007, 73, 452–462. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; Chawla, K.; Umoh, N.A.; Cousins, V.M.; Ketegou, A.; Reddy, M.G.; AlRubaiee, M.; Haddad, G.E.; Burke, M.W. Alcohol and Apoptosis: Friends or Foes? Biomolecules 2015, 5, 3193–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menk, M.; Graw, J.A.; Poyraz, D.; Möbius, N.; Spies, C.D.; von Haefen, C. Chronic Alcohol Consumption Inhibits Autophagy and Promotes Apoptosis in the Liver. Int. J. Med Sci. 2018, 15, 682–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, J.B.; Pastorino, J.G. Cellular signaling mechanisms in alcohol-induced liver damage. Semin. Liver Dis. 2004, 24, 257–272. [Google Scholar] [CrossRef] [PubMed]
- He, J.; de la Monte, S.; Wands, J.R. Acute ethanol exposure inhibits insulin signaling in the liver. Hepatology 2007, 46, 1791–1800. [Google Scholar] [CrossRef]
- Mason, E.F.; Rathmell, J.C. Cell metabolism: An essential link between cell growth and apoptosis. Biochim. Biophys. Acta. 2011, 1813, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Marte, B.M.; Downward, J. PKB/Akt: Connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem. Sci. 1997, 22, 355–358. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.; Choi, S.J.; Park, K.H.; Lee, C.-K.; Moon, J.-S. Impaired Glycolysis Promotes Alcohol Exposure-Induced Apoptosis in HEI-OC1 Cells via Inhibition of EGFR Signaling. Int. J. Mol. Sci. 2020, 21, 476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020476
Kang H, Choi SJ, Park KH, Lee C-K, Moon J-S. Impaired Glycolysis Promotes Alcohol Exposure-Induced Apoptosis in HEI-OC1 Cells via Inhibition of EGFR Signaling. International Journal of Molecular Sciences. 2020; 21(2):476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020476
Chicago/Turabian StyleKang, Hyunsook, Seong Jun Choi, Kye Hoon Park, Chi-Kyou Lee, and Jong-Seok Moon. 2020. "Impaired Glycolysis Promotes Alcohol Exposure-Induced Apoptosis in HEI-OC1 Cells via Inhibition of EGFR Signaling" International Journal of Molecular Sciences 21, no. 2: 476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020476